Third Department of Pediatrics, University of Athens School of Medicine, “Attikon” University Hospital, Athens, Greece
Cerebral hypothyroidism, MCT8, MCT8 deficiency, Mental retardation, OATP1C1
INTRODUCTION
It has been more than 120 years since a committee of the Royal College of Physicians in London linked the paramount role of the thyroid gland to normal brain development.1 Currently, all developed countries and an increasing number of the developing ones have implemented the neonatal hypothyroidism screening test for early diagnosis of congenital hypothyroidism, since the latter is the most common cause of preventable mental retardation.2 Thyroid hormones (TH) are not only crucial for the maturation and function of the central nervous system (CNS), but also exert a broad range of effects on virtually all tissues.3
All thyroid hormone actions are mediated by the binding of 3,5,3’-triiodothyronine (T3) to specific nuclear receptors. T3 is the principal bioactive form of TH, whereas T4 acts as a prohormone to T3. T3 is produced by outer ring deiodination of T4, a reaction catalyzed by type 1 and type 2 deiodinases (D1 and D2, respectively) in a tissue-specific manner. D1 is mainly found in peripheral tissues, such as liver, kidney and the thyroid, and is responsible for the conversion of the majority of T4 to T3 in circulation. D2 is mainly found in the brain, pituitary, thyroid and skeletal muscle. The major function of D2 lies in the conversion of T4 to T3 in tissues, such as the brain and pituitary.4 Type 3 deiodinase (D3) converts T4 to the inactive reverse (r) T3 (3,3’,5’-triiododothyronine) and T3 to 3,3’-T2 (3,3’-diiodothyronine) by inner ring deiodination.5 D3 is expressed in the adult brain, in fetal brain and other fetal tissues, in the placenta as well as the pregnant uterus. Most of the effects of T3 are mediated by thyroid receptor (TR) regulation of target gene transcription in the nucleus. Therefore, TH has to cross cell membranes in order to be metabolized and to interact with its receptors.
Thyroid hormone receptors belong to the superfamily of ligand-dependent transcription factors. Alternative splicing of primary transcripts gives rise to four T3-binding TR isoforms (β1, β2, β3 and α1) and two TRs (α2 and α3) that do not bind T3. TRα1 is expressed predominantly in the heart, bone and brain, whereas TRβ1 is more abundant in the liver, kidney and thyroid. TRβ2 is limited to the pituitary, hypothalamus, retina and inner ear and TRβ3 is expressed mainly in the heart and kidney.
It was generally believed that TH passes through cell membranes passively. This notion was based on the fact that TH being lipophilic could easily cross the lipid-rich bilayer of the cell membrane.6 However, robust evidence has accumulated to suggest that the transport of TH into their target-cells is facilitated by saturable, stereospecific and energy-dependent specific transport proteins. Several TH transporters have been identified: these include Na-taurocholate co-transporting polypeptide (NTCP), different members of the Na-independent organic anion-transporting polypeptide (OATP) family and the L-type aminoacid transporters LAT1 and LAT2,7 and fatty acid translocate (CD36).8 The molecules that have been identified as transporters for thyroid hormones in the human belong mainly to two families: the organic anion-transporting peptides (OATP) and the monocarboxylate anion transporters (MCT).9
The aim of this review is to present the current knowledge on the most important thyroid hormone transporters.
ORGANIC ANION-TRANSPORTING PEPTIDES (OATPs)
OATPs represent a large family of homologous proteins that transport different iodothyronines, as well as various other organic compounds including bromosulfophthaleine, bile acids, bilirubin and its derivatives, estrogen conjugates, oligopeptide, etc. The corresponding genes are referred to as the SLCO (solute carrier OATP) family. OATPs are large proteins of 652 to 868 amino acids, containing 12 transmembrane domains, whose N- and C-terminal domains are located inside the cell. To date, 40 OATPs have been characterized in man, rats and mice. OATP1B1 and OATP1B3 are expressed specifically in the liver, OATP1C1 is expressed solely in the brain and testis and OATP1A2 is expressed in brain, liver and kidney.10,11
The genes encoding for OATP1A2, OATP1B1, OATP1B3 and OATP1C1 are located on chromosome 12p,12,13 forming a gene cluster together with a related pseudogene. The other OATP genes are located on individual chromosomes.
From the thyroid hormone transport point of view, OATP1C1 is the most interesting because it shows a high specificity and affinity for iodothyronines, especially for T4 and rT3, but not for T3. Furthermore, its localization preferentially in the endothelium of brain capillaries suggests that OATP1C1 is important for the transport of TH across the blood-brain barrier (BBB). OATP1C1 polymorphisms have been associated with fatigue and depression in hypothyroid patients, this underlining its crucial role for T4 transport across the BBB.14 To date, no association between OATP1C1 polymorphisms and TH levels has been documented.
Regarding OATP1B1, it enhances uptake of iodothyronine sulfates T4S, T3S, rT3S, but has little activity towards nonsulfated T4, T3, rT3.15 Like OATP1B1, OATP1B3 transports sulfated iodothyronines, but also has an affinity for rT3.16 Finally, OATP1A2 transports both iodothyronines and their sulfates, it is expressed in multiple tissues, among which liver, brain and kidney,12,17 and it could therefore play a role in the delivery of TH across the BBB. Actions of primary thyroid hormone transporters are depicted in Table 1 .
MONOCARBOXYLATE TRANSPORTER (MCT) FAMILY
The first members of the family were characterized as transporters of monocarboxylates, such as lactate, pyruvate and ketone bodies. To date, 14 members of the family have been recognized, but only in 6 of them has a ligand been identified. MCTs transport metabolites across plasma membranes with direction controlled by proton and metabolite concentration independently of energy input, but they may also function in subcellular membranes. They are implicated in leucocyte-mediated immunity, hypoxia induced cellular responses and partitioning of the energy supply in several tissues.18 Monocarboxylates are the ligands for MCT1-4, and aromatic aminoacid derivatives are ligands for MCT8 and MCT10. The proper expression and function of MCT1-4 in the plasma membrane depends on the oligomerization (e.g. heterotetramer formation) with the ancillary proteins basigin (CD147) or embigin (gp70).19,20 MCT6 has been shown to transport the diuretic bumatadine, but its natural ligands have not been identified yet.21 The functions of the orphan transporters MCT5-7, MCT9 and MCT11-14 remain to be determined. Herein, we focus on MCT8 and MCT10 as specific TH transporters.
MCT8
The human MCT8 gene resides on chromosome Xq13.2 and consists of six exons. It encodes a protein of 613 aminoacids with a molecular mass of 67 kD. The protein contains 12 hydrophobic predicted transmembrane domains that are characteristic of transporter proteins. The N-terminal of the protein contains a PEST domain, consisting mainly of proline, glutamate, serine and threonine repeats, suggesting that the protein may be rapidly or conditionally degraded. MCT8 is expressed in many tissues, such as liver, kidney, heart, brain, bone, placenta, lung and skeletal muscle. It is considered to be crucial for normal thyroid hormone-dependent development of the central nervous system, involving processes such as migration, dendritic outgrowth, formation of synapses and myelination.22 Moreover, it enhances T3 uptake into neuronal cells of the hippocampus, amygdalae, basal ganglia and cerebellar Purkinje cells, where transcript levels have been detected.23 Neurons are the major target cells for TH, expressing T3 receptors,24 D3 and MCT8.23 Loss-of-function mutations in MCT8 lead to a reduced or absent supply of T3 to neurons, resulting in impaired neurological development, as well as a reduced clearance of T3 by neuronal D3. Moreover, MCT8 represses neuronal cell growth by modulating cell proliferation in the developing brain, independently of T3 uptake, as this was shown by James et al.25 A schematic presentation of thyroid hormone transport in the CNS is shown in Figure 1.
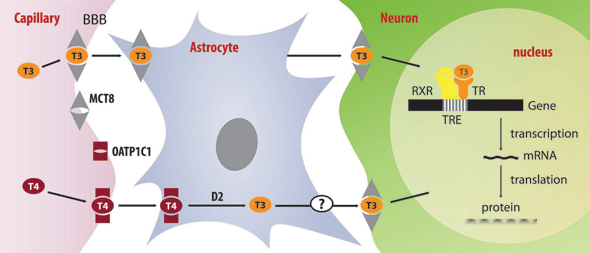
Figure 1. T4 is transported across the BBB by OATP1C1 and uptaken by astrocytes by an unknown transporter. In astrocytes, T4 is converted to T3 by the action of D2 and then T3 is released via an unknown mechanism. T3 uptake by neurons is facilitated by MCT8, which may also be involved in the transport of T3 through BBB (BBB: Blood-Brain-barrier; D2: type 2 deiodinase).
Although the cloning of MCT8 was achieved in 1994, it was not until 2003 that Friesema et al identified rat MCT8 as a specific thyroid hormone transporter by in vitro expression in Xenopus oocytes. A 10-fold increase in the uptake of the iodothyronines T4, T3, rT3 and 3,3’-T2 was observed. Furthermore, the amino acids tyrosine, tryptophan, phenylalanine and leucine did not compete with the transport of iodothyronines. Subsequent studies in mammalian cells indicated that net uptake of T4 and T3 was greatly stimulated by hMCT8 expression in mammalian cells in the presence of CRYM (cytoplasmic thyroid hormone-binding protein μ-crystallin).26
In addition, a recent study by Di Cosmo et al suggested a putative role of MCT8 in thyroid hormone secretion. Thyroid hormones are released across the basolateral membrane of thyroid follicular cells, adjacent to the capillary bed. MCT8 immunohistochemical localization at the basolateral membrane of thyrocytes and the impairment in the efflux of T4 from the thyroid gland in MCT8KO mice implies that MCT8 mediates, at least in part, the transport of thyroid hormones from the thyrocytes into the bloodstream. Of the studied thyroid hormone transporters, MCT8 is the most abundantly expressed, followed by MCT10 and, to a lesser extent, the secondary TH transporters Lat1 and Lat2.27 According to Nishimura and Naito, MCT8 is abundantly expressed in the human thyroid gland and therefore the molecular mechanism regulating thyroid hormone secretion in mice could also exist in humans.28
Dramatic evidence of the importance of MCT8 on thyroid hormone transport derives from the identification of a novel syndrome that affects boys and is characterized by severe neonatal hypotonia and developmental delay. Several patients have been described; the phenotype of the patients is that of severe mental retardation, profound hypotonia, spastic quadriplegia, these essentially being the phenotypic findings of the Allan-Herndon-Dudley (AHDS) syndrome.29 This syndrome was first described in 1944 and was among the first X-linked mental retardation syndromes to be recognized. Clinical manifestations in infancy and childhood in the AHDS syndrome are characterized by marked hypotonia (the infant is unable to hold his head up), weakness, generalized muscular atrophy and delay of developmental milestones. Although facial manifestations are not distinctive, the facies tends to be elongated, while some patients have myopathic facies and large, simple ears.
Head circumference is normal; however, a number of patients present acquired microcephaly usually manifesting after the 7th month of life. Generalized weakness is manifested by drooling, failure to walk independently or ataxia in those who manage to walk. Speech is dysarthric or absent altogether. Other neurological manifestations include spastic paraplegia with hyperreflexia, clonus and Babinski reflexes. In adult life hypotonia turns to spasticity. The hands exhibit dystonic and athetoid posturing and fisting. Cognitive development is severely impaired. Behavior tends to be passive, with little evidence of aggressive or disruptive behavior. Schwartz et al found that each of 6 large families with AHDS had mutations in the MCT8 gene. Moreover, in a large Brazilian family with AHDS, affected members were observed to have a mutation in the MCT8 gene.30 Clinical characteristics of MCT8 mutations are summarized in Table 2 .
THYROID ABNORMALITY AND NEUROLOGICAL DEFICITS RELATED TO MCT8 LOSS-OF-FUNCTION MUTATIONS
The underlying mechanisms leading to the combination of abnormal thyroid hormone levels and severe neurological deficits are not yet understood. Studies performed on skin fibroblasts in MCT8 deficient patients demonstrated impaired uptake of both T4 and T3 and increased D2 activity. However, these results only partially explained the thyroid phenotype. Therefore, several research groups undertook laboratory animal studies in order to elucidate the pathophysiologic mechanism responsible for the clinical phenotype in loss-of-function mutations of the MCT8 gene. Dumitrescu et al were the first to generate MCT8 knockout mice through homologous recombination of embryonic stem cells.31 The thyroid phenotype of male MCT8-null (Mct8-/y) mice was similar to the one observed in the hemizygous human male. Female MCT8-/- mice present the same thyroid phenotype as the males. However, the neurological abnormalities manifesting in the human male are not replicated in the mouse model. This has tentatively been attributed to species-specific regulation of intracellular thyroid hormone availability and thyroid hormone demands for normal function of the central nervous system. It seems that an increase in local T3 production is sufficient to provide certain neuronal cells, such as cerebellar Purkinje cells, with adequate amounts of TH thereby preventing serious neurological damage.32 At 6 weeks of life, male wild type (WT) and Mct8-/y mice had similar levels of TSH, but by the 16th week, TSH levels were slightly increased in Mct8-/y mice. When the authors tested pituitary sensitivity, 6-fold higher doses of L-T3 were needed to suppress serum TSH levels in the Mct8-/y mice, suggesting a central resistance due to impaired uptake of L-T3. Suppression of endogenous T4 resulted in similar serum T3 levels in both genotypes, indicating that T4 is responsible for the maintenance of the high T3 levels in the MCT8 deficient. When the authors examined tissue availability of T3, they found that liver T3 concentration was higher in Mct8-/y mice, reflecting the circulating T3 levels, whereas T3 concentration in the cerebrum was 10 times lower in Mct8-/y mice as a consequence of the reduced uptake of T3 in this tissue. Taken together, these data suggest that in MCT8 deficiency, hepatic thyrotoxicosis results from T3 uptake by the liver through MCT8 independent TH transporters. Increased hepatic T3 stimulates D1 enzymatic activity which increases the conversion of T4 to T3, resulting in further increase in T3. Increased D1 activity stimulates the metabolism of rT3 and, together with the consumption of T4 by 5’ deiodination, results in the low rT3 characteristic of MCT8 deficiency.
Recently, Trajkovic et al reported on the renal thyroid state in MCT8-null mice. They showed that renal D1 enzymatic activity is strongly up-regulated in the absence of MCT8, together with enhanced T4 and T3 renal uptake. Renal hyperthyroid status in MCT8 deficiency reflects either direct interference with the renal efflux of thyroid hormones or indirect activation of other renal thyroid hormone transporters.33 The same group also demonstrated that MCT8-null mice developed normally similarly to their WT or heterozygous littermates and did not present any motor deficits or brain weight differences compared to control mice.34 The authors explained the discrepancy between the neurological phenotype of humans and mice by the existence of other thyroid hormone transporters, like LAT2 in mice but not in humans that compensate for the absence of MCT8 in the animals, or the fact that in humans MCT8 facilitates the transport of other molecules important for brain development but not in mice. This hypothesis arises from the observation that LAT2 is expressed in neurons throughout murine brain development, while in human brain it is expressed in microglia during gestation, but not in neurons.35 In addition to hepatic and renal thyrotoxicosis, entry of T3 into the brain was impaired but did not affect significantly the CNS of the mice. In an effort to restore T3 levels, T3 production within the CNS was stimulated by increased D2 enzymatic activity in astrocytes, and inactivation of T3 was reduced by decreased D3 activities in neurons. As a result, sufficient T3 was produced locally so that cerebellar neurons were able to differentiate normally. Furthermore, the study showed that in MCT8-null mice the TRH transcript levels were strongly elevated in the hypothalamic PVN neurons to levels comparable to those observed in athyroid Pax8–/– mice. Injection of a high dose of T4 suppressed the increased TRH expression in MCT8-null animals, indicating that the PVN neurons in general still respond to locally produced T3, whereas peripheral injection of a supraphysiological dose of T3 did not affect TRH expression. These data suggest that the hypothalamic TRH expression in WT mice is not only dependent on T4 supply but is also influenced by the serum T3 levels. The authors concluded that the increase in TRH expression in the PVN of MCT8-null mice may not only reflect insufficient local T4 to T3 conversion but might also indicate an impaired uptake of T3 from the circulation, which may well represent the predominant mechanism underlying the central resistance to T3 of MCT8-null mice. Moreover, while the hypothalamus was in a hypothyroid state, the pituitary of mutant animals was found to be in a euthyroid state but, remarkably, the pituitary appeared not to have sensed adequately the high serum T3 levels. The mechanism underlying T3 insensitivity is unclear.
The current explanation for the pathophysiologic mechanism that leads to cerebral hypothyroidism, the resultant hypotonia and mental retardation, and the abnormal thyroid function tests is the following. MCT8 deficiency results in dramatic reduction of T3 uptake by the neurons in the brain and subsequent increase of T3 in the circulation. Circulating T3 is taken up by the liver and kidney by thyroid hormone transporters other than MCT8, producing thyrotoxicity. This leads to increase in D1 enzymatic activity and increased conversion of T4 to T3, resulting in T4 consumption and further increase in T3 levels. The low rT3 are accounted for by the increased rT3 metabolism due to increased D1 activity in liver and kidney and the low precursor (T4) levels. Given that the biochemical clue that leads to early diagnosis is the increased T3 concentration, it is suggested that in boys with early postnatal hypotonia it is important to determine TH levels, especially T3.36
THERAPEUTIC POTENTIALS IN PATIENTS WITH MCT8 LOSS-OF-FUNCTION MUTATIONS
Administration of L-thyroxine, even at high doses, alone or in combination with L-T3, was unsuccessful in improving the neurological condition of patients.37,38
Recently Di Cosmo et al tested the effect of 3,5-diiodothyropropionic acid (DITPA), a thyroid hormone receptor agonist, on thyroid hormone tissue levels in both WT and MCT8KO mice.39 DITPA is a TH agonist that binds with the same affinity to TR-a and TR-b. Its advantage compared to TH is its lower metabolic activity. In particular, DITPA induces mild stimulation of hepatic a-glycerolphosphate dehydrogenase activity when given to rats in equivalent doses as T4.40 Moreover, DITPA was used in randomized controlled trials for the treatment of heart failure and was found to reduce the serum TSH, T4, cholesterol, triglycerides, lipoproteins and body weight, without increasing heart rate or blood pressure.41
In Di Cosmo’s experiment, after nutritional and pharmaceutical induction of hypothyroidism, 2.5-fold more L-T4 and 6-fold more T3 than the physiological dose were required to normalize the serum TSH concentration in MCT8KO mice compared to the WT phenotype. On the other hand, serum TSH was normalized in both genotypes with the same dose of DITPA, suggesting that its action is MCT8 independent. It is also of note that DITPA contrary to L-T4 was able to normalize D3 expression in the MCT8KO mice, indicating that it can enter different brain cell types to act as a TH analog. Another point that must be taken into consideration is that administration of high doses of TH produces a thyrotoxic liver effect on both genotypes, while DITPA corrected all tested parameters of TH action in the brain of MCT8KO mice, without producing toxic liver effects, even when a higher dose was administered. This animal model has prompted researchers to examine the usefulness of DITPA in humans lacking MCT8 expression.
ALTERATION OF MCT8 EXPRESSION DURING CRITICAL ILLNESS
During critical illness, the circulating and tissue TH levels are low and this is called the low T3 syndrome or non-thyroidal illness syndrome. Reduced expression of TRH in the hypothalamus appears to play a key role in the prolonged phase of critical illness in an attempt to compensate for low circulating TH levels.42 These changes are considered to be adaptive, effecting postponement of anabolism and at the same time activation of the immune response.43 Regarding iodothyronine deiodination during critical illness, there is a cytokine-mediated decrease in hepatic D1 expression and activity.44,45 D1 activity correlates positively with the T3/rT3 ratio, with the latter being a marker of tissue hypoperfusion in critically ill patients.46 In contrast, although D2 adapts appropriately to the low T3 levels, it does not seem to contribute to the “low T3 syndrome” in the prolonged phase of critical illness.47 Finally, as regards D3, Peeters et al demonstrated that D3, normally absent in adult tissue, is reactivated in liver and muscle of critically ill patients.48 Tissue response to low circulating T3 levels involves increasing expression of TH transporters, this facilitating cellular uptake. In the study on the role of TH transporters in critical illness, Mebis et al found that the expression of MCT8 is up-regulated in skeletal muscle and liver of prolonged critically ill patients, whereas MCT10 expression is not altered. Moreover, the authors reported an inverse correlation between circulating TH and MCT8 gene expression in skeletal muscle, suggesting that patients with the lowest serum T3 and T4 levels show the highest up-regulation of MCT8 mRNA. Inversely, in animal models, MCT8 expression is down-regulated when circulating TH levels are elevated and vice versa.47 These data suggest that in critically ill patients, TH transporter alteration is the result of compensation to hypothyroidism.
MCT10
The human MCT10 gene is located on chromosome 6q21-q22. It has the same structure as the hMCT8 gene, coding for a protein of 515 amino acids with amino acid sequence homologous to that of MCT8, and it contains 12 putative transmembrane domains. MCT10 and MCT8 have an amino acid identity of 49%,46 which is the highest in the transmembrane domains (TMD) and the lowest in the N-terminal and C-terminal domains, both of which are located intracellularly.16 MCT10 is expressed in different human and rodent tissues, in particular intestine, liver, skeletal muscle, heart and placenta, acting as a T-type aminoacid transporter which facilitates cellular uptake and efflux of aromatic aminoacids, such as Trp, Phe, Tyr and 3,4-dihydroxyphenylalanine (DOPA).32 MCT10 is more important for the export than for the import of aromatic amino acids;50,51 it is speculated that cellular uptake of thyroid hormone is driven by cellular efflux of aromatic amino acid through this transporter.16 Like MCT8, MCT10 induces iodothyronine uptake, which is augmented in the presence of CRYM (cytoplasmic thyroid hormone-binding protein μ-crystallin), and also stimulates the deiodination of iodothyronines in cells cotransfected with the different deiodinases through an increase in the intracellular availability of these subtrates.49
THYROID HORMONE TRANSPORT IN HYPOTHALAMUS AND PITUITARY
Our knowledge of the pathway(s) by which transport of the thyroid hormones to the hypothalamus or pituitary takes place is still very limited, this problem being compounded by the fact that the central feedback mechanism of the TH has not as yet been examined extensively.
MCT8 has been found to be expressed in the human hypothalamus. Areas of the hypothalamus that are densely stained are the paraventricular nucleus (PVN), the suprachiasmatic nucleus (SON) and the infundibular nucleus (IFN), but the areas in which MCT8 is expressed most prominently are the perifornicular and the lateral hypothalamus (LHA).
It is well known that TRH neurons, predominantly those in PVN, play a key role in the neuroendocrine regulation of thyroid hormone. PVN also expresses all four TR isoforms and D3. Co-expression of MCT8, TRs and D3 with TRH suggests their possible involvement in TH feedback on the hypothalamus. A candidate pathway requires uptake of T4 by astrocytes across the BBB, a process facilitated by OATP1C1, which indeed exhibits considerably higher transport capacity for T4 than for T3 after release from endothelial cells, probably also mediated by OATP1C1.52 T4 is transformed to T3 by the enzymatic action of D2 in the astrocytes and subsequently T3 is transported to neurons by MCT8. Although MCT8 is predominantly expressed in neurons, after studying the expression of MCT8 mRNA in the murine central nervous system, Heuer et al noted that tanycytes, specialized glial cells lining the third ventricle, were labelled for MCT8. Because these cells are mainly involved in the transport of hormones or other substances into the hypothalamus, this MCT8 localization might offer a possible scenario for the entrance of TH into hypothalamic regions.23 Regarding TH transport through BBB, although previous studies in mice have shown that MCT8 mRNA is primarily expressed in neurons and the choroid plexus, Roberts et al studied the BBB expression of the TH transporters MCT8 and OATP1C1 in human, rat and mouse and demonstrated that MCT8 mRNA and protein are expressed in microvessels of the BBB in human, mouse and rat brain, while OATP1C1 mRNA and protein are expressed in BBB microvessels of human brain to much lesser extent than that of mouse and rat.53 These findings suggest that an alternative explanation for the milder clinical phenotype of the MCT8-null mice may be the increased BBB-OATP1C1 mediated TH transport. Additional studies have shown that the main restriction to T3 action in the absence of MCT8 is at the level of the blood-brain barrier, while TH transport role of MCT8 in the plasma membrane of neurons, at least in the striatum and cerebellum, seems to be minimal.54
In the human anterior pituitary MCT8 expression was found exclusively in folliculostellate (FS) cells. Pituitary FS cells are usually located between the secretory cells in the anterior pituitary. They form a functional network of interconnecting cells and they produce many paracrine factors that act on hormone-producing pituitary cells. FS cells, under certain conditions, may serve as a pool of progenitor cells capable of differentiating into specialized endocrine cells. The observed both MCT8 and D2 immunoreactivity in these cells suggests that a subset of FS cells produce T3 from T4, stimulated by TSH via TSH receptor binding and also that they may be able to transport T3 to other cells. The mechanism by which T4 is taken up by FS cells is not known; however, it has been proposed that MCT8 may have such a role. Also, it has been suggested that MCT8 may be involved in the transport of T3 from FS cells. It is very interesting that MCT8, deiodinases and TRs are not co-expressed, this implying the existence of yet another transporter that is involved in the transport of T3 to hormone secreting cells.
CONCLUSION
From the several transporting peptides that have been identified to date, the most important molecules that have been shown to act, in the human, as thyroid hormone transporters are OATP1C1 and MCT8. The former is important for the transport of thyroid hormones across the blood-brain barrier (BBB) and the latter acts as a specific transporter for T3. Mutations of the MCT8 result in dramatic reduction of T3 uptake by the neurons in the brain causing cerebral hypothyroidism. The clinical manifestations of MCT8 deficiency include marked hypotonia, weakness, generalized muscular atrophy and delay of developmental milestones. Thyroid-function tests in MCT8 deficiency are characterized by a high serum total and free T3, low total and free T4, as well as low rT3. MCT8 resides on the X chromosome, therefore, in boys with early postnatal hypotonia, it is important to determine thyroid hormone levels, especially T3. Until the time of the writing of this review there is no effective treatment for MCT8 deficiency. However, recently the administration of an MCT8 independent thyroid hormone receptor agonist DITPA in MCT8 knock-out mice restored thyroid hormone abnormality, suggesting its future potential use in the treatment of human MCT8 deficiency.
REFERENCES
1. Ord WM, 1898 The Bradshaw Lecture on Myxoedema and Allied Disorders: Delivered before the Royal College of Physicians. Br Med J 2: 1473-1479.
2. Bhatara V, Sankar R, Unutzer J, Peabody J, 2002 A review of the case for neonatal thyrotropin screening in developing countries: the example of India. Thyroid
12: 591-598.
3. Brent GA, 1994 The Molecular Basis of Thyroid Hormone Action. N Eng J Med 331: 847-853.
4. Yen PM, 2001 Physiological and molecular basis of thyroid hormone action. Physiol Rev 81: 1097-1142.
5. Tu HM, Legradi G, Bartha T, Salvatore D, Lechan RM, Larsen PR, 1999 Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the
rat central nervous system and its regulation by thyroid hormone. Endocrinology 140: 784-790.
6. Hennemann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ, 2001 Plasma membrane transport of thyroid hormones and its role in thyroid hormone
metabolism and bioavailability. Endocr Rev 22: 451-476.
7. Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ, 2003 Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem 278: 40128-40135.
8. van der Deure WM, Peeters RP, Visser TJ, 2007 Genetic variation in thyroid hormone transporters. Best Pract Res Clin Endocrinol Metab 21: 339-350.
9. Friesema EC, Jansen J, Milici C, Visser TJ, 2005 Thyroid hormone transporters. Vit Horm 70: 137-167.
10. Hagenbuch B, 2007 Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrinol Metab 21: 209-221.
11. Svoboda M, Riha J, Wlcek K, Jaeger W, Thalhammer T, 2011 Organic anion transporting polypeptides (OAT Ps): regulation of expression and function. Curr Drug Metab 12: 139-153.
12. Kullak-Ublick GA, Hagenbuch B, Stieger B, et al, 1995 Molecular and functional characterization of an organic anion transporting polypeptide cloned from human liver. Gastroenterology 109: 1274-1282.
13. Tamai I, Nezu J, Uchino H, et al 2000 Molecular identification and characterization of novel members of the human organic anion transporter (OAT P) family. Biochem Biophys Res Commun 273: 251-260.
14. van der Deure WM, Appelhof BC, Peeters RP, et al, 2008 Polymorphisms in the brain-specific thyroid hormone transporter OAT P1C1 are associated with fatigue and depression in hypothyroid patients. Clin Endocrinol 69: 804-811.
15. van der Deure M, Hansen PS, Peeters RP, et al, 2008 Thyroid hormone transport and metabolism by organic ion transporter 1C1 and consequences of genetic variation. Endocrinology 149: 5307-5314.
16. van der Deure WM, Peeters RP, Visser TJ, 2010 Molecular aspects of thyroid hormone transporters, including MCT8, MCT10, and OAT Ps, and the effects of genetic variation in these transporters. J Mol Endocrinol 44: 1-11.
17. Gao B, Hagenbuch B, Kullak-Ublick GA , Benke D, Aguzzi A, Meier PJ, 2000 Organic ion-transporting polypeptides mediate transport of opioid peptides across
blood-brain barrier. J Pharmacol Exp Ther 294: 73-79.
18. Merezhinskaya N, Fishbein WN, 2009 Monocarboxylate transporters: past, present, and future. Histol Histopathol 24: 243-264.
19. Halestrap AP, Meredith D, 2004 The SLC16 gene family from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch 447: 619-628.
20. Wilson MC, Meredith D, Fox JE, Manoharan C, Davies AJ, Halestrap AP, 2005 Basigin (CD147) is the target for organomercurial inhibition of monocarboxylate
transporter isoforms 1 and 4: the ancillary protein for the insensitive MCT2 is EMBIGIN (gp70). J Biol Chem 280: 27213-27221.
21. Myrakami Y, Kohyama N, Kobayashi Y, et al, 2005 Functional characterization of human monocarboxylate transporter 6 (SLC16A5). Drug Metab Dispos 33: 1845-1851.
22. Bernal J, 2005 Pathophysiology of thyroid hormone deficiency during fetal development. J Pediatr Endocrinol Metab 18: Suppl 1: 1253-1256.
23. Heuer H, Maier MK, Iden S, et al, 2005 The monocarboxylate transporter 8 linked to psychomotor retardation is highly expressed in thyroid hormone-sensitive neuronal populations. Endocrinology 146: 1701-1706.
24. Leonard JL, Farwell AP, Yen PM, Chin WW, Stula M, 1994 Differential expression of thyroid hormone receptor isoforms in neurons and astroglial cells. Endocrinology
135: 548-555.
25. James SR, Franklyn JA, Reaves BJ, et al, 2009 Monocarboxylate transporter 8 in neuronal cell growth. Endocrinology 150: 1961-1969.
26. Friesema EC, Kuiper GG, Jansen J, Visser TJ, Kester MH, 2006 Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in
intracellular metabolism. Mol Endocrinol 20: 2761-2772.
27. Di Cosmo C, Liao X-H, Dumitrescu AM, Philp NJ, Weiss RE, Refetoff S, 2010 Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin
Invest 120: 3377-3388.
28. Nishimura M, Naito S, 2008 Tissue-specific mRNA expression profiles of human solute carrier transporters superfamilies. Drug Metab Pharmacokinet 23: 22-24.
29. Schwartz CE, May MM, Carpenter NJ, et al, 2005 Allan- Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet 77: 41-53.
30. Maranduba CM, Friesema EC, Kok F, et al, 2006 Decreased cellular uptake and metabolism in Allan-Herndon-Dudley syndrome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter. J Med Genet 43: 457-460.
31. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S, 2004 A novel syndrome combining thyroid and neurological abnormalities is associated with mutations
in a monocarboxylate transporter gene. Am J Hum Genet 74: 168-175.
32. Heuer H, Visser TJ, 2009 Minireview: Pathophysiological importance of thyroid hormone transporters. Endocrinology 150: 1078-1083.
33. Trajkovic-Arsic M, Visser TJ, Darras VM, et al, 2010 Consequences of monocarboxylate transporter 8 deficiency for renal transport and metabolism of thyroid
hormones in mice. Endocrinology 151: 802-809.
34. Trajkovic M, Visser TJ, Mittag J, et al, 2007 Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest 117: 627-635.
35. Wirth EK, Roth S, Blechschmidt C, et al, 2009 Neuronal 3?, 3, 5- Triiodothyronine( T3) uptake and behavioral phenotype of mice deficient in MCT8, the neuronal T3
transporter mutated in Allan-Herndon-Dudley syndrome. J Neuroscience 29: 9439-9449.
36. Papadimitriou A, Dumitrescu AM, Papavasiliou A, Fretzayas A, Nicolaidou P, Refetoff S, 2008 A novel monocarboxylate transporter 8 gene mutation as a cause
of severe neonatal hypotonia and developmental delay Pediatrics 121: 199-202.
37. Biebermann H, Ambrugger P, Tarnow P, von Moers A, Schweizer U, Grueters A, 2005 Extended clinical phenotype, endocrine investigations and functional studies
of a loss-of-function mutation A150V in the thyroid hormone specific transporter MCT8. Eur J Endocrinol 153: 359-366.
38. Zung A, Visser TJ, Uitterlinden AG, Rivadeneira F, Friesema EC, 2011 A child with a deletion in the monocarboxylate transporter 8 gene: 7-year follow-up and effects of thyroid hormone treatment. Eur J Endocrinol 165: 823-830.
39. Di Cosmo C, Liao XH, Dumitrescu AM, Weiss RE, Refetoff S, 2009 A thyroid hormone analog with reduced dependence on the monocarboxylate transporter 8 for
tissue transport. Endocrinology 150: 4450-4458.
40. Pennock GD, Raya TE, Bahl JJ, Goldman S, Morkin E, 1992 Cardiac effects of 3,5- diiodothyroninepropionic acid, a thyroid hormone analog with inotropic selectivity.
J Pharmacol Exp Ther 263: 163-169.
41. Morkin E, Pennock GD, Spooner PH, et al, 2002 Clinical and experimental studies on the use of 3,5- diiodothyropropionic acid, a thyroid hormone, in heart failure.
Thyroid 12: 527-533.
42. Mebis L, van den Berghe G, 2009 The hypothalamuspituitary- thyroid axis in critical illness. Neth J Med 67: 332-340.
43. Utiger RD, 1980 Decreased extrathyroidal triiodothyronine production in non-thyroidal illness: benefit or harm? Am J Med 67: 807-810.
44. Hennermann G, Docter R, Friesema EC, de Jong M, Krenning EP, Visser TJ, 2001 Plasma membrane transThyroid hormone transporters 279
port of thyroid hormones and its role in thyroid hormone metabolism and bioavailability. Endocr Rev 22: 451-476.
45. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR, 2002 Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 23: 38-89.
46. Fliers E, Alkemade A, Wiersinga WM, 2001 The hypothalamic- pituitary-thyroid-axis in critical illness. Best Pract Res Clin Endocrinol Metab 15: 453-464.
47. Mebis L, Paletta D, Debaveye Y, et al, 2009 Expression of thyroid hormone transporters during critical illness. Eur J Endocrinol 61: 243-250.
48. Peeters RP, Wooters PJ, Kaptein E, van Toor H, Visser TJ, van den Berghe G, 2003 Reduced activation and increased inactivation of thyroid hormone in tissues
of critically ill patients. J Clin Endocrinol Metab 88: 3202-3211.
49. Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ, 2008 Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate
transporter 10. Mol Endocrinol 22: 1357-1369.
50. Ramadan T, Camargo SM, Summa V, et al, 2006 Basolateral aromatic aminoacid transporter TAT (Slc16a10) functions as an efflux pathway. J Cell Physiol 206:
771-779.
51. Ramadan T, Camargo SM, Herzog B, Bordin M, Pos KM, Verrey F, 2007 Recycling of aromatic aminoacids via TAT 1 allows efflux of neutral amino acids via LAT 2-
4f2hc exchanger. Pflugers Arch 454: 507-516.
52. Sugiyama D, Kusuhara H, Taniguchi H, et al, 2003 Functional characterization of the rat brain-specific organic ion transporter (OAT P14) at the blood-brain barrier. J Biol Chem 278: 43489-43495.
53. Roberts LM, Woodford K, Zhou M, et al, 2008 Expression of the thyroid hormone transporters, monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology 149: 6251-6261.
54. Ceballos A, Belinchon MM, Sanchez-Mendoza E, et al, 2009 Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3?-triiodo-L-thyronine. Endocrinology 150: 2491-2496.
Corresponding Author:
Anastasios Papadimitriou, MD, 3rd Department of Pediatrics,
University of Athens School of Medicine, “Attikon”
University Hospital, 1 Rimini Street, 124 64 Athens, Greece,
Tel: +30-210-5832046, Fax:+30-210-5832229, e-mail:anpapad@med.uoa.gr
Received 31-08-11, Revised 08-09-11, Accepted 25-09-11