1. 引言
随着基因组测序工作的完成,蛋白质组的相关工作也随之展开,蛋白质的研究有利于人类对疾病的深入了解和相关药物研发,其中的重点工作便是蛋白质的结构研究。蛋白质结构可划分为一级结构、二级结构、三级结构和四级结构,其中具有生物活性的是三级结构。获取三级结构最直接的方法是使用X射线和核磁共振进行观察 [1],但是这种方式效率低,成本高,因此从1951年Pauling和Corey预测了蛋白质多肽骨架的螺旋和片状构象开始,研究者便展开了对蛋白质结构的预测工作。尽管蛋白质结构的信息隐藏在氨基酸序列中,但直接利用氨基酸序列进行三级结构预测难度非常大,因此研究者通常先进行蛋白质二级结构预测,基于该结果做进一步的研究。
早期的蛋白质二级结构预测使用统计学方法和启发式规则 [2],支持向量机 [3] [4],贝叶斯分类算法,马尔可夫模型 [5],前馈神经网络 [6] [7] 等均被应用在了蛋白质二级结构预测中。近年来,随着深度学习方法在自然语言处理、机器视觉和语音识别等方向取得了巨大的进展,生物信息学领域也开始了对深度学习方法的广泛使用 [8],深度学习通过多层特征变换,可以更好的刻画出数据的隐藏信息,捕捉氨基酸的局部和长距离相互作用。例如Fang等提出了MOUFOLD方法,使用了由两个嵌套的可进行卷积操作的初始模块、卷积层以及完全联通的致密层组成的Deep3I网络(Deep Inception-Inside-Inception Networks, Deep3I)进行蛋白质二级结构预测 [9]。WANG等结合了深度卷积神经网络(Deep Convolutional Neural Networks, DCNN)和条件神经场(Conditional Neural Fields, CNF),提出名为Raptor X的预测方法 [10]。SPOT-1D在Spider [11] 基础结合了双向长短时记忆循环网络(Bidirectional long-short-term memory recurrent network,Bi-LSTM)和残余卷积网络(Residual Convolutional Networks, ResNets)用来识别和传播整个序列中的短期和长期依赖关系 [12]。值得注意的是,上述方法中MOFOLD,Raptor X,Spider3和SPOT-1D对特征输入均有细致的设计,MOFOLD和Spider3网络输入为氨基酸理化性质、PSSM和隐马尔可夫特征,Raptor X网络输入为PSSM+21个元素的二进制向量,SPOT-1D在Spider3的特征基础上增加了SPOT-Contact预测接触图。另外,大数据集的使用也为模型更好的提取隐藏特征起到了重要的作用。
在本实验中,我们采用双向LSTM作为训练模型,为了充分发挥LSTM网络对序列数据的处理能力,对训练集取消了滑动窗口限制,以捕捉氨基酸长距离相互作用。同时对训练集特征重新进行了设计,在常用的PSSM基础上,增加了蛋白质42基团的新编码特征,经过大数据集的训练,网络模型可以充分的提取序列隐藏特征。这种基于大数据和新编码方式的模型预测能力在公共测试集CASP9,CASP10,CASP11和CASP12中进行了评估实验,Q3准确率分别达到了85.74%,86.83%,84.73%和83.79%。
2. 实验模型
2.1. 模型结构
LSTM在自然语言处理领域中应用广泛,对序列数据的分类有良好的实验效果,蛋白质二级结构预测是在给定的蛋白质序列下对每一个氨基酸的结构归属做出分类,同样可以看作对序列数据的分类问题,因此本次实验选择了双向LSTM作为训练模型。
模型训练示意图如图1所示,为了充分发挥LSTM对长序列数据的学习优势,实验未设置滑动窗口,而是将训练集拆分成小批量并补零填充序列,使它们具有同一小批量中最长序列的长度。为了防止训练过程中添加过多填充,在训练集输入网络前,根据蛋白质长度对数据进行了排序。在结构预测中,每一个氨基酸的输出状态不仅与它之前的序列信息有关,位置靠后的部分同样会产生影响,而双向LSTM可以看作两层标准的LSTM网络,分别以开头和结尾作为输入,可以为输出层提供输入序列中每一个氨基酸完整的上下文信息,真正考虑到前后位置的相互作用。

Figure 1. Schematic diagram of network structure and bidirectional LSTM training process
图1. 网络结构和双向LSTM训练示意图
2.2. 模型原理
双向LSTM可看作由循环神经网络(Recurrent Neural Network, RNN)逐步演化而来的网络结构,RNN可较好的针对序列变化的数据进行训练 [13],循环神经网络具有重复的模块单元,展开后的RNN单元如图2所示。
其中
,t,
表示序列的时间位置,X表示输入的样本,X经过简单的激活函数得到输出Y和记忆数据S,数学表达式为:
(1)
(2)
W为输入的权重,U表示t时刻下的输入样本权重,O表示输出样本的权重。
传统的RNN面对长序列数据会产生梯度消失和梯度爆炸问题,LSTM的提出有效的降低了这些情况的风险。在RNN的基础上对循环单元做了细致的设计,增设了三种门结构 [14],这种设计方式使LSTM单元具备将重要的信息进一步记忆,将不重要信息适当丢弃的能力。标准LSTM单元示意图如图3所示。
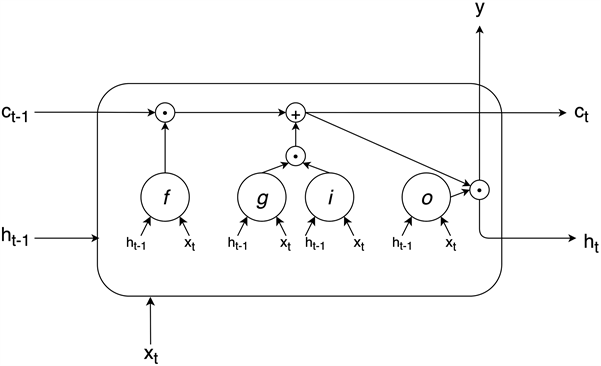
Figure 3. Standard LSTM unit schematic
图3. 标准LSTM单元示意图
单元输入是t时刻下的样本
和
时刻单元的输出
(也称为隐藏状态),其输出可作为下一个单元的输入
以及本单元的输出y。C叫做细胞状态,相比于隐藏状态它的变化很少,通过遗忘门f,候选门g和输入门i进行信息更新,信息在这条线路中传输只有少量的相乘和相加操作,梯度更加稳定,因此LSTM对远距离的相互作用有更强大的学习能力。LSTM单元的数学表达式如下:
(3)
(4)
(5)
(6)
其中,W为输入权值,R为递归权值,b为偏置,f代表遗忘门,g代表候选门,i代表输入门,o代表输出门。首先公式3说明隐藏状态
和输入
一同输入至遗忘门,通过激活函数
输出
,0代表全部遗忘,1代表全部记忆。公式4表示候选门产生候选向量,公式5代表输入门来决定更新哪些值,两者的结果进行相乘操作,然后相加操作更新至细胞状态
如公式7所示。最后通过输出门决定最后的输出内容,可作为当前单元的隐藏状态
或输出y如公式8所示。
(7)
(8)
最后一层Bi-LSTM后是全连接层,全连接层中的每一个神经元都与上一层的神经元连接,将Bi-LSTM学习到的高维特征进行整合,最后经过Softmax分类器进行分类,计算每一个氨基酸所在的C、E、H三种结构的概率,最终完成分类。
3. 实验数据
3.1. 数据集
本次实验的训练集为CULLPDB数据集 [15] 共15,125条蛋白质,同源性低于25%,涵盖类别丰富。我们剔除了同测试集中重复的蛋白质,最终训练集数量为14,199条蛋白质。CASP数据集为全球蛋白质结构预测实验采用的公共测试集,我们选取了其中的4组作为测试集,分别为122条蛋白质的CASP9,99条蛋白质的CASP10,81条蛋白质的CASP11和19条蛋白质的CASP12。
3.2. 输入特征
实验对网络的输入特征进行了新的设计,一共有62维,包含20维PSSM矩阵和42维的基团特征。位置特异性打分矩阵(Position-Specific Scoring Matrix, PSSM)富含生物进化进信息,极大提高了蛋白质二级结构预测的精度,是一种广泛使用的特征信息 [16],本次实验的PSSM是通过多序列对比nr数据库中的蛋白质,设置PSI-BLAST参数阈值为0.001和3次迭代生成。PSSM形式为20*L矩阵,20是特征维度,L代表了不同蛋白质的长度。基团是指蛋白质序列中氢原子和非氢原子之间,或者非氢原子之间形成的官能团,具有稳定的结构特点,根据基团中原子相互连接的共价键进行分类,生成基团信息表 [17],共42种基团编号,每个氨基酸都可以用长度42的二进制编码唯一表示,氨基酸出现在哪些基团中就将相应位置用1表示,其余位置为0,最终氨基酸的编码结果如表1所示。相比于传统的正交编码,基团编码包含了稳定的氨基酸结构信息,对于氨基酸的表示是直观的,用于蛋白质二级结构预测具有更好的效果 [18]。因此模型的输入为一条完整的蛋白质序列,形式为62*L特征矩阵(L为序列长度),模型的输出为该序列对应长度的C、E、H序列。

Table 1. Amino acid radical group coding table
表1. 氨基酸基团编码表
4. 实验结果与分析
针对本次蛋白质二级结构预测,我们对模型中的超参数设置了多组对比试验,分别进行了Bi-LSTM层数和隐藏单元的调整,模型学习率的调整,Dropout值的调整以及正则化系数的调整。实验调参过程体现均以CASP10作为测试集。
4.1. 网络层数和隐藏单元
在实验模型中,Bi-LSTM层的数量会直接影响到网络的复杂程度以及网络的学习能力,不合理的隐藏单元数也会导致模型过拟合,在网络结构上我们希望设计一个复杂程度合理的模型,在适当的训练时间下拥有良好的泛化性。因此我们针对网络层数和隐藏单元数进行了调整实验,U1代表第一层Bi-LSTM的隐藏单元数,U2代表第二层Bi-LSTM的隐藏单元数。
Table 2. Experimental results of setting up one layer of Bi-LSTM
表2. 设置一层Bi-LSTM的实验结果
Table 3. Experimental results of setting up two layers of Bi-LSTM
表3. 设置两层Bi-LSTM的实验结果
从表2可以看出,只设置一层Bi-LSTM的情况下,网络的学习能力是较为欠缺的,隐藏单元的改变并不会对实验结果有太大的影响,因此需要增加网络层数,以第一层每一时间步的隐藏单元输出作为下一层相应时间步隐藏单元的输入,网络进行了更加深层次的特征学习。实验结果表明,这种操作对结果有一定的提升,但隐藏单元数的确定便十分重要,由于是双层网络,因此两层隐藏单元数之间的选择是相互影响的,如表3所示,单纯的使用第一层网络下最好的方案去确定第二层的单元数,得到的结果并不是最优解,因此我们需要找到最优的组合,结果表明,两层隐藏单元为1000的组合下,可到的最优组合。考虑到实验的可行性和模型复杂度,最终的网络结构确定为两层Bi-LSTM,隐藏单元都别为1000。
4.2. 学习率
网络模型采用了Adam优化算法,相比于传统的随机梯度下降方式,Adam通过计算梯度的一阶矩估计和二阶矩估计为不同的参数设计独立的自适应性学习率,适合解决含大规模数据和参数的优化问题 [19]。学习率的初始值会对结果产生较大影响,因此本组实验为确定合适的初始学习率,实验结果如图4所示,首先以10倍缩量进行粗调节,然后在合适的量级下进行细调节,最终确定为0.0006,Q3结果可达84.64%,训练过程中学习率以每4个epoch进行10倍缩放的方式训练。

Figure 4. Experimental results at different learning rates
图4. 不同学习率下的实验结果
4.3. Dropout
由于模型采用的双层Bi-LSTM的设计,同时加入了大量的隐藏单元,模型可能会存在过拟合且训练时间过长的问题,因此在每一层后引入了Dropout,以一定的概率使部分神经元失活,我们将两层Dropout设置为相同值进行同步调整,从图5中可以看出,在数据集CASP10上,两个Dropout值同为0.2时,结果最优,且明显高于其他值,分析原因可能是因为Dropout在0.2的状态下失活神经元的概率更符合本模型的训练方式,这样网络模型可以获得更好的泛化性。表4中D1代表第一层Dropout,D2代表第二层Dropout,可以看出,设置两层Dropout是更好的设计方案。

Figure 5. Experimental results of different Dropout values
图5. 不同Dropout值的实验结果
Table 4. The impact of different amounts of Dropout
表4. 不同数量Dropout的影响
4.4. 正则化系数
为防止模型过拟合,实验过程中加入了参数正则化方式,选择L2正则化,在损失函数中增加惩罚项,L2正则化的惩罚项是指权值向量w中各个元素的平方和然后再求平方根,可表示为
,调节
系数控制惩罚项的大小。实验结果表明,调整L2正则化系数,对实验结果有进一步的提升,在CASP10上结果可达86.83%,实验结果如图6所示,L2正则化系数最终确定为0.000005。

Figure 6. Experimental results of different L2 regularization coefficients
图6. 不同L2正则化系数的实验结果
经过上述的参数调整,网络最终确定的结构如表5所示。
Table 5. Finalized network structure parameters
表5. 最终确定的网络结构参数
4.5. 预测评估
1983年,Wolfgang Kabsch提出DSSP,将二级结构被划分为α螺旋(H)、β折叠(E)、310螺旋(G)、β桥(B)、π螺旋(I)、转角(S)、β转角(T)和无规则卷曲(C)8种结构 [20],实验中将上述8态结构以“GHI→H”,“BE→E”和“其他结构→C”的规则转为3态结构,进行3态二级结构预测。CULLPDB中的DSSP来自于蛋白质的PDB文件,评估标准为Q3精度 [3] [7] [8] [10] [11],Q3为正确预测的氨基酸数占所有氨基酸的比例,计算公式如下:
将测试集输入模型,得出预测数据,同真实DSSP进行对比。其中,QC 为正确预测的转角数,QE为正确预测的折叠数,QH为正确预测的螺旋数,S为总的氨基酸数。文献 [10] 在五折交叉验证下,使用CULLPDB数据集获得了73.2%的八态分类结果。文献 [21] 使用TR9993数据集进行十折交叉验证,Q3准确率为72.42%。文献 [22] 使用25PDB数据集进行三折交叉验证,Q3准确率可达80.18%。表6为本实验使用CULLPDB做十折交叉验证的结果,为了保证不同长度的蛋白质均匀分布,在划分训练集和测试集之前未将CULLPDB根据蛋白质长度排序,数据集均分为10份,每次取9份作为训练集,1份作为测试集,十次训练的数据集数量见表6,十折交叉验证的平均值可达83.08%,比CASP测试集结果低0.7%~2.7%,结果表明,实验避免了数据集划分不合理而出现过拟合现象。
Table 6. 10-fold cross validation of CULLPDB
表6. CULLPDB十折交叉验证
表7列出了在测试集CASP9,CASP10,CASP11和CASP12 [23] [24] 上Q3准确率以及转角、折叠和螺旋结构的预测准确率,其中Q3的识别率分别有85.74%,86.83%,84.73%和83.79%。值得注意的是,模型对折叠E的预测准确值较低,分析后发现实验选取的CASP数据有部分蛋白质没有转角结构,相比于预测效果更加稳定的螺旋和转角结构,总体基数较少。为了验证加入42基团特征是有效的,我们进行了对比试验,如图7所示,在最终确定的网络结构下,分别以PSSM和PSSM+42基团为输入。单纯使用PSSM作为输入特征,在CASP9-12上结果分别为85.17%,85.83%,83.54%和82.71%。实验结果表明,在四组CASP测试集上,加入42基团特征对模型分类能力有进一步的提升,Q3提升约1%。
Table 7. Experimental results under the CASP test set
表7. CASP测试集下实验结果

Figure 7. The influence of two characteristics of PSSM and PSSM+42 radical groups on the model
图7. PSSM和PSSM+42基团两种特征对模型的影响
实验选取了SPINE-X [25],SSpro [26],PSIPred,DeepCNF和MUFOLD五种预测方法,在测试集CASP10,CASP11和CASP12上进行对比,其中SPINE-X采用的多步神经网络,SSpro模型是双向递归神经网络,PSIpred采用的两层前馈神经网络,Jpred使用两层来自SNNS神经网络包的人工神经网络,MUFOLD采用深度神经网络(五种方法结果均摘自文献 [2] )。对比结果如表8所示。相比与上述模型方法,本模型使用双向长短时记忆循环网络和多维特征融合的预测方式有效的提高了3态蛋白质二级结构预测结果。
Table 8. Comparison of prediction results
表8. 预测结果对比
5. 结论
蛋白质二级结构预测在蛋白质结构研究领域意义重大,不断有新的模型和方法被提出,本文采用了双向长短时记忆循环网络,在蛋白质序列特征构建上融合了新的42基团编码方式,数据包含更多特征,同时使用大数据集进行模型训练,取消了滑动窗口的设计,最大化的捕捉氨基酸之间的长距离相互作用,双向LSTM又可以考虑到氨基酸序列前后文的影响,提高了3态二级结构的预测结果,相比于多种深度学习方法融合的预测方式,模型更加简洁有效,但取消滑动窗口的训练方式人为增加了训练集的噪声,下一步可进一步改进多维特征构建方式,构建更加有效的特征编码,在网络训练过程方面参数的自动优化将是更加高效的方式,搭建新的网络模型提高预测结果。
基金项目
国家自然科学基金(No. 61375013),山东省自然科学基金(No. ZR2013FM020)资助。