





Abstract
Chordomas are rare embryogenetic tumors, arising from remnants of the notochord, characterized by local invasiveness and variable tendency for recurrence. No molecular markers are currently used in a clinical setting to distinguish chordomas with an indolent or an aggressive pattern. Among the genetic lesions observed in this tumor, one of the most commonly detected is 1p loss. In a previous study we observed 1p36 loss of heterozygosity (LOH) in 85% of the analyzed chordomas. We studied a group of 16 homogeneously treated skull base chordomas (SBCs), reporting 1p36 LOH in 75% of them and determining the expression pattern of eight apoptotic genes mapped at 1p36. No tumors shared a common expression profile with nucleus pulposus, which is considered the only adult normal tissue deriving from notochord. In particular, tumor necrosis factor receptor superfamily genes TNFRSF8, TNFRSF9, and TNFRSF14 were differently expressed compared with control in a higher percentage of tumors (40%-53%) than were the remaining analyzed genes, suggesting that the deregulation of these three genes might have a role in chordoma tumorigenesis. The presence/absence of LOH and the expression/nonexpression of each apoptotic gene were studied in a survival analysis. Our results suggest that the lack of 1p36 LOH or the presence of TNFRSF8 expression might be associated with a better prognosis in patients with SBCs.
Chordomas are rare tumors that originate from embryonic remnants of the primitive notochord. Their incidence in the population is around 0.05/100,000. They account for 0.1%-0.25% of intracranial tumors1 and for less than 5% of all bone tumors. The most common locations are the sacrococcygeal region (50%), the skull base (35%), and the spinal axis (15%).2 No racial predilection was reported for chordomas, whereas male-to-female ratio is generally reported to be 2:1, with slight variation.3 Chordomas occur in every age group, with predominance for the third and fourth decades for intracranial localization, whereas spinal chordomas are generally observed in older age groups because of late signs and symptoms.4
Chordomas lie in the bone, so they initially grow extradurally with bone destruction and secondary extension into the adjacent soft tissues5; they have common characteristics of a malignant tumor, with local invasiveness, tendency for recurrence, and the potential to metastasize.3 Two histological variants of chordoma have been described: classic and chondroid. Clinically, chordomas are slow-growing tumors. The tumor spread mainly occurs locally. Symptoms manifest late, even after years, and therefore the local extent of disease is often quite large at diagnosis.6
Given the natural history and histological characteristics of the disease, treatment is based on local modalities. Surgery, when feasible, is the treatment of choice. Aggressive surgical treatment followed by postoperative radiotherapy seems to be related with a better progression-free survival.7,8
If surgery is unfeasible, radiation therapy alone is the second choice of treatment. Chordomas are generally viewed as chemoresistant, low-grade tumors, for which standard cytotoxic chemotherapy lacks an established role. A report of a chordoma expressing platelet-derived growth factor receptor β, a target of tyrosine kinase inhibitor imatinib, prompted the compassionate use of imatinib in a few patients, with partial success.9
Common genetic lesions in chordoma are 1p36, 3p, and 12p losses and 1q, 7q, and 9q gains.10 1q gain might be related to the formation of an isochromosome 1q, as previously reported.11 In particular, a loss of heterozygosity (LOH) study centered on the 1p36 region revealed an interestingly high incidence of 1p36 losses among sporadic chordomas.12,13 Genetic lesions in 1p are described in many tumors, such as neuroblastomas,14 gliomas,15 stomach, colon and rectum, lung, breast, endometrium, ovary, testis, kidney, thyroid, and sarcomas.16 In other cancers, 1p losses were correlated with prognosis and in a few cases may even change the therapeutic approach, as in Wilms's tumor.17
Until now, no molecular characteristics have proven conclusive in establishing prognosis in skull base chordomas (SBCs), although evidence was found for a set of cadherins and catenins18; E-cadherin and MIB-1 labeling were correlated with disease-free survival and recurrence in pediatric SBCs.19 Expression of matrix metalloproteinases and nuclear pleomorphism were reported as indicators of unfavorable clinical outcome in non-skull base chordomas (NSBCs),20 and an additional study including both SBCs and NSBCs confirmed that p53 overexpression is associated with unfavorable prognosis in patients with chordoma.21
In this study, we analyzed 16 samples from 15 patients for 1p36 LOH and determined the expression profile of genes mapping in this region and involved in the apoptotic pathway, thus being possibly relevant to tumor progression and aggressiveness, as reported for other cancers. We also compared these findings in a survival analysis, attempting to identify molecular prognostic markers of chordoma.
A possible correlation among absence of 1p36 LOH or expression of tumor necrosis factor receptor superfamily gene TNFRSF8 and progression-free and overall survival of patients is proposed.
Materials and Methods
Patients
This study includes 15 patients with SBC (Table 1), each of whom underwent surgery at the Department of Neurosurgery of the San Raffaele Scientific Institute in Milan between August 1997 and December 2005. All patients gave informed consent to be in the study.
Clinical data of patients with skull base chordomas
| Patient | Age | Sex | Histology | Recurrencea | Deatha |
|---|---|---|---|---|---|
| 12 | 67 | M | CH | — | — |
| 13 | 55 | M | CL | — | — |
| 20 | 55 | M | CL | — | — |
| 21 | 46 | M | CL | — | — |
| 22 | 40 | M | CL | 22 | 30 |
| 24 | 29 | F | CH | — | — |
| 25 | 27 | M | CH | 24 | 53 |
| 26 | 52 | M | CL | — | — |
| 35 | 25 | F | CL | — | — |
| 37 | 41 | F | CL | — | — |
| 38 | 31 | M | CL | — | — |
| 39 | 32 | F | CH | — | — |
| 40 | 30 | F | CH | 16 | — |
| 45 | 52 | M | CH | 5 | 7 |
| 49 | 46 | M | CL | — | — |
| Patient | Age | Sex | Histology | Recurrencea | Deatha |
|---|---|---|---|---|---|
| 12 | 67 | M | CH | — | — |
| 13 | 55 | M | CL | — | — |
| 20 | 55 | M | CL | — | — |
| 21 | 46 | M | CL | — | — |
| 22 | 40 | M | CL | 22 | 30 |
| 24 | 29 | F | CH | — | — |
| 25 | 27 | M | CH | 24 | 53 |
| 26 | 52 | M | CL | — | — |
| 35 | 25 | F | CL | — | — |
| 37 | 41 | F | CL | — | — |
| 38 | 31 | M | CL | — | — |
| 39 | 32 | F | CH | — | — |
| 40 | 30 | F | CH | 16 | — |
| 45 | 52 | M | CH | 5 | 7 |
| 49 | 46 | M | CL | — | — |
Abbreviations: M, male; CH, chondroid chordoma; CL, classic chordoma; F, female.
Months after surgery.
Clinical data of patients with skull base chordomas
| Patient | Age | Sex | Histology | Recurrencea | Deatha |
|---|---|---|---|---|---|
| 12 | 67 | M | CH | — | — |
| 13 | 55 | M | CL | — | — |
| 20 | 55 | M | CL | — | — |
| 21 | 46 | M | CL | — | — |
| 22 | 40 | M | CL | 22 | 30 |
| 24 | 29 | F | CH | — | — |
| 25 | 27 | M | CH | 24 | 53 |
| 26 | 52 | M | CL | — | — |
| 35 | 25 | F | CL | — | — |
| 37 | 41 | F | CL | — | — |
| 38 | 31 | M | CL | — | — |
| 39 | 32 | F | CH | — | — |
| 40 | 30 | F | CH | 16 | — |
| 45 | 52 | M | CH | 5 | 7 |
| 49 | 46 | M | CL | — | — |
| Patient | Age | Sex | Histology | Recurrencea | Deatha |
|---|---|---|---|---|---|
| 12 | 67 | M | CH | — | — |
| 13 | 55 | M | CL | — | — |
| 20 | 55 | M | CL | — | — |
| 21 | 46 | M | CL | — | — |
| 22 | 40 | M | CL | 22 | 30 |
| 24 | 29 | F | CH | — | — |
| 25 | 27 | M | CH | 24 | 53 |
| 26 | 52 | M | CL | — | — |
| 35 | 25 | F | CL | — | — |
| 37 | 41 | F | CL | — | — |
| 38 | 31 | M | CL | — | — |
| 39 | 32 | F | CH | — | — |
| 40 | 30 | F | CH | 16 | — |
| 45 | 52 | M | CH | 5 | 7 |
| 49 | 46 | M | CL | — | — |
Abbreviations: M, male; CH, chondroid chordoma; CL, classic chordoma; F, female.
Months after surgery.
Ten patients were male (66.7%), and five were female (33.3%); ages ranged from 25 to 67 years (average 41 years; SD = 12.65). Six patients (40%) had been treated previously. Previous biopsy had been performed at another institution in five patients; one patient had been given gamma-knife radiosurgery.
All patients underwent computed tomography (CT) and magnetic resonance imaging (MRI) scans both preoperatively and postoperatively. Twenty-three surgical procedures were performed; three staged operations were carried out. All operations were performed by the same surgeon (P.M.).
The extent of resection was classified as total, near total, subtotal, or partial, according to criteria of Gay et al.22 The histological specimens were reviewed in each case by the same pathologist, and the tumors were classified as classic or chondroid chordoma. All patients received postoperative radiotherapy.
Follow-up
The mean duration of follow-up was 38.9 months (range, 7-89 months). Both postoperative CT and MRI scans were carried out every 3 months after surgery and then every 6 months. All postoperative MRI and CT scans were reviewed to assess the extent of tumor and bony resection and the current status of the disease.
LOH Analysis
An LOH analysis was carried out on 11 patients and completed on five previously characterized patients using 33 microsatellite markers localized in the 1p36.33-1p36.12 region (Fig. 1), five microsatellites (D1S2841, D1S1172, D1S2868, D1S2726, and D1S2696) mapped at 1p, and six microsatellites (D1S498, D1S400, D1S238, D1S413, D1S1175, and D1S2800) mapped at 1q, as indicated in the GDB Human Genome Database (http://www.gdb.org). DNA was obtained from one formalin-fixed, paraffin-embedded chordoma by deparaffination with xylene, rehydration with decreasing ethanol scale, and incubation with proteinase K in the lysis buffer consisting of Tris-HCl 50 mM (pH 8), EDTA 1 mM, and 0.5% Tween; and from 15 fresh or frozen chordomas using Trizol reagent (Life Technologies, Carlsbad, CA, USA) according to the producer's instructions. Constitutional DNA was obtained from peripheral blood cells by means of QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The tumor and peripheral blood genomic DNA was amplified by PCR using a carboxyfluorescein- or hexachlorofluorescein-labeled forward primer for each marker (Life Technologies), and the DNA fragments were separated by means of capillary electrophoresis (ABI 3100, Applied Biosystems, Foster City, CA, USA).
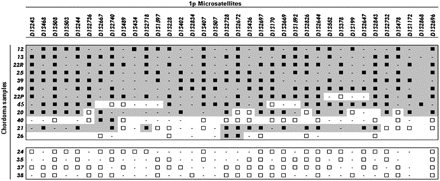
Loss of heterozygosity (LOH) analysis of 16 chordoma samples (listed in rows) from 15 patients using 33 microsatellite markers mapped to 1p36.33-1p36.12 (listed in columns from the most telomeric to the most centromeric). Black squares indicate LOH; white squares, retention of heterozygosity; dashes, uninformative markers. Gray shades indicate LOH regions.
To evaluate LOH, the peak areas of both alleles were measured using Genescan software (Applied Biosystems, version 3.1), and the ratios of the blood (N) and tumor (T) samples were compared: a ratio (T2XN1)/(T1XN2) or (T1XN2)/(T2XN1) of <0.7 is indicative of allele loss, ratios between 0.71 and 0.79 require evaluation of surrounding regions, and ratios >0.8 are considered to retain heterozygosity as reported in detail by Nishimura et al.23 Five of the 16 samples were evaluated in a previous LOH study.13 Because of paucity of tumor material, the LOH analysis of patient 26 was restricted to few markers.
1p36 Apoptotic Gene Expression with Reverse Transcriptase PCR Analysis
The CASP9, DFFA, DFFB, TP73, TNFRSF1B, TNFRSF8, TNFRSF9, and TNFRSF14 genes were selected from the genomic region under study by means of in silico physical mapping analysis (http://www.ncbi.nlm.nih.gov). The RNA analysis was carried out on total RNA isolated from 15 fresh or frozen chordomas, normal adult nucleus pulposus, placenta, and an Epstein-Barr virus-immortalized lymphoblastoid cell line using Trizol reagent (Life Technologies) according to the producer's instructions. Total RNA from fetal heart, fetal brain, fetal aorta, fetal skeletal muscle, and fetal and adult liver was obtained from Stratagene (La Jolla, CA, USA).
Reverse transcription was carried out on 1 μg of total RNA using the ThermoScript reverse transcriptase (RT)-PCR System and oligo-dT primers for first-strand cDNA synthesis according to the instructions of the manufacturer (Invitrogen, Carlsbad, CA, USA). The gene-specific PCR primers were designed on different exons for each amplimer, thus making it possible to distinguish the cDNA-specific band from genomic PCR products (sequences are reported in Table 2). The PCR conditions were 95°C for 5 min, 1 cycle; 95°C for 30 sec, specific annealing temperature for 30 sec, and 72°C for 30 sec, 35 cycles.
Sequences and annealing temperatures of primers used in reverse transcriptase PCR analysis
| Forward | Reverse | AT | |
|---|---|---|---|
| TNFRSF14 | GGAACTGCTCCAGGACAGAG | TACATCACCCCTTGGCTTTC | 56°C |
| TP73 | GATTCCAGCATGGACGTCTT | TTCTTCAAGAGCGGGGAGTA | 56°C |
| DFFB | CAATGGCAGCTACTTCGACA | GTTGAGCTTGTGGGTGGTTT | 56°C |
| TNFRSF9 | CACTCTGTTGCTGGTCCTCA | AGTTTGTCCAGGGTCGACAG | 56°C |
| DFFA | AGTCCCTGACACCAGTCACC | GACGCACTTCCTCTCTTTGG | 56°C |
| TNFRSF8 | GGACACCTGTCATGGAAACC | GTGCCTGGGAACTTGACAAT | 56°C |
| TNFRSF1B | TTCGCTCTTCCAGTTGGACT | TTCACGATGCAGGTGACATT | 56°C |
| CASP9 | CCAGAGATTCGCAAACCAGAG | CAGGGGACTCGTCTTCAGGG | 58°C |
| TNFRSF8-M | GAGAATCGTCGGGATCGTAC | CCGCTTACCTATAAAAAAACTCGTA | 58°C |
| TNFRSF8-U | TTGAGAATTGTTGGGATTGTATGT | CCCACTTACCTATAAAAAAACTCATA | 56°C |
| Forward | Reverse | AT | |
|---|---|---|---|
| TNFRSF14 | GGAACTGCTCCAGGACAGAG | TACATCACCCCTTGGCTTTC | 56°C |
| TP73 | GATTCCAGCATGGACGTCTT | TTCTTCAAGAGCGGGGAGTA | 56°C |
| DFFB | CAATGGCAGCTACTTCGACA | GTTGAGCTTGTGGGTGGTTT | 56°C |
| TNFRSF9 | CACTCTGTTGCTGGTCCTCA | AGTTTGTCCAGGGTCGACAG | 56°C |
| DFFA | AGTCCCTGACACCAGTCACC | GACGCACTTCCTCTCTTTGG | 56°C |
| TNFRSF8 | GGACACCTGTCATGGAAACC | GTGCCTGGGAACTTGACAAT | 56°C |
| TNFRSF1B | TTCGCTCTTCCAGTTGGACT | TTCACGATGCAGGTGACATT | 56°C |
| CASP9 | CCAGAGATTCGCAAACCAGAG | CAGGGGACTCGTCTTCAGGG | 58°C |
| TNFRSF8-M | GAGAATCGTCGGGATCGTAC | CCGCTTACCTATAAAAAAACTCGTA | 58°C |
| TNFRSF8-U | TTGAGAATTGTTGGGATTGTATGT | CCCACTTACCTATAAAAAAACTCATA | 56°C |
Abbreviation: AT, annealing temperature.
Sequences and annealing temperatures of primers used in reverse transcriptase PCR analysis
| Forward | Reverse | AT | |
|---|---|---|---|
| TNFRSF14 | GGAACTGCTCCAGGACAGAG | TACATCACCCCTTGGCTTTC | 56°C |
| TP73 | GATTCCAGCATGGACGTCTT | TTCTTCAAGAGCGGGGAGTA | 56°C |
| DFFB | CAATGGCAGCTACTTCGACA | GTTGAGCTTGTGGGTGGTTT | 56°C |
| TNFRSF9 | CACTCTGTTGCTGGTCCTCA | AGTTTGTCCAGGGTCGACAG | 56°C |
| DFFA | AGTCCCTGACACCAGTCACC | GACGCACTTCCTCTCTTTGG | 56°C |
| TNFRSF8 | GGACACCTGTCATGGAAACC | GTGCCTGGGAACTTGACAAT | 56°C |
| TNFRSF1B | TTCGCTCTTCCAGTTGGACT | TTCACGATGCAGGTGACATT | 56°C |
| CASP9 | CCAGAGATTCGCAAACCAGAG | CAGGGGACTCGTCTTCAGGG | 58°C |
| TNFRSF8-M | GAGAATCGTCGGGATCGTAC | CCGCTTACCTATAAAAAAACTCGTA | 58°C |
| TNFRSF8-U | TTGAGAATTGTTGGGATTGTATGT | CCCACTTACCTATAAAAAAACTCATA | 56°C |
| Forward | Reverse | AT | |
|---|---|---|---|
| TNFRSF14 | GGAACTGCTCCAGGACAGAG | TACATCACCCCTTGGCTTTC | 56°C |
| TP73 | GATTCCAGCATGGACGTCTT | TTCTTCAAGAGCGGGGAGTA | 56°C |
| DFFB | CAATGGCAGCTACTTCGACA | GTTGAGCTTGTGGGTGGTTT | 56°C |
| TNFRSF9 | CACTCTGTTGCTGGTCCTCA | AGTTTGTCCAGGGTCGACAG | 56°C |
| DFFA | AGTCCCTGACACCAGTCACC | GACGCACTTCCTCTCTTTGG | 56°C |
| TNFRSF8 | GGACACCTGTCATGGAAACC | GTGCCTGGGAACTTGACAAT | 56°C |
| TNFRSF1B | TTCGCTCTTCCAGTTGGACT | TTCACGATGCAGGTGACATT | 56°C |
| CASP9 | CCAGAGATTCGCAAACCAGAG | CAGGGGACTCGTCTTCAGGG | 58°C |
| TNFRSF8-M | GAGAATCGTCGGGATCGTAC | CCGCTTACCTATAAAAAAACTCGTA | 58°C |
| TNFRSF8-U | TTGAGAATTGTTGGGATTGTATGT | CCCACTTACCTATAAAAAAACTCATA | 56°C |
Abbreviation: AT, annealing temperature.
Statistical Analysis
The Kaplan-Meier method24 was used for the overall survival and progression-free survival analyses. Progression-free survival was measured from the date of surgery to the date of relapse and was censored at the date of the last MRI. All calculations were performed using the statistical package StatView 5.0 (Abacus Concepts Inc., Berkeley, CA, USA).
Methylation-Specific PCR
Genomic DNA (800 ng) extracted from 14 patients and from nucleus pulposus and lymphocytes from two healthy controls was converted with sodium bisulfite (EpiTect Bisulfite Kit, Qiagen) according to the producer's protocol and amplified for methylation-specific PCR (MSP). Primers were designed with the software MethPrimer (http://www.urogene.org/methprimer). The PCR conditions were 95°C for 5 min, one cycle; 95°C for 30 sec, specific annealing temperature for 30 sec, and 72°C for 20 sec, 40 cycles. Primers and annealing temperatures are reported in Table 2.
Results
Clinical and Follow-up Results
The 15 patients underwent 23 surgical operations for primary tumors and recurrences. The extent of resection was near total in nine cases and subtotal in six cases. The histological examination revealed nine cases (60%) of classic chordoma and six cases (40%) of chondroid chordoma.
MRI scans showed evidence of tumor recurrence in four patients (26.7%). Three patients died from progression of disease; the other patient underwent surgery for the recurrent tumor and had stable disease at the last follow-up. The mean time of recurrence was 31.4 months after surgery.
LOH Analysis
We performed an LOH study in 16 tumors from the 15 patients described above, in order to verify the loss of 1p36 in our chordoma samples, a genetic lesion frequently observed in chordomas. We tested 33 microsatellite markers spanning a region from D1S243 (1p36.33) to D1S478 (1p36.12). As represented in Fig. 1, 12 chordoma patients (75%) showed LOH, six of whom displayed a wide region of LOH involving all the tested markers. The remaining six patients displayed a variable region of LOH that was different in LOH extent and/or localization. Within the last group, three patients showed segmental LOH.
In order to determine whether the LOH was confined to 1p36 or extended across 1p, we tested further microsatellite markers (D1S2841, D1S1172, D1S2868, D1S2726, and D1S2696). Among tumor samples exhibiting LOH at 1p36, nine also exhibited LOH at a more centromeric location, suggesting total 1p loss, while two of them showed no allelic imbalance. We could exclude chromosome 1 monosomy in every tumor showing 1p36 LOH, with the exception of patient 39, by assessing the heterozygosity of 1q microsatellites (data not shown). The remaining four patients (25%) displayed no LOH for any of the tested markers.
1p36 Gene Expression Analysis
Since failure of apoptosis is known to be a key mechanism for the induction and maintenance of the neoplastic phenotype and is believed to be involved in notochord regression, we performed a database search for apoptotic genes mapping in 1p36, which is a commonly shared region of LOH in chordomas and harbors a high concentration of apoptotic genes. Genes were selected according to their involvement in either regulation or execution of apoptosis.
In particular, we identified eight genes with the above-mentioned characteristics: four tumor necrosis factor receptors superfamily, TNFRSF1B, TNFRSF8, TNFRSF9, and TNFRSF14, involved in cell death or survival signaling; CASP9, involved in the activation cascade of caspases and responsible for apoptosis execution; DFFA and DFFB, effectors of DNA fragmentation after apoptotic stimuli; and TP73, encoding a member of the p53 family.
We evaluated the presence/absence of the transcripts of the above genes in the 15 fresh or frozen chordoma samples by RT-PCR and compared the expression profiles with that of adult nucleus pulposus. The control tissue was selected as the most appropriate counterpart of chordoma because it originates from the involution of notochord. In order to define an expression profile specific for nucleus pulposus for this set of genes, we also investigated other normal tissues: lymphocytes, placenta, fetal brain, fetal heart, fetal aorta, fetal skeletal muscle, and fetal and adult liver (Fig. 2).
Most of the studied genes were expressed evenly in nucleus pulposus compared to the other tissues. A notable exception was TP73, which was peculiarly nonexpressed in nucleus pulposus, whereas TNFRSF9 was expressed only in nucleus pulposus and lymphocytes. A third gene that did not show a similar expression profile in normal tissues is TNFRSF8 which was not detected in nucleus pulposus.
Considering all the selected genes, tumor samples showed variable expression profiles, none of which matched that of nucleus pulposus. The TNFRSF14 gene was differently expressed in six samples in comparison with nucleus pulposus, TP73 was differently expressed in two tumors, DFFB in four, TNFRSF9 in eight, DFFA in one, TNFRSF8 in seven, TNFRSF1B in three, and CASP9 in two chordomas (Fig. 2). None of the analyzed samples showed the same expression profile observed in the reference tissue for all of the tested genes.
Statistical Analysis
The Kaplan-Meier analysis showed a predicted overall survival rate of 93% at 2 years (95% confidence interval [95% CI], 80.7-100%) and 69% at 5 years (95% CI, 38.2-100%). The progression-free survival estimates were 68.4% at both 2 and 5 years (95% CI, 42.5-94.4%).

Reverse transcriptase PCR analysis of the eight apoptotic genes mapped to 1p36, positioned from the telomere (left) to the centromere (right), and the housekeeping gene GAPDH in 15 chordoma samples (a), in the reference nucleus pulposus tissue (b), and in eight adult and fetal normal tissues (c). Abbreviations: NP, nucleus pulposus; Ly, lymphoblastoid cell line; a-L, adult liver; f-L, fetal liver; f-SM, fetal skeletal muscle; f-Ao, fetal aorta; f-H, fetal heart; f-Br, fetal brain; Pl, placenta.
We then grouped the patients according to LOH status at 1p36 and observed that all events (recurrence/death) clustered in the group of 1p36 LOH-positive patients as shown by the Kaplan-Meier plot (Fig. 3).
We also grouped patients according to expression/nonexpression for each of the mentioned genes. We observed that all events (recurrence or death) clustered in the group of TNFRSF8 expression-negative patients. We found no correlations related to the expression/nonexpression of the other studied genes and clinical outcome (data not shown).
Methylation-Specific PCR
In order to verify whether TNFRSF8 silencing was due to an epigenetic mechanism, we performed a bisulfite treatment and an MSP on a CpG island located at the 5' of the TNFRSF8 gene. The analysis was performed on 13 samples for which genomic DNA of sufficient quality and gene expression data were available and on nucleus pulposus and peripheral blood DNA from two healthy individuals used as negative controls for methylation. We detected no methylation-specific band for control peripheral blood or nucleus pulposus, or for six of the patients, whereas seven other patients did show a methylation-specific band (Fig. 4).
Discussion
Chordomas are rare tumors, and SBCs account for 36%-39% of them.25 Surgical treatment has a definitive role in the management of SBCs in association with highdose radiation therapy.22,26,27
Clival chordomas represent a challenge for the neurosurgeon because of their proximity to cranial nerves and major blood vessels. Macroscopically near-total resection often does not result in oncologically complete microscopic resection; negative resection margins are rarely accomplished. In two reported series, no significant difference was found in recurrence intervals between chordomas in which a near-total resection was achieved and those in which a subtotal removal was performed.7,28 Ultimately, the majority of patients will experience recurrence and will die of local progression. It also appears, however, that chordomas that have been resected to the same extent and that received postoperative radiotherapy might exhibit different rates of regrowth. This result supports the hypothesis that the recurrence rate of chordomas might depend on variables other than the extent of resection and postoperative radiotherapy. Previous studies investigated histological, cell cycle, and apoptotic markers in relation to the biological and clinical behavior of clival chordomas; the results suggested that the pathological features studied are poor predictors of outcome.7,8,22,28-33 Recently, the decreased expression of E-cadherin and the increased expression of N-cadherin have been correlated with SBC aggressiveness,18 and a study including both SBCs and NSBCs confirmed that p53 and MDM2 overexpression is associated with unfavorable prognosis.21
In our study, 15 patients underwent surgery for clival chordomas by the same surgeon (P.M.) at the same neurosurgical department. On this selected group of patients, we performed a molecular genetic characterization of 16 chordoma samples (in one patient we were able to study both the primary tumor and its recurrence). The genetic characterization was centered on the LOH analysis on 1p36 and the determination of the expression profile of apoptotic genes mapped to 1p36. The LOH analysis showed complete LOH in six samples in the genomic region under study. The finding of segmental LOH involving different 1p36 subregions in the same patient suggests the coexistence of clonal cell lines with different LOH extents in the same tumor sample. Four patients showed no LOH for any of the tested informative markers.
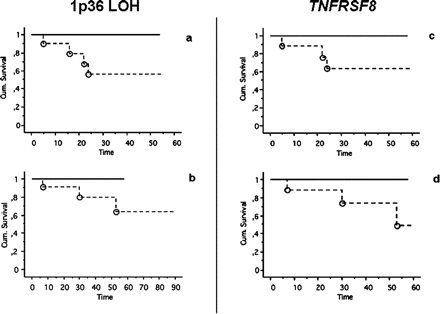
Kaplan-Meier progression-free (a, c) and overall survival (b, d) according to presence (continuous line) or absence (dashed line) of 1p36 loss of heterozygosity (LOH) (a, b) at one or more investigated markers, and according to expression (continuous line) or nonexpression (dashed line) of TNFRSF8 (c, d) in patients with skull base chordomas. Survival times are expressed in months at the most recent census. Events (circles) were observed only in the group of patients displaying 1p36 LOH and in the group of patients not expressing TNFRSF8.
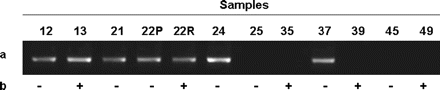
Methylation-specific PCR on the CpG island at the 5' of the TNFRSF8 gene. A 164-bp methylation-specific band was identified in 7 of 12 patients (a). In seven samples, the methylation status was in accord with the expression status by reverse transcriptase PCR (b). +, TNFRSF8 expression; -, no expression.
The high incidence of 1p36 LOH detected in this study (75%) confirms the findings of a previous study on a larger group of chordomas,13 suggesting that this genetic lesion might be a favorable condition for cellular proliferation in SBCs. The 1p36 LOH profile identified in our SBC sample evidenced the presence of large overlapping regions of LOH, hampering the definition of a minimal deleted region (MDR), but including the previously identified MDR centered on the D1S2697 marker.13 The apparent discrepancy between our presented investigation and the previous one is likely due to a more complete LOH analysis here performed, allowing us to better define LOH intervals.
We also verified, by 1q LOH analysis, that 1p losses in our samples are not related to chromosome 1 monosomy or to the formation of isochromosome 1q, the latter being previously observed by other authors in 3 of 11 samples from a different group of chordomas.11
The 1p region is one of the most frequently affected genomic regions by tumor-specific rearrangements, indicating the presence of loci of general importance for cancer development and progression. Tumors showing high percentages of 1p LOH (>70%) are classic oligodendrogliomas (83%),15 yolk sac tumors of infants (75%),34 malignant melanomas (86%),35 pheochromocytomas (72%),36 oral squamous cell carcinomas (73%),37 and endometrial carcinomas (72%).16 In some instances, 1p36 losses have been related to tumor progression; in particular, they have been correlated with poor prognosis in neuroblastomas14 and with the evolution of myelodysplastic syndrome to acute myeloid leukemia.38 Similarly, 1p36 losses do not seem to be random events in chordomas.
Evidence on 1p losses in chordomas has also been obtained by classic cytogenetics, fluorescence in situ hybridization, and comparative genomic hybridization,10 without attributing a prognostic significance in SBCs.
The presence of several genes mapped to 1p36 and involved in the apoptotic pathways prompted us to determine their expression profile in our chordoma samples. A transcription profile for the same genes was also obtained in a reference tissue, nucleus pulposus, considered the only normal adult tissue derived from notochord.39 By a comparison of the nucleus pulposus expression pattern with that of other eight normal tissues, we found that TP73 is peculiarly nonexpressed in nucleus pulposus. The TP73 gene product shares significant homology with p53, although recent data suggest that p73 has distinct biological activities; in particular, it has been shown to have a specific role during embryo development. TP73 harbors two different promoters that yield two different classes of proteins (TAp73 and ΔNp73). TAp73 can induce cell growth arrest and/or apoptosis; ΔNp73, on the contrary, elicits opposite biological effects.40 By using a primer pair that amplifies TAp73 transcript, we were able to exclude TP73 gene expression both in chordoma samples and in nucleus pulposus. We therefore speculate that the p73-mediated proapoptotic pathway is not physiologically active in nucleus pulposus and in most chordomas.
TNFRSF9 was found to be expressed only in nucleus pulposus and lymphocytes, where it is known to exert a role in T-cell responses,41 even if little is known on its specific biological function. The other genes were found to be expressed in our normal control as well as in most analyzed tissues.
Tumor samples can be subgrouped for each gene according to their overlapping or nonoverlapping expression with nucleus pulposus. The expression data for tumor necrosis factor receptor superfamily genes suggests that the widespread deregulation of TNFRSF8, TNFRSF9, and TNFRSF14, which is higher than that detected for the other analyzed genes, points out their possible role in chordoma behavior. The tumor necrosis factor receptor superfamily has unique structural attributes that couple these receptors directly to signaling pathways for cell proliferation, survival, and differentiation.41
In this study, we confirmed the absence of CASP9 in one of four previously characterized chordomas13 and in an additional specimen, indicating that its different expression with respect to nucleus pulposus in a limited number of chordomas might imply a minor role in tumorigenesis. Since all these genes are involved in apoptosis in several ways, we can speculate that the remnants of notochord, from which chordomas arise, might derive from an inefficient notochord regression due to apoptotic defects.42
Furthermore, our results underline that no analyzed chordoma showed the same expression profile as nucleus pulposus for every considered gene, pinpointing 1p36 as a region with a pathogenetic significance.
The present study allowed us to assess the expression/nonexpression of the selected genes in SBCs and in its normal counterpart, highlighting different expression patterns of genes involved in apoptosis/proliferation, mapping in the critical 1p36 region. The limited availability of tumor RNA hampered the quantitative determination of each transcript level and the investigation of gene expression modulation among the tumors and the control tissue, also in relation to LOH.
We then compared molecular findings and patients' clinical outcome, considering tumor recurrence and patient death as clinical end points. By applying the Kaplan-Meier method, we obtained overall survival and progression-free survival curves in relation to the presence/absence of LOH and the expression/nonexpression of TNFRSF8. No events of recurrence or death occurred in the group of patients without LOH in 1p36 or with TNFRSF8 gene expression. This evidence has no statistical significance because of the lack of events in one of the two groups, but suggests a possible correlation between a better clinical outcome and the absence of LOH in 1p36 or the expression of TNFRSF8. This hypothesis should be confirmed on a larger series of patients, pointing to new markers possibly predicting a different recurrence rate.
The TNFRSF8 gene has a 660-bp CpG island located in its first exon downstream of the transcription initiation site.43 Such localization of CpG islands is not uncommon in genes with a limited expression pattern;44 moreover, dense methylation of a CpG island, which begins in the first exon of the MCJ gene, has been observed to be associated with transcriptional silencing in cancer cells.45 We therefore searched for an association between methylation status of the CpG island and TNFRSF8 expression in our chordoma samples. The nucleus pulposus did not express TNFRSF8 and had no methylation-specific band, indicating that other mechanisms are physiologically responsible for gene silencing. Analysis of methylation status of CpG islands in human adult somatic tissues determined that they are almost always methylation free even in tissues where the associated gene was not expressed.46 We detected in 9 out of 13 analyzed samples an accord between methylation status of the CpG island and gene expression by RT-PCR. In two instances we observed lack of methylation and no expression, suggesting that the gene may be silenced by other mechanisms that might act also in nucleus pulposus. Two further samples expressing TNFRSF8 are methylated: a possible contamination with TNFRSF8-positive cells, such as activated lymphocytes, can be excluded by simultaneous lack in chordomas of TP73 transcript, typically present in lymphocytes. The alteration of DNA methylation status seems to play a key role in cancer, and in most of our analyzed SBCs, it is associated with TNFRSF8 gene silencing.
The low incidence of chordoma limits the size of the samples included in correlation studies, making validation of results difficult at the epidemiological level. The evidence reported until now points to a few markers, including proliferative/apoptotic effectors and adhesion molecules, that seem to be related to prognosis. If the data on a single marker cannot be considered conclusive for prognosis, the evaluation of a panel of prognostic markers may be more efficient in predicting chordoma behavior, addressing suitable therapeutic plans.
We thank Dr. Mario Morosi (Fatebenefratelli Hospital, Milan) for providing human nucleus pulposus, Gianluca Facchi for his support, and Dr. Marco Losa for his valuable contribution to the correlation analysis. This work was supported by a Fondo Interno per la Ricerca Scientifica e Tecnologica, University of Milan (FIRST) grant to P.R. and by a FIRST University of Brescia grant to P.M.
References
Watkins L, Khudados ES, Kaleoglu M, Revesz T, Sacares P, Crockard HA. Skull base chordomas: a review of 38 patients, 1958-88.
Forsyth PA, Cascino TL, Shaw EG, et al. Intracranial chordomas: a clinicopathological and prognostic study of 51 cases.
Higinbotham NL, Phillips RF, Farr HW, Hustu HO. Chordoma: thirty-five year study at the Memorial Hospital.
Unni KK.
Oikawa S, Kyoshima K, Goto T, et al. Histological study on local invasiveness of clival chordoma. Case report of autopsy.
Colli B, Al-Mefty O. Chordomas of the craniocervical junction: follow-up review and prognostic factors.
Crockard HA, Steel T, Plowman N, et al. A multidisciplinary team approach to skull base chordomas.
Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma.
Larizza L, Mortini P, Riva P. Update on the cytogenetics and genetics of chordoma.
Sawyer JR, Husain M, Al-Mefty O. Identification of isochromosome 1q as a recurring chromosome aberration in skull base chordomas: a new marker for aggressive tumors?
Miozzo M, Dalprà L, Riva P, et al. A tumor suppressor locus in familial and sporadic chordoma maps to 1p36.
Riva P, Crosti F, Orzan F, et al. Mapping of candidate region for chordoma development to 1p36.13 by LOH analysis.
Attiyeh EF, London WB, Mosse YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma.
Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M. Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene.
Ragnarsson G, Eiriksdottir G, Johannsdottir JT, Jonasson JG, Egilsson V, Ingvarsson S. Loss of heterozygosity at chromosome 1p in different solid human tumours: association with survival.
Perlman EJ. Pediatric renal tumors: practical updates for the pathologist.
Triana A, Sen C, Wolfe D, Hazan R. Cadherins and catenins in clival chordomas: correlation of expression with tumor aggressiveness.
Saad AG, Collins MH. Prognostic value of MIB-1, E-cadherin, and CD44 in pediatric chordomas.
Naka T, Boltze C, Kuester D, et al. Expression of matrix metalloproteinase (MMP)-1, MMP-2, MMP-9, cathepsin B, and urokinase plasminogen activator in non-skull base chordoma.
Naka T, Boltze C, Kuester D, et al. Alteration of G1-S checkpoint in chordoma: the prognostic impact of p53 overexpression.
Gay E, Sekhar LN, Rubinstein E, et al. Chordomas and chondrosarcomas of the cranial base: results and follow-up of 60 patients.
Nishimura T, Nishida N, Itoh T, et al. Comprehensive allelotyping of well-differentiated human hepatocellular carcinoma with semiquantitative determination of chromosomal gain or loss.
Kaplan EL, Meier P. Nonparametric estimation from incomplete observations.
Thieblemont C, Biron P, Rocher F. Prognostic factors in chordoma: role of postoperative radiotherapy.
Zorlu F, Gurkaynak M, Yildiz F, Oge K, Atahan IL. Conventional external radiotherapy in the management of clivus chordomas with overt residual disease.
Pallini R, Maira G, Pierconti F, et al. Chordoma of the skull base: predictors of tumor recurrence.
Deniz ML, Kilic T, Almaata I, Kurtkaya O, Sav A, Pamir MN. Expression of growth factors and structural proteins in chordomas: basic fibroblast growth factor, transforming growth factor alpha, and fibronectin are correlated with recurrence.
Matsuno A, Sasaki T, Nagashima T, et al. Immunohistochemical examination of proliferative potentials and the expression of cell cycle-related proteins of intracranial chordomas.
Mitchell A, Scheithauer BW, Unni KK, Forsyth PJ, Wold LE, McGivney DJ. Chordoma and chondroid neoplasms of the spheno-occiput: an immunohistochemical study of 41 cases with prognostic and nosologic implications.
O'Connell JX, Renard LG, Liebsch NJ. Base of skull chordoma: a correlative study of histologic and clinical features of 62 cases.
Schoedel KE, Martinez AJ, Mahoney TM, Contis L, Becich MJ. Chordomas: pathological features; ploidy and silver nucleolar organizing region analysis. A study of 36 cases.
Kato N, Tamura G, Fukase M, Shibuya H, Motoyama T. Hypermethylation of the RUNX3 gene promoter in testicular yolk sac tumor of infants.
Poetsch M, Dittberner T, Woenckhaus C. Microsatellite analysis at 1p36.3 in malignant melanoma of the skin: fine mapping in search of a possible tumour suppressor gene region.
Edstrom Elder E, Nord B, Carling T, et al. Loss of heterozygosity on the short arm of chromosome 1 in pheochromocytoma and abdominal paraganglioma.
Araki D, Uzawa K, Watanabe T, et al. Frequent allelic losses on the short arm of chromosome 1 and decreased expression of the p73 gene at 1p36.3 in squamous cell carcinoma of the oral cavity.
Mori N, Morsetti R, Mizoguchi H, Koeffler HP. Progression of myelodysplastic syndrome: allelic loss on chromosomal arm 1p.
Stemple DL. Structure and function of the notochord: an essential organ for chordate development.
Benard J, Douc-Rasy S, Ahomadegbe JC. TP53 family members and human cancers.
Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology.
Vleesch Dubois VN, Quan Qi B, Beasley SW, Williams A. Abnormal branching and regression of the notochord and its relationship to foregut abnormalities.
UCSC Genome Browser. Santa Cruz: Genome Bioinformatics Group of UC Santa Cruz. Last updated March 2006 (NCBI Build 36.1). Available at http://genome.ucsc.edu/cgi-bin/hgGateway. Accessed November 27, 2006.
Strathdee G, Davies BR, Vass JK, Siddiqui N, Brown R. Cell type-specific methylation of an intronic CpG island controls expression of the MCJ gene.



{kind=link}
{kind=link}
{kind=link}
{kind=link}