





Abstract
Karenitecin is a highly lipophilic camptothecin analogue with a lactone ring that is relatively resistant to inactivating hydrolysis under physiologic conditions. This phase I clinical trial was conducted to determine the maximum tolerated dose (MTD) of karenitecin in adults with recurrent malignant glioma (MG), to describe the effects of enzyme-inducing antiseizure drugs (EIASDs) on its pharmacokinetics, and to obtain preliminary evidence of activity. Karenitecin was administered intravenously over 60 min daily for 5 consecutive days every 3 weeks to adults with recurrent MG who had no more than one prior chemotherapy regimen. The continual reassessment method was used to escalate doses, beginning at 1.0 mg/m2/day, in patients stratified by EIASD use. Treatment was continued until disease progression or treatment-related dose-limiting toxicity (DLT). Plasma pharmacokinetics was determined for the first daily dose of karenitecin. Thirty-two patients (median age, 52 years; median KPS score, 90) were accrued. Seventy-eight percent had glioblastoma, and 22% had anaplastic glioma. DLT was reversible neutropenia or thrombocytopenia. The MTD was 2.0 mg/m2 in †EIASD patients and 1.5 mg/m2 in -EIASD patients. The mean (±SD) total body clearance of karenitecin was 15.9 ± 9.6 liters/h/m2 in †EIASD patients and 10.2 ± 3.5 liters/h/m2 in -EIASD patients (p = 0.02). No objective responses were observed in 11 patients treated at or above the MTD. The total body clearance of karenitecin is significantly enhanced by the concurrent administration of EIASDs. This schedule of karenitecin, a novel lipophilic camptothecin analogue, has little activity in recurrent MG.
The prognosis of patients with high-grade glioma remains poor despite recent improvements in adjuvant therapy. Even the most promising approach for treating glioblastoma multiforme yields median survival of just over 1 year, a 2-year mortality of 75%, and a cure rate that approaches zero.1 As a result, more effective chemotherapeutic agents are required for additional progress to be made in this disease.
The camptothecins have emerged as an important class of antitumor drugs during the last two decades.2 Currently, topotecan and irinotecan are the only camptothecins approved for clinical use as anticancer drugs. Topotecan is indicated as second-line therapy against metastatic ovarian cancer and small-cell lung cancer and for the treatment of recurrent cervical carcinoma when combined with cisplatin. Irinotecan is approved as a first-line treatment for advanced colorectal cancer in combination with fluoropyrimidines and as a single agent following failure of 5-fluorouracil-based chemotherapy. Clinical trials have been performed to evaluate the activity of both agents against malignant gliomas (MGs). Although preliminary findings were suggestive of promising activity,3 several phase II trials consistently demonstrated that single-agent irinotecan has minimal activity in newly diagnosed, recurrent, and progressive MGs in adult patients.4-6 Topotecan has not shown substantial clinical activity against MGs, even when given as first-line treatment in combination with standard cranial radiotherapy, although it appears to penetrate the blood-brain barrier more easily than does irinotecan.7-12
Chemical structure of karenitecin.
Efforts to improve the pharmacologic properties of the first-generation camptothecins resulted in the identification of highly lipophilic derivatives that exhibit considerably greater stability of the biologically active form of the compound in human blood.13 These have the potential to provide better tumor penetration and efficacy in patients with primary brain tumors.14 Karenitecin (Fig. 1) is a 7-alkylsilane derivative of camptothecin that was selected for clinical development from a series of rationally designed compounds based upon its spectrum of antitumor activity, potency, and other advantageous properties.14 Intraperitoneal karenitecin has been shown to significantly inhibit the growth of human brain tumor xenografts, including cell lines derived from adult and pediatric high-grade gliomas, implanted either subcutaneously or intracranially in athymic mice.15 These findings generated interest in undertaking a phase I trial to evaluate karenitecin in adults with recurrent MGs. The route of administration, schedule, and starting dose were determined from dose escalation trials in systemic cancers.16 Although enzyme-inducing antiseizure drugs (EIASDs) have a striking effect on the pharmacokinetics of the approved camptothecins, no data existed on their impact on karenitecin.
This study was designed to determine the effect of EIASDs on the maximum tolerated dose (MTD), toxicity profile, and pharmacokinetic behavior of karenitecin. A secondary objective of the study was to obtain a preliminary assessment of the activity of single-agent karenitecin in recurrent gliomas.
Patients and Methods
Patient Selection
Adult patients (age ≥18 years) with histologically proven MG (anaplastic astrocytoma, anaplastic oligodendroglioma, glioblastoma multiforme) that was progressive or recurrent after radiation therapy were eligible for the study. Conditions required for entry into the study included (1) measurable disease by contrast-enhanced MRI or CT, (2) prior radiotherapy, (3) no more than two previous treatments, (4) KPS score ≥60, and (5) life expectancy >2 months. Minimum permitted time intervals from prior treatments were 3 months for radiation, 6 weeks for chloroethylnitrosoureas, and 3 weeks for all other chemotherapeutic agents. Full recovery from the effects of any earlier intervention was required. Eligibility also required demonstrating acceptable hematologic parameters (absolute neutrophil count ≥1,500/μl, platelet count ≥100,000/μl, hemoglobin ≥8 g/dl), renal function (serum creatinine ≤1.7 mg/dl), and hepatic function (total bilirubin ≤1.5 mg/dl, serum levels of aspartate and alanine aminotransferase ≤4× the upper limit of normal). Exclusion criteria included (1) a prior malignancy other than curatively treated basal cell carcinoma or cervical carcinoma in situ; (2) a serious concurrent infection, illness, or medical condition; (3) females who were pregnant or nursing; and (4) any other condition that would compromise treatment with reasonable safety or result in noncompliance with prescribed medical care. Agreement to practice adequate birth control methods was required for fertile patients. The protocol for this study was reviewed and approved by the Cancer Therapy and Evaluation Program of the National Cancer Institute (Bethesda, MD, USA) and the institutional review board of each participating institution. All patients signed a written informed consent document, satisfying all federal and institutional policies and regulations, as a condition of registering for participation in the study.
Drug Administration and Dose Escalation
Patients were assigned to one of two treatment groups based on preexisting use of antiseizure drugs. Patients taking phenytoin, carbamazepine, phenobarbital, primidone, or oxcarbazepine were assigned to the †EIASD group. Patients assigned to the -EIASD group were either not being treated with an antiseizure drug or taking one that does not significantly induce hepatic enzymes, such as gabapentin, lamotrigine, valproic acid, felbamate, levetiracetam, tiagabine, topiramate, and zonisamide. Inclusion in the -EIASD group also required discontinuation of any EIASDs for at least 10 days. Patients who required treatment with a corticosteroid, such as dexamethasone, had to be receiving the lowest clinically appropriate daily dose for at least 5 days before beginning the first cycle of therapy. Efforts were made to maintain the same dose until the radiographic tumor measurement was performed after completing the second cycle.17
Karenitecin was supplied by BioNumerik Pharmaceuticals, Inc. (San Antonio, TX, USA) in amber glass vials containing at least 0.5 mg of karenitecin lactone as a 0.1 mg/ml pale yellow solution in a vehicle composed of N-methyl-2-pyrrolidinone, polyethylene glycol 300, polysorbate 80, ethyl alcohol, and citric acid. This was diluted with 25-100 parts by volume of 5% dextrose for injection, USP in a non-polyvinyl chloride infusion bag for administration. Stability of the final dosing solution was documented for up to 24 h at room temperature. The dosing solution was administered as a constant i.v. infusion over 60 min using a non-polyvinyl chloride administration set, once a day, for 5 consecutive days every 21 days in an outpatient setting. The use of ondansetron to control vomiting was permitted. The prophylactic use of hematopoietic growth factors was prohibited during any cycle of therapy. Treatment with any other approved or investigational chemotherapeutic agents was not permitted.
The starting dose for both treatment groups was 1.0 mg/m2/day. The dose was independently escalated in each group to estimate the MTD using the modified continual reassessment method, as previously described, with minor modifications.18 Three patients were entered into each new dose level and monitored for treatment-related toxicities, as described below. Only toxicity associated with the first cycle was used for the dose-finding determination. After all patients completed the first cycle, all of the available toxicity data were used to develop and update a two-parameter logistic dose-response model. The next dose level to be evaluated was determined by using the model to calculate the dose associated with a dose-limiting toxicity (DLT) rate of 33%, with the maximum increment permitted being 50% of the previous dose level. The entire process was repeated until the recommended dose remained within 10% of the preceding dose for two consecutive iterations. This dose was considered to be the MTD.
Treatment with the same daily dose of karenitecin was repeated every 3 weeks for patients who did not experience a DLT, in the absence of tumor progression, if all eligibility requirements continued to be satisfied. Retreatment in the event of a DLT during any course was permitted with a 25% dose reduction after all toxicities recovered to baseline values or grade 0 or 1. Further decreases in the dose, in this same manner, were allowed upon the occurrence of a DLT after treatment with a reduced dose. A maximum of three dose reductions were permitted before the patient was removed from the study. Patients were also removed from the study because of disease progression, circumstances for which continued treatment could be detrimental to the health of a patient, noncompliance, or upon the decision of a patient to discontinue treatment for any reason.
Patient Evaluations
Evaluations performed within 14 days of initiating therapy included a medical history, physical and neurologic examinations, Mini-Mental Status Exam, KPS determination, electrocardiogram, chest X-ray, vital signs, complete blood count with differential and platelet counts, blood coagulation parameters, serum chemistry profile, urinalysis, and pregnancy test for women of child-bearing potential. After initiating treatment, a complete blood count with differentials and platelet count were performed weekly, or more often if significant myelosuppression was observed. A history and physical examination, KPS determination, complete blood count with differentials and platelet count, and serum electrolyte and creatinine assays were performed before beginning each cycle of therapy. In addition to these tests, a Mini-Mental Status Exam and serum chemistry profile were repeated before every odd-numbered cycle of therapy. Toxicities were evaluated during each monthly cycle and graded according to the National Cancer Institute's Common Toxicity Criteria, version 2.0 ((http://ctep.cancer.gov/forms/CTCv20_4-30-992.pdf). DLT was defined as any of the following treatment-related adverse events: (1) absolute neutrophil count ≤500/μl, (2) platelet count ≤25,000/μl, (3) febrile neutropenia, (4) any grade 3 or 4 nonhematologic toxicity with the exception of nausea and vomiting, and (5) a delay in starting a subsequent course of treatment for more than 14 days because of incomplete recovery from toxicity.
Response to therapy was determined by MRI or CT imaging and neurologic examinations. The use of CT was restricted to patients who were unable to undergo MRI for physical or medical reasons. Imaging studies to provide tumor measurements were performed within 14 days of beginning treatment (baseline) and after each second cycle of therapy until relapse. A confirmatory scan was obtained 1 month after the initial detection of a complete or partial response. Standard response criteria of the New Approaches to Brain Tumor Therapy CNS Consortium (NABTT) were used as described previously.19 The pathology and MRI scans for all patients responding to therapy were to be centrally reviewed centrally reviewed. All patients were followed for survival. Survival time was calculated from start of treatment until death or the last follow-up. Survival was estimated by the method of Kaplan and Meier. Confidence intervals (CIs) were calculated using standard methods. Analysis was performed using SAS, version 9.1.3 (SAS Institute, Inc., Cary, NC, USA).
Pharmacokinetic Studies
Sampling to characterize the plasma pharmacokinetics of karenitecin was performed during treatment with the first daily dose of the first cycle of therapy. Blood specimens (7 ml) were drawn from a peripheral vein in the arm contralateral to the site of drug administration and collected directly in tubes containing freeze-dried sodium heparin before dosing, coincident with the end of the 60 min infusion of karenitecin, and then at 1, 2, 4, 6, and 23 h after the end of the infusion. Sample tubes were mixed by inversion and placed on wet ice until centrifuged (1,000g, 10 min, 4°C) within 15 min. Plasma was transferred into a polypropylene cryovial and stored at -70°C until assayed.
The total concentration (i.e., lactone plus carboxylate forms) of karenitecin in plasma specimens was determined by reversed-phase high-performance liquid chromatography (HPLC) with fluorescence detection. The sample preparation procedure, chromatographic conditions, method of detection, and procedures for the validation of the assay were adapted from previously reported analytical methods for other camptothecin analogues that had been developed in our laboratories.19-21 An analytical reference sample of karenitecin was provided by BioNumerik. The internal standard (IS), acetylkarenitecin, was synthesized by derivatizing karenitecin (0.13 mg) with acetic anhydride in pyridine containing a catalytic amount of 4-dimethylaminopyridine.22,23
Samples were prepared for chromatographic analysis by pipetting 100 μl plasma and 5 μl IS working solution (1 μg/ml in acetonitrile) into a polypropylene microcentrifuge tube and vortexing. Proteins were precipitated by adding acetonitrile (400 μl) to the tube and vortexing the mixture for 5 sec. After centrifugation (13,000g, 2 min), 400 μl of the supernatant was diluted with 100 μl 100 mM trifluoroacetic acid; 100 μl of the solution was loaded onto on a 15 cm × 4.6 mm inner-diameter Luna Phenyl-Hexyl (5-μm particle size) HPLC column (Phenomenex, Torrance, CA, USA). Chromatography was performed at ambient temperature using acetonitrile/0.1 M ammonium acetate buffer (pH 5.0; 65:35, vol/vol) as the mobile phase at 1.0 ml/min. Elution of the drug and IS was monitored using an Agilent Technologies (Palo Alto, CA, USA) 1100 series fluorescence detector with a xenon flash lamp and an 8-μl flow cell (operating parameters: lamp flash frequency, 74 Hz; excitation wavelength, 372 nm; emission wavelength, 435 nm; photomultiplier tube gain, 10; response time, 2.0 s). Chromatograms were integrated to provide peak areas.
Each study sample was independently assayed in duplicate, on different days, together with a set of eight calibration standards of the drug in human plasma at concentrations ranging from 1 to 100 ng/ml and three quality control samples (3, 45, 90 ng/ml). The relationship between the drug-to-IS peak area ratio and known concentration of the calibration standards was analyzed by linear regression, with weighting in proportion to the reciprocal of the drug concentration normalized to the number of standards. Values of the slope and y-intercept for the best-fit line of the standard curve were used to calculate the drug concentration in study samples. Specimens with an estimated drug concentration exceeding the upper range of the standard curve were reassayed upon dilution with drug-free human donor plasma. The average of the two determinations of each study sample was calculated. Samples were reassayed in cases where the individual determinations differed from their average by more than 10%.
Plasma concentration-time curves for each patient were analyzed by standard noncompartmental methods using WinNonlin Professional, version 4.0.1 (Pharsight Corp., Cary, NC, USA). Area under the plasma concentration-time curve from time 0 to infinity was estimated using the log-linear trapezoidal algorithm to the last data point, with extrapolation to infinity using the slope of the terminal log-linear disposition phase. Pharmacokinetic parameters estimated by the program included the total body clearance (CL), half-life of the terminal disposition phase (t½,z), and the apparent volume of distribution at steady state (Vss). Mean values of the pharmacokinetic variables were calculated as the geometric mean of the individual patient values and reported with the standard deviation estimated by the jackknife technique.24-26 Linear regression was used to assess dose-dependent trends in pharmacokinetic parameters. Parametric statistical tests were used to compare mean pharmacokinetic variables between the two treatment groups after logarithmic transformation of the data. All tests were two-sided, and p <0.05 was the criterion for significance.
Results
Patient Characteristics
A total of 32 patients with recurrent high-grade gliomas were enrolled in this clinical trial from April 2002 to July 2004. Two patients enrolled in the †EIASD group became inevaluable for toxicity assessments during the first cycle of therapy and were replaced. One patient experienced toxicity attributable to phenytoin, and the other refused to complete the initial week of treatment. Characteristics of the 30 evaluable patients are summarized in Table 1. Twenty-seven of the patients had undergone surgical resection of the tumor, and all 30 completed radiation therapy. Three patients had no prior chemotherapy, and 25 patients had one prior regimen of chemotherapy. Fifty-eight percent of the 12 patients in the -EIASD arm were not taking an anticonvulsant. Phenytoin was used by 72% of the 18 patients in the †EIASD arm. A stable daily dose of dexamethasone was being given to 40% of the patients in the -EIASD arm and 72% of the patients in the †EIASD treatment arm.
Patient characteristics
| Characteristic | Patient Data |
|---|---|
| No. of evaluable patients | 30 |
| Age (years) | |
| Median | 54 |
| Range | 36-74 |
| Gender | |
| Male | 16 (53%) |
| Female | 14 (47%) |
| KPS score | |
| Median | 90 |
| Range | 60-100 |
| Histologic diagnosis | |
| Glioblastoma multiforme | 23 (77%) |
| Anaplastic astrocytoma | 4 (13%) |
| Anaplastic oligodendroglioma | 1 (3%) |
| Anaplastic mixed glioma | 2 (7%) |
| Prior chemotherapy (number of regimens) | |
| 0 | 3 (10%) |
| 1 | 25 (83%) |
| >1 | 2 (7%)a |
| Concomitant medications | |
| −EIASD arm | 12 (40%) |
| Dexamethasone | 5 (42%) |
| No anticonvulsant | 7 (58%) |
| Levetiracetam | 4 (33%) |
| Valproic acid | 1 (8%) |
| +EIASD arm | 18 (60%) |
| Dexamethasone | 13 (72%) |
| Phenytoin | 13 (72%) |
| Carbamazepine | 4 (22%) |
| Phenobarbitalb | 1 (6%) |
| Characteristic | Patient Data |
|---|---|
| No. of evaluable patients | 30 |
| Age (years) | |
| Median | 54 |
| Range | 36-74 |
| Gender | |
| Male | 16 (53%) |
| Female | 14 (47%) |
| KPS score | |
| Median | 90 |
| Range | 60-100 |
| Histologic diagnosis | |
| Glioblastoma multiforme | 23 (77%) |
| Anaplastic astrocytoma | 4 (13%) |
| Anaplastic oligodendroglioma | 1 (3%) |
| Anaplastic mixed glioma | 2 (7%) |
| Prior chemotherapy (number of regimens) | |
| 0 | 3 (10%) |
| 1 | 25 (83%) |
| >1 | 2 (7%)a |
| Concomitant medications | |
| −EIASD arm | 12 (40%) |
| Dexamethasone | 5 (42%) |
| No anticonvulsant | 7 (58%) |
| Levetiracetam | 4 (33%) |
| Valproic acid | 1 (8%) |
| +EIASD arm | 18 (60%) |
| Dexamethasone | 13 (72%) |
| Phenytoin | 13 (72%) |
| Carbamazepine | 4 (22%) |
| Phenobarbitalb | 1 (6%) |
Abbreviations: −EIASD, patients not taking enzyme-inducing antiseizure drugs; +EIASD, patients taking enzyme-inducing antiseizure drugs. Unless indicated otherwise, data are reported as number of patients, with percentage of the total in parentheses.
Two patients were found to have received more than one prior regimen of chemotherapy, in violation of the eligibility criteria, but not until after completing the first cycle of therapy; they were not replaced.
The patient was also receiving phenytoin.
Patient characteristics
| Characteristic | Patient Data |
|---|---|
| No. of evaluable patients | 30 |
| Age (years) | |
| Median | 54 |
| Range | 36-74 |
| Gender | |
| Male | 16 (53%) |
| Female | 14 (47%) |
| KPS score | |
| Median | 90 |
| Range | 60-100 |
| Histologic diagnosis | |
| Glioblastoma multiforme | 23 (77%) |
| Anaplastic astrocytoma | 4 (13%) |
| Anaplastic oligodendroglioma | 1 (3%) |
| Anaplastic mixed glioma | 2 (7%) |
| Prior chemotherapy (number of regimens) | |
| 0 | 3 (10%) |
| 1 | 25 (83%) |
| >1 | 2 (7%)a |
| Concomitant medications | |
| −EIASD arm | 12 (40%) |
| Dexamethasone | 5 (42%) |
| No anticonvulsant | 7 (58%) |
| Levetiracetam | 4 (33%) |
| Valproic acid | 1 (8%) |
| +EIASD arm | 18 (60%) |
| Dexamethasone | 13 (72%) |
| Phenytoin | 13 (72%) |
| Carbamazepine | 4 (22%) |
| Phenobarbitalb | 1 (6%) |
| Characteristic | Patient Data |
|---|---|
| No. of evaluable patients | 30 |
| Age (years) | |
| Median | 54 |
| Range | 36-74 |
| Gender | |
| Male | 16 (53%) |
| Female | 14 (47%) |
| KPS score | |
| Median | 90 |
| Range | 60-100 |
| Histologic diagnosis | |
| Glioblastoma multiforme | 23 (77%) |
| Anaplastic astrocytoma | 4 (13%) |
| Anaplastic oligodendroglioma | 1 (3%) |
| Anaplastic mixed glioma | 2 (7%) |
| Prior chemotherapy (number of regimens) | |
| 0 | 3 (10%) |
| 1 | 25 (83%) |
| >1 | 2 (7%)a |
| Concomitant medications | |
| −EIASD arm | 12 (40%) |
| Dexamethasone | 5 (42%) |
| No anticonvulsant | 7 (58%) |
| Levetiracetam | 4 (33%) |
| Valproic acid | 1 (8%) |
| +EIASD arm | 18 (60%) |
| Dexamethasone | 13 (72%) |
| Phenytoin | 13 (72%) |
| Carbamazepine | 4 (22%) |
| Phenobarbitalb | 1 (6%) |
Abbreviations: −EIASD, patients not taking enzyme-inducing antiseizure drugs; +EIASD, patients taking enzyme-inducing antiseizure drugs. Unless indicated otherwise, data are reported as number of patients, with percentage of the total in parentheses.
Two patients were found to have received more than one prior regimen of chemotherapy, in violation of the eligibility criteria, but not until after completing the first cycle of therapy; they were not replaced.
The patient was also receiving phenytoin.
Dose Escalation and Toxicities
The dose levels evaluated in both treatment arms and the toxicities experienced by patients during the first cycle of therapy are listed in Table 2. Five dose levels ranging from 1.0 to 2.1 mg/m2 were evaluated in the †EIASD arm. In the -EIASD group, daily doses of 1.0, 1.5, and 1.8 mg/m2 were evaluated. Myelosuppression presenting as reversible neutropenia and thrombocytopenia were the major severe toxicities observed. Grade 3-4 neutropenia occurred in 23% of patients, and thrombocytopenia in 13%. The MTD was determined to be 1.5 mg/m2 in the -EIASD patients and 2.0 mg/m2 in the †EIASD group.
Dose levels evaluated and summary of clinical toxicities for the first cycle of therapy
| Karenitecin Treatment Group (Daily Dose, mg/m2) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −EIASD | +EIASD | ||||||||||||||
| Toxicity | 1.0 | 1.5 | 1.8 | 1.0 | 1.5 | 1.7 | 1.9 | 2.1 | |||||||
| No. of patients evaluated | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 3 | |||||||
| Dose-limiting toxicitya | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | |||||||
| Hematologic toxicity (grade 2/3/4)b | |||||||||||||||
| Neutropenia | 1/0/0 | 1/0/2 | 0/0/2 | 0/0/1 | 1/1/0 | 0/1/0 | |||||||||
| Thrombocytopenia | 0/1/0 | 0/2/0 | 0/1/0 | ||||||||||||
| Nonhematologic toxicity (grade 2/3/4) | |||||||||||||||
| Constipation | 1/0/0 | ||||||||||||||
| Diarrhea | 0/1/0 | ||||||||||||||
| Dyspnea | 1/0/0 | 0/0/1 | 1/0/0 | ||||||||||||
| Fatigue | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | ||||||||||
| Nausea/vomiting | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | |||||||||||
| Karenitecin Treatment Group (Daily Dose, mg/m2) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −EIASD | +EIASD | ||||||||||||||
| Toxicity | 1.0 | 1.5 | 1.8 | 1.0 | 1.5 | 1.7 | 1.9 | 2.1 | |||||||
| No. of patients evaluated | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 3 | |||||||
| Dose-limiting toxicitya | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | |||||||
| Hematologic toxicity (grade 2/3/4)b | |||||||||||||||
| Neutropenia | 1/0/0 | 1/0/2 | 0/0/2 | 0/0/1 | 1/1/0 | 0/1/0 | |||||||||
| Thrombocytopenia | 0/1/0 | 0/2/0 | 0/1/0 | ||||||||||||
| Nonhematologic toxicity (grade 2/3/4) | |||||||||||||||
| Constipation | 1/0/0 | ||||||||||||||
| Diarrhea | 0/1/0 | ||||||||||||||
| Dyspnea | 1/0/0 | 0/0/1 | 1/0/0 | ||||||||||||
| Fatigue | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | ||||||||||
| Nausea/vomiting | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | |||||||||||
Abbreviations: −EIASD, patients not taking enzyme-inducing antiseizure drugs; +EIASD, patients taking enzyme-inducing antiseizure drugs.
Number of patients experiencing a dose-limiting toxicity.
Number of patients experiencing toxicity; entries were not made for dose levels where the indicated toxicity was not observed.
Dose levels evaluated and summary of clinical toxicities for the first cycle of therapy
| Karenitecin Treatment Group (Daily Dose, mg/m2) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −EIASD | +EIASD | ||||||||||||||
| Toxicity | 1.0 | 1.5 | 1.8 | 1.0 | 1.5 | 1.7 | 1.9 | 2.1 | |||||||
| No. of patients evaluated | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 3 | |||||||
| Dose-limiting toxicitya | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | |||||||
| Hematologic toxicity (grade 2/3/4)b | |||||||||||||||
| Neutropenia | 1/0/0 | 1/0/2 | 0/0/2 | 0/0/1 | 1/1/0 | 0/1/0 | |||||||||
| Thrombocytopenia | 0/1/0 | 0/2/0 | 0/1/0 | ||||||||||||
| Nonhematologic toxicity (grade 2/3/4) | |||||||||||||||
| Constipation | 1/0/0 | ||||||||||||||
| Diarrhea | 0/1/0 | ||||||||||||||
| Dyspnea | 1/0/0 | 0/0/1 | 1/0/0 | ||||||||||||
| Fatigue | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | ||||||||||
| Nausea/vomiting | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | |||||||||||
| Karenitecin Treatment Group (Daily Dose, mg/m2) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| −EIASD | +EIASD | ||||||||||||||
| Toxicity | 1.0 | 1.5 | 1.8 | 1.0 | 1.5 | 1.7 | 1.9 | 2.1 | |||||||
| No. of patients evaluated | 3 | 6 | 3 | 3 | 6 | 3 | 3 | 3 | |||||||
| Dose-limiting toxicitya | 0 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | |||||||
| Hematologic toxicity (grade 2/3/4)b | |||||||||||||||
| Neutropenia | 1/0/0 | 1/0/2 | 0/0/2 | 0/0/1 | 1/1/0 | 0/1/0 | |||||||||
| Thrombocytopenia | 0/1/0 | 0/2/0 | 0/1/0 | ||||||||||||
| Nonhematologic toxicity (grade 2/3/4) | |||||||||||||||
| Constipation | 1/0/0 | ||||||||||||||
| Diarrhea | 0/1/0 | ||||||||||||||
| Dyspnea | 1/0/0 | 0/0/1 | 1/0/0 | ||||||||||||
| Fatigue | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | ||||||||||
| Nausea/vomiting | 1/0/0 | 1/0/0 | 1/0/0 | 1/0/0 | |||||||||||
Abbreviations: −EIASD, patients not taking enzyme-inducing antiseizure drugs; +EIASD, patients taking enzyme-inducing antiseizure drugs.
Number of patients experiencing a dose-limiting toxicity.
Number of patients experiencing toxicity; entries were not made for dose levels where the indicated toxicity was not observed.
Response and Survival
No complete or partial responses were observed, even though 11 patients (seven with glioblastoma multiforme, three with anaplastic astrocytoma, and one with a mixed high grade) were treated at or above the MTD. Thirtyone of the 32 (97%) patients enrolled have died. Patients did not remain on karenitecin long, because 23 of 32 patients were withdrawn at the time of their first MRI (6 weeks), and 30 of 32 by their second MRI (12 weeks). One patient, who survived after 53 months of follow-up, discontinued treatment during the first cycle because of toxicity. Median survival time after entering this study was 6.0 (95% CI, 3.9-9.7) months.
Pharmacokinetics
The analytical method was thoroughly validated according to current recommendations.27 Peaks that interfered with detection of the drug or IS were not evident in chromatograms of drug-free plasma from anonymous donors and plasma samples obtained shortly before beginning the karenitecin infusion to patients participating in this clinical trial. Calibration curves exhibited excellent linearity with correlation coefficients ranging from 0.998 to 1.000. During application to this study, interday accuracy and precision of the assay were assessed by analyzing the interpolated drug concentrations from 11 independently prepared standard curves and sets of quality control samples that were run over a period of 14 months. The lowest concentration of karenitecin included in the standard curve, 1.0 ng/ml, was measured with an accuracy of 112.1% and a precision of 3.9%. Quality control samples of karenitecin in human plasma at concentrations of 3.0, 45.6, and 91.2 ng/ml were assayed with an accuracy ranging from 100.1% to 109.5% of the known concentrations and the precision, calculated as the coefficient of variation, was 4.6%-10.3%.
Pharmacokinetic data for the first daily dose of karenitecin were obtained from all 12 patients in the -EIASD group and 16 patients in the †EIASD group. Mean plasma concentration-time profiles of karenitecin for the cohort of patients evaluated in the 1.5 mg/m2 dose level of both treatment groups are shown in Fig. 2. Linear regression analyses revealed that the CL, Vss, and t½,z values for karenitecin in individual patients were independent of the administered dose, for both treatment groups, indicative of linear pharmacokinetic behavior across the range of doses evaluated in this study. Mean pharmacokinetic parameters for karenitecin at each dose level and for the entire cohort of patients evaluated in the two treatment arms are presented in Table 3. For the entire group of 12 patients who were not receiving EIASDs, karenitecin exhibited a mean CL of 10.2 ± 3.5 liters/h/m2, a Vss of 136 ± 41 liters/m2, and a mean t½,z of 13.2 ± 4.1 h. As depicted in Fig. 3, the concurrent administration of EIASDs was associated with a 56% increase in the mean CL (p = 0.019) and a 70% increase in the mean Vss (p < 0.001), although the mean t½,z of the drug was not significantly different between the two groups. The magnitude of the influence of EIASDs on the pharmacokinetic behavior of karenitecin is evident in the mean plasma profiles shown in Fig. 2.
Mean pharmacokinetic parameters for the first daily dose of karenitecin
| Dose (mg/m2) | Number of patients | Cmax (ng/ml) | AUC0-24 (ng·h/ml) | t½,z (h) | CL (liters/h/m2) | Vss (liters/m2) |
|---|---|---|---|---|---|---|
| −EIASD cohort | ||||||
| 1.0 | 3 | 19.4 ± 6.8 | 101.9 ± 1.7 | 13.5 ± 2.6 | 7.7 ± 0.6 | 116 ± 22 |
| 1.5 | 6 | 35.8 ± 20.9 | 114.7 ± 34.0 | 13.3 ± 5.5 | 10.5 ± 3.5 | 136 ± 51 |
| 1.8 | 3 | 24.4 ± 7.3 | 119.9 ± 15.6 | 12.7 ± 2.8 | 11.9 ± 2.5 | 161 ± 21 |
| All | 12 | 13.2 ± 4.1 | 10.2 ± 3.5 | 136 ± 41 | ||
| +EIASD cohort | ||||||
| 1.0 | 2 | 16.4 ± 2.2 | 52.7 ± 20.9 | 18.3 ± 19.3 | 12.2 ± 11.0 | 240 ± 73 |
| 1.5 | 5 | 19.1 ± 4.2 | 73.2 ± 30.8 | 18.9 ± 9.3 | 13.5 ± 11.2 | 271 ± 80 |
| 1.7 | 2 | 20.9 ± 2.7 | 58.3 ± 2.6 | 18.6 ± 0.8 | 21.6 ± 1.7 | 389 ± 26 |
| 1.9 | 4 | 39.6 ± 20.3 | 111.± 6 21.8 | 13.1 ± 1.6 | 13.8 ± 4.0 | 161 ± 29 |
| 2.1 | 3 | 32.1 ± 5.5 | 102.4 ± 22.9 | 12.4 ± 3.1 | 16.6 ± 5.4 | 202 ± 34 |
| All | 16 | 15.8 ± 6.6 | 15.9 ± 9.6 | 232 ± 81 |
| Dose (mg/m2) | Number of patients | Cmax (ng/ml) | AUC0-24 (ng·h/ml) | t½,z (h) | CL (liters/h/m2) | Vss (liters/m2) |
|---|---|---|---|---|---|---|
| −EIASD cohort | ||||||
| 1.0 | 3 | 19.4 ± 6.8 | 101.9 ± 1.7 | 13.5 ± 2.6 | 7.7 ± 0.6 | 116 ± 22 |
| 1.5 | 6 | 35.8 ± 20.9 | 114.7 ± 34.0 | 13.3 ± 5.5 | 10.5 ± 3.5 | 136 ± 51 |
| 1.8 | 3 | 24.4 ± 7.3 | 119.9 ± 15.6 | 12.7 ± 2.8 | 11.9 ± 2.5 | 161 ± 21 |
| All | 12 | 13.2 ± 4.1 | 10.2 ± 3.5 | 136 ± 41 | ||
| +EIASD cohort | ||||||
| 1.0 | 2 | 16.4 ± 2.2 | 52.7 ± 20.9 | 18.3 ± 19.3 | 12.2 ± 11.0 | 240 ± 73 |
| 1.5 | 5 | 19.1 ± 4.2 | 73.2 ± 30.8 | 18.9 ± 9.3 | 13.5 ± 11.2 | 271 ± 80 |
| 1.7 | 2 | 20.9 ± 2.7 | 58.3 ± 2.6 | 18.6 ± 0.8 | 21.6 ± 1.7 | 389 ± 26 |
| 1.9 | 4 | 39.6 ± 20.3 | 111.± 6 21.8 | 13.1 ± 1.6 | 13.8 ± 4.0 | 161 ± 29 |
| 2.1 | 3 | 32.1 ± 5.5 | 102.4 ± 22.9 | 12.4 ± 3.1 | 16.6 ± 5.4 | 202 ± 34 |
| All | 16 | 15.8 ± 6.6 | 15.9 ± 9.6 | 232 ± 81 |
Abbreviations; Cmax, maximum drug concentration in plasma; AUC0-24, area under the plasma concentration-time curve from 0 to 24 h; t½,z, half-life of the apparent terminal disposition phase; CL, total body clearance; Vss, steady-state apparent volume of distribution ; −EIASD cohort, patients not taking enzyme-inducing antiseizure drugs; +EIASD cohort, patients taking enzyme-inducing antiseizure drugs. Data are presented as the geometric mean ± SD.
Mean pharmacokinetic parameters for the first daily dose of karenitecin
| Dose (mg/m2) | Number of patients | Cmax (ng/ml) | AUC0-24 (ng·h/ml) | t½,z (h) | CL (liters/h/m2) | Vss (liters/m2) |
|---|---|---|---|---|---|---|
| −EIASD cohort | ||||||
| 1.0 | 3 | 19.4 ± 6.8 | 101.9 ± 1.7 | 13.5 ± 2.6 | 7.7 ± 0.6 | 116 ± 22 |
| 1.5 | 6 | 35.8 ± 20.9 | 114.7 ± 34.0 | 13.3 ± 5.5 | 10.5 ± 3.5 | 136 ± 51 |
| 1.8 | 3 | 24.4 ± 7.3 | 119.9 ± 15.6 | 12.7 ± 2.8 | 11.9 ± 2.5 | 161 ± 21 |
| All | 12 | 13.2 ± 4.1 | 10.2 ± 3.5 | 136 ± 41 | ||
| +EIASD cohort | ||||||
| 1.0 | 2 | 16.4 ± 2.2 | 52.7 ± 20.9 | 18.3 ± 19.3 | 12.2 ± 11.0 | 240 ± 73 |
| 1.5 | 5 | 19.1 ± 4.2 | 73.2 ± 30.8 | 18.9 ± 9.3 | 13.5 ± 11.2 | 271 ± 80 |
| 1.7 | 2 | 20.9 ± 2.7 | 58.3 ± 2.6 | 18.6 ± 0.8 | 21.6 ± 1.7 | 389 ± 26 |
| 1.9 | 4 | 39.6 ± 20.3 | 111.± 6 21.8 | 13.1 ± 1.6 | 13.8 ± 4.0 | 161 ± 29 |
| 2.1 | 3 | 32.1 ± 5.5 | 102.4 ± 22.9 | 12.4 ± 3.1 | 16.6 ± 5.4 | 202 ± 34 |
| All | 16 | 15.8 ± 6.6 | 15.9 ± 9.6 | 232 ± 81 |
| Dose (mg/m2) | Number of patients | Cmax (ng/ml) | AUC0-24 (ng·h/ml) | t½,z (h) | CL (liters/h/m2) | Vss (liters/m2) |
|---|---|---|---|---|---|---|
| −EIASD cohort | ||||||
| 1.0 | 3 | 19.4 ± 6.8 | 101.9 ± 1.7 | 13.5 ± 2.6 | 7.7 ± 0.6 | 116 ± 22 |
| 1.5 | 6 | 35.8 ± 20.9 | 114.7 ± 34.0 | 13.3 ± 5.5 | 10.5 ± 3.5 | 136 ± 51 |
| 1.8 | 3 | 24.4 ± 7.3 | 119.9 ± 15.6 | 12.7 ± 2.8 | 11.9 ± 2.5 | 161 ± 21 |
| All | 12 | 13.2 ± 4.1 | 10.2 ± 3.5 | 136 ± 41 | ||
| +EIASD cohort | ||||||
| 1.0 | 2 | 16.4 ± 2.2 | 52.7 ± 20.9 | 18.3 ± 19.3 | 12.2 ± 11.0 | 240 ± 73 |
| 1.5 | 5 | 19.1 ± 4.2 | 73.2 ± 30.8 | 18.9 ± 9.3 | 13.5 ± 11.2 | 271 ± 80 |
| 1.7 | 2 | 20.9 ± 2.7 | 58.3 ± 2.6 | 18.6 ± 0.8 | 21.6 ± 1.7 | 389 ± 26 |
| 1.9 | 4 | 39.6 ± 20.3 | 111.± 6 21.8 | 13.1 ± 1.6 | 13.8 ± 4.0 | 161 ± 29 |
| 2.1 | 3 | 32.1 ± 5.5 | 102.4 ± 22.9 | 12.4 ± 3.1 | 16.6 ± 5.4 | 202 ± 34 |
| All | 16 | 15.8 ± 6.6 | 15.9 ± 9.6 | 232 ± 81 |
Abbreviations; Cmax, maximum drug concentration in plasma; AUC0-24, area under the plasma concentration-time curve from 0 to 24 h; t½,z, half-life of the apparent terminal disposition phase; CL, total body clearance; Vss, steady-state apparent volume of distribution ; −EIASD cohort, patients not taking enzyme-inducing antiseizure drugs; +EIASD cohort, patients taking enzyme-inducing antiseizure drugs. Data are presented as the geometric mean ± SD.
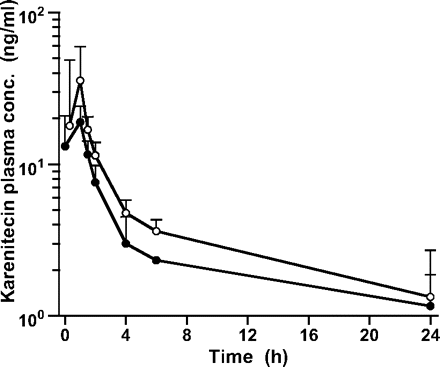
Mean plasma concentration-time profiles of karenitecin for the first daily dose of 1.5 mg/m2 given as a 1-h i.v. infusion to the -EIASD (open circles) and †EIASD (solid circles) treatment groups. Data points are connected sequentially with line segments and shown together with 1 SD unit error bars.
Discussion
The camptothecins continue to be studied in clinical trials in patients with newly diagnosed and recurrent high-grade gliomas.3 The anticancer activity of the camptothecins is dependent upon the intrinsic chemical reactivity of an α-hydroxy-δ-lactone ring that is susceptible to reversible hydrolysis, resulting in biologically inactive agent.2 Whereas topotecan is intrinsically cytotoxic, irinotecan is a prodrug that is inefficiently converted by human carboxylesterases, predominantly in the liver, to the biologically active compound 7-ethyl-10-hydroxycamptothecin (SN-38).28 The percentage of these compounds present in the active lactone form at equilibrium in human whole blood in vitro is only 12% for topotecan, 21% for irinotecan, and 20% for SN-38.29
Investigations undertaken to improve the pharmacologic properties of the first generation of clinically approved camptothecins revealed that the lactone-carboxylate equilibrium position can be shifted in favor of the active lactone form in human blood by the presence of a lipophilic functional group on the quinoline subunit, specifically, at the 7-position of the camptothecin ring system.13 In addition to greater lactone stability in blood, highly lipophilic camptothecin analogues may exhibit further enhanced therapeutic effectiveness by better tissue penetration and oral bioavailability compared with the water-soluble camptothecins.14 Karenitecin is a highly lipophilic, 7-alkylsilane derivative of camptothecin that exists almost entirely as the active lactone form in plasma, and it exhibits good oral bioavailability.14,30 Furthermore, karenitecin does not require metabolic activation, and it is not a substrate of UDP glucuronosyltransferase or the various ATP-binding cassette membrane transport proteins associated with drug resistance in cancer cells.
Several clinical trials have been performed with karenitecin given as a daily 1-h i.v. infusion for 5 consecutive days, with additional cycles of therapy repeated every 21 days. The MTD was found to be 1.0 mg/m2/day in a phase I trial involving patients with advanced solid malignancies.16 Reversible neutropenia and thrombocytopenia were the DLTs observed, and gastrointestinal toxicities were minimal. Phase II trials employing this same dose regimen were undertaken to assess the activity of karenitecin as second-line therapy in patients with relapsed or refractory non-small-cell lung cancer and heavily pretreated patients with malignant melanoma.31,32 Disease stabilization was the primary indication of clinical benefit in both studies. Objective responses occurred in only 2 of 52 patients with non-small-cell lung cancer (partial responses) and in none of the 43 melanoma patients evaluated.
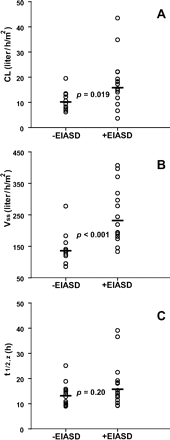
Pharmacokinetic parameters for karenitecin in patients taking enzyme-inducing antiseizure drugs (†EIASD) and not taking EIASDs (-EIASD): (A) total body clearance (CL), (B) apparent volume of distribution at steady state (Vss), and (C) terminal-phase half-life (t½,z). Circles represent the observed values in individual patients, and horizontal bars depict the geometric mean value for each group. Statistical comparison of the log-transformed data between the treatment groups was performed using a two-tailed t-test, assuming unequal variances.
The study reported here was designed to determine the MTD of karenitecin, to evaluate the influence of concurrent EIASD administration on its pharmacokinetics, and to estimate the activity in patients with recurrent high-grade gliomas who had minimal prior chemotherapy. All three of these goals were achieved upon enrolling 32 patients into this dose escalation trial that employed the modified continual reassessment method to minimize patient numbers. The MTD in -EIASD patients was noted to be 1.5 mg/m2 rather than 1.0 mg/m2 as noted in other solid tumors, for reasons that are not understood. As noted with other camptothecin analogues, karenitecin CL was significantly enhanced by EIASDs. Consistent with the 29% higher karenitecin CL in the †EIASD patients, the MTD was greater for patients who were taking EIASDs (2.0 mg/m2) compared with those who were not (1.5 mg/m2). Even though the effect of EIASDs on the MTD of this agent is significant, it is far less striking than with irinotecan, for which the MTD is nearly 400% higher when EIASDs are given concomitantly.19
Clinical trials exploring the efficacy of other camptothecins in patients with high-grade gliomas have been disappointing.4-6 Response rates have ranged from 5% to 15% in most studies, and the toxicities, which include myelosuppression and life-threatening diarrhea, can be substantial. It was hoped that this novel, lipid-soluble camptothecin analogue would provide a more acceptable side effect profile and higher response rates in this patient population. The observed toxicities were modest and reversible. However, no responses were noted in 11 patients treated at or above the MTD who had previously received only one or fewer prior chemotherapy regimens. If encouraging efficacy data are defined as a 30% response rate in minimally pretreated recurrent disease, then with 0 out of 11 responses, the probability of achieving a 30% response rate is less than 2%. Using an exact binomial CI, we are 95% confident that the true response rate is not higher than 24%. As a result, it appears that karenitecin administered on this schedule is unlikely to contribute to the treatment of patients with recurrent MGs.
This study was supported by grants U01-CA62475, U01-CA105689, and P30-CA0516 from the National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (Bethesda, MD, USA), and by BioNumerik Pharmaceuticals, Inc. (San Antonio, TX, USA).
References
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma.
Garcia-Carbonero R, Supko JG. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins.
Reardon DA, Friedman HS, Powell JB Jr, Gilbert M, Yung WK. Irinotecan: promising activity in the treatment of malignant glioma.
Raymond E, Fabbro M, Boige V, et al. Multicentre phase II study and pharmacokinetic analysis of irinotecan in chemotherapy-naive patients with glioblastoma.
Batchelor TT, Gilbert MR, Supko JG, et al. Phase 2 study of weekly irinotecan in adults with recurrent malignant glioma: final report of NABTT 97-11.
Prados MD, Lamborn K, Yung WK, et al. A phase 2 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: a North American Brain Tumor Consortium Study.
Macdonald D, Cairncross G, Stewart D, et al. Phase II study of topotecan in patients with recurrent malignant glioma. National Clinical Institute of Canada Clinical Trials Group.
Friedman HS, Kerby T, Fields S, et al. Topotecan treatment of adults with primary malignant glioma. The Brain Tumor Center at Duke.
Burch PA, Bernath AM, Cascino TL, et al. A North Central Cancer Treatment Group phase II trial of topotecan in relapsed gliomas.
Fisher B, Won M, Macdonald D, Johnson DW, Roa W. Phase II study of topotecan plus cranial radiation for glioblastoma multiforme: results of Radiation Therapy Oncology Group 9513.
Blaney SM, Takimoto C, Murry DJ, et al. Plasma and cerebrospinal fluid pharmacokinetics of 9-aminocamptothecin (9-AC), irinotecan (CPT-11), and SN-38 in nonhuman primates.
Baker SD, Heideman RL, Crom WR, Kuttesch JF, Gajjar A, Stewart CF. Cerebrospinal fluid pharmacokinetics and penetration of continuous infusion topotecan in children with central nervous system tumors.
Bom D, Curran DP, Kruszewski S, et al. The novel silatecan 7-tertbutyldimethylsilyl-10-hydroxycamptothecin displays high lipophilicity, improved human blood stability, and potent anticancer activity.
Van Hattum AH, Pinedo HM, Schluper HM, Hausheer FH, Boven E. New highly lipophilic camptothecin BNP1350 is an effective drug in experimental human cancer.
Keir ST, Hausheer F, Lawless AA, Bigner DD, Friedman HS. Therapeutic activity of 7-[(2-trimethylsilyl)ethyl)]-20 (S)-camptothecin against central nervous system tumor-derived xenografts in athymic mice.
Schilsky R, Hausheer F, Bertucci D, Berghorn E, Kindler H, Ratain M. Phase I trial of karenitecin (KT) administered intravenously daily for five consecutive days in patients with advanced solid tumor using accelerated dose titration [abstract].
Watling CJ, Lee DH, Macdonald DR, Cairncross JG. Corticosteroid-induced magnetic resonance imaging changes in patients with recurrent malignant glioma.
Piantadosi S, Fisher JD, Grossman S. Practical implementation of a modified continual reassessment method for dose-finding trials.
Gilbert MR, Supko JG, Batchelor T, et al. Phase I clinical and pharmacokinetic study of irinotecan in adults with recurrent malignant glioma.
Supko JG, Malspeis LA. A reversed phase HPLC method for determining camptothecin in plasma with specificity for the intact lactone form of the drug.
Supko JG, Malspeis LA. Liquid chromatographic analysis of 9-aminocamptothecin in plasma monitored by fluorescence induced upon postcolumn acidification.
Wall MF, Wani MC, Cook CE, Palmer KK, Mcphail AT, Sim GA. Plant antitumor agents. 1. The isolation and structure of campothecin, a novel alkaloid leukemia and tumor inhibitor from Camptotheca acuminata.
Hassner A, Krepski I, Alexanian V. Aminopyridines as acylation catalysts for tertiary alcohols.
Mizuta E, Tsubotani A. Preparation of mean drug concentration-time curves in plasma. A study on the frequency distribution of pharmacokinetic parameters.
Lacey LF, Keene ON, Pritchard JF, Bye A. Common noncompartmental pharmacokinetic variables: are they normally or log-normally distributed?
Shah VP, Midha KK, Findlay JW, et al. Bioanalytical method validation—a revisit with a decade of progress.
Mathijssen RH, van Alphen RJ, Verweij J, et al. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11).
Burke TG, Munshi CB, Mi Z, Jiang Y. The important role of albumin in determining the relative human blood stabilities of the camptothecin anticancer drugs.
Thompson PA, Berg SL, Aleksic A, et al. Plasma and cerebrospinal fluid pharmacokinetic study of BNP1350 in nonhuman primates.
Miller AA, Herndon JE 2nd, Gu L, Green MR, Cancer and Leukemia Group B. Phase II trial of karenitecin in patients with relapsed or refractory non-small cell lung cancer (CALGB 30004).



{kind=link}
{kind=link}
{kind=link}