





Abstract
Chemotherapy or irradiation treatment induces breast cancer cell apoptosis, but this can be limited by estradiol (E2) through unknown mechanisms. To investigate this, we subjected estrogen receptor-expressing human breast cancer cells (MCF-7 and ZR-75–1) to paclitaxel (taxol) or to UV irradiation. Marked increases in cell apoptosis were induced, but these were significantly reversed by incubation with E2. Taxol or UV stimulated c-Jun N-terminal kinase (JNK) activity, which was inhibited by E2. Expression of a dominant-negative Jnk-1 protein strongly prevented taxol- or UV-induced apoptosis, whereas E2 inhibition of apoptosis was reversed by expression of constituitively active Jnk-1. As targets for participation in apoptosis, Bcl-2 and Bcl-xl were phosphorylated in response to JNK activation by taxol or UV; this was prevented by E2. Taxol or UV activated caspase activity in a JNK-dependent fashion and caused the cleavage of procaspase-9 to caspase-9, each inhibited by E2. Independently, the steroid also activated extracellular signal-regulated protein kinase activity, which contributed to the anti-apoptotic effects. We report novel and rapid mechanisms by which E2 prevents chemotherapy or radiation-induced apoptosis of breast cancer, probably mediated through the plasma membrane estrogen receptor.
INTRODUCTION
Estrogen receptors are expressed in approximately 65% of human breast cancer, implying that this sex steroid plays an important role in the development and propagation of the disease (1, 2). Approximately one third of women with breast cancer respond to ablative endocrine therapy (3, 4), and anti-estrogens positively influence the course of established breast cancer (5) or prevent the development of primary disease (6). Based upon in vitro and in vivo data, estrogen probably acts as both a growth factor and a survival factor for breast cancer (1, 2).
The ability of estradiol (E2) to act as a survival factor for breast cancer is not well understood, but a substantial part of the effects probably occur through the prevention of programmed cell death, apoptosis (7). Apoptosis is often initiated when a cell is exposed to a stressful stimulus, which then triggers a transmembrane signal to an intracellular protease cascade, primarily composed of the caspase family (8). As a result, intracellular enzymes are activated that cleave DNA and cause cell shrinkage, chromatin condensation, and membrane blebbing. In the early course of establishment of breast cancer, a cytokine response could induce apoptosis of the cancer cells via the activation of cell surface receptors for tumor necrosis factor, as an example (9). In established breast cancer, treatment with chemotherapy or irradiation induces apoptosis. In vitro, breast cancer treatment with chemotherapy is markedly less effective in the setting of estrogen (10, 11). Thus, E2 may establish a survival advantage in this setting, but the mechanisms of this effect are not well understood.
The actions of E2 are traditionally thought to be mediated by the nuclear estrogen receptor (ER), through the regulation of target gene transcription (12). This occurs when ER either binds estrogen response elements on the promoters of target genes, or acts through protein-protein interactions involving a variety of coactivators, corepressors, and the basal transcriptional machinery protein complex. Emerging evidence, however, has implicated a second distinct mechanism of E2 action, where this steroid binds a putative plasma membrane ER and enacts signal transduction (13, 14). Each mechanism could work cooperatively or distinctly to effect cell biological actions. Conceivably, the ability of E2 to prevent apoptosis in several target cells (15, 16) could be initiated through signal transduction mediated through the plasma membrane receptor.
As the anti-apoptotic effects of E2 are poorly understood, we investigated the mechanism of action and whether the membrane receptor mediates these effects of the sex steroid.
RESULTS
Apoptosis is Inhibited by Membrane ER Ligation
MCF-7 and ZR-75–1 cells were subjected either to a brief (1-min) UV irradiation, followed by 4-h incubation, or to 20μ m taxol treatment for 4 h. As shown in Fig. 1A, UV (panel b) induced 13% of the MCF-7 breast cancer cells to undergo apoptosis, compared with 1% in the control cells (panel a). Preincubation with 10 nm E2 or 100 nm E2-BSA (panels c and d), significantly prevented the effect of UV, lowering cell death to 6% and 7%, respectively. The protective effects of either estrogen compound were reversed by ICI182,780, the specific ER antagonist (panels e and f). Similar effects were seen for taxol-induced apoptosis (Fig. 1B), although taxol was not as potent in this regard as UV exposure. The data are summated in the bar graphs below each composite figure. These short exposures were chosen to support the idea that a rapid, nongenomic effect of E2 might be involved. In contrast, neither 100 nm testosterone nor progesterone had any significant effect on UV or taxol-induced apoptosis in MCF-7 cells (data not shown).
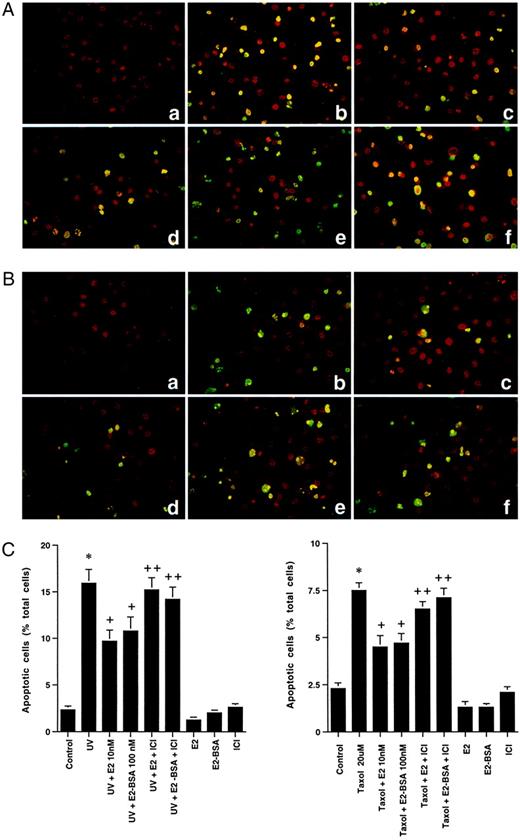
A, Apoptosis of MCF-7 Cells Is Induced by UV Irradiation Exposure for 1 Min (b), as Demonstrated by Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling Staining Compared with Control Cells (a) This is inhibited by 10 nm E2 (c) or 100 nm E2-BSA (d), which is reversed by 1 μm ICI182,780, an ER antagonist (e and f, respectively). B, Taxol (20 μm) exposure for 4 h induces apoptosis in MCF-7 cells (b), inhibited by E2 or E2-BSA (c and d), again reversed by ICI182,780 (e and f). The study shown here was repeated three times. Bar graphs show the mean ± sem number of apoptotic cells in each condition, based on combined data, which are shown below the composites (C). Apoptotic cells are stained yellow/green compared with viable cells (red) stained with propidium iodide.
These results indicate that E2 is rapidly acting through a specific plasma membrane ER to inhibit apoptosis. This is supported by the similar effects of E2-BSA, a compound that has been shown by several laboratories to neither enter the cell nor bind/activate the nuclear ER (17–19). E2-BSA has previously been shown to be less potent than E2, perhaps due to the steric hindrance of E2 accessing its receptor, when conjugated to BSA (17–19). We have previously shown that BSA by itself does not affect signaling (18).
To further support the idea that E2 and E2-BSA are acting through a membrane ER, we transiently transfected the cells with an estrogen response element (ERE)-luciferase reporter construct, as previously described (19). Over 8 h, E2 (10 nm) significantly stimulated the reporter activity, but E2-BSA did not (Fig. 2). This indicates that E2-BSA does not enter the cell to bind the nuclear ER by 8 h, determined at several times later than the inhibition of apoptosis shown here (4 h). These results also indicate that E2 does not substantially dissociate from the BSA, although this has recently been called into question (see Discussion).
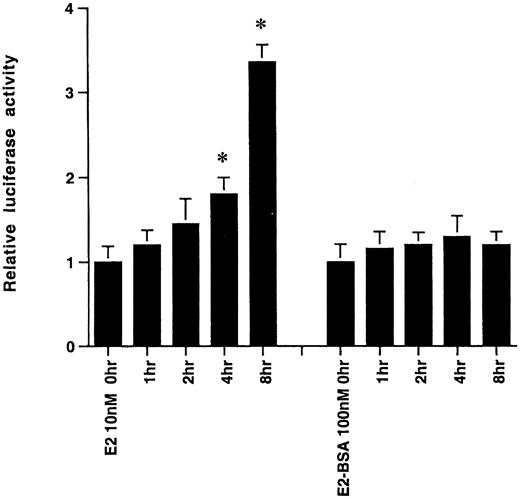
Activation of an ERE-Luciferase Reporter Over Time by E2 (10 nm), but Not by E2-BSA (100 nm) The results are the mean ± sem luciferase activity, determined from triplicate determinations per time point in each experiment. All data were combined from three separate experiments. *, P < 0.05, by ANOVA and Scheffe’s test for time zero vs. 4 or 6 h.
We also assessed apoptosis by labeling MCF-7 and another ER-positive breast cancer cell, ZR-75–1, with bromodeoxyuridine and propidium iodide; we then determined cell death by fluorescent-activated cell sorting (FACS) analysis. As shown in Table 1, UV and taxol induced at least, 6- and 4-fold respective increases in programmed death in either cell line. E2 afforded between a 66–80% protection against apoptosis across both conditions and both cell lines. The effects of E2-BSA were comparable to those of E2, and all steroid actions were reversed 70–90% by ICI182,780. In contrast, E2 had no effect on UV- or taxol-induced apoptosis in an ER-negative cell line, HCC1569 (data not shown).
Apoptosis in Breast Cancer Cells after Exposure to Ultraviolet (UV) Irradiation or Taxol, in the Presence or Absence of Estrogen
| Treatment | % Apoptosis (BrdU incorporation) | |
|---|---|---|
| MCF-7 cells | ZR-75-1 cells | |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| UV | 13.0 ± 1.0a | 12.1 ± 0.9a |
| UV+ E2 10 nm | 4.1 ± 0.5b | 3.0 ± 0.3b |
| UV+ E2+ ICI 1 μm | 10.1 ± 0.8a | 9.3 ± 0.7a |
| UV+ E2-BSA 100 nm | 5.9 ± 0.9b | — |
| UV+ E2-BSA+ ICI 1 μm | 11.3 ± 1.2a | — |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| Taxol 20 μm | 7.6 ± 0.8a | 6.5 ± 0.5a |
| Taxol+ E2 10 nm | 3.4 ± 0.4b | 2.7 ± 0.3b |
| Taxol+ E2+ ICI 1 μm | 6.6 ± 0.5a | 5.3 ± 0.5a |
| Taxol+ E2-BSA 100 nm | 4.1 ± 0.3b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 6.1 ± 0.4a | — |
| Treatment | % Apoptosis (BrdU incorporation) | |
|---|---|---|
| MCF-7 cells | ZR-75-1 cells | |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| UV | 13.0 ± 1.0a | 12.1 ± 0.9a |
| UV+ E2 10 nm | 4.1 ± 0.5b | 3.0 ± 0.3b |
| UV+ E2+ ICI 1 μm | 10.1 ± 0.8a | 9.3 ± 0.7a |
| UV+ E2-BSA 100 nm | 5.9 ± 0.9b | — |
| UV+ E2-BSA+ ICI 1 μm | 11.3 ± 1.2a | — |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| Taxol 20 μm | 7.6 ± 0.8a | 6.5 ± 0.5a |
| Taxol+ E2 10 nm | 3.4 ± 0.4b | 2.7 ± 0.3b |
| Taxol+ E2+ ICI 1 μm | 6.6 ± 0.5a | 5.3 ± 0.5a |
| Taxol+ E2-BSA 100 nm | 4.1 ± 0.3b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 6.1 ± 0.4a | — |
Apoptosis was determined by FACS analysis of cells stained with propidium iodide and BrdU. Data are the mean ± sem from three experiments combined.
P < 0.05 by ANOVA plus Scheffe’s for control vs. condition or UV or taxol versus UV or taxol + ICI.
P < 0.05 for UV or taxol vs. UV or taxol + E2. ICI alone was not different from control (data not shown).
Apoptosis in Breast Cancer Cells after Exposure to Ultraviolet (UV) Irradiation or Taxol, in the Presence or Absence of Estrogen
| Treatment | % Apoptosis (BrdU incorporation) | |
|---|---|---|
| MCF-7 cells | ZR-75-1 cells | |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| UV | 13.0 ± 1.0a | 12.1 ± 0.9a |
| UV+ E2 10 nm | 4.1 ± 0.5b | 3.0 ± 0.3b |
| UV+ E2+ ICI 1 μm | 10.1 ± 0.8a | 9.3 ± 0.7a |
| UV+ E2-BSA 100 nm | 5.9 ± 0.9b | — |
| UV+ E2-BSA+ ICI 1 μm | 11.3 ± 1.2a | — |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| Taxol 20 μm | 7.6 ± 0.8a | 6.5 ± 0.5a |
| Taxol+ E2 10 nm | 3.4 ± 0.4b | 2.7 ± 0.3b |
| Taxol+ E2+ ICI 1 μm | 6.6 ± 0.5a | 5.3 ± 0.5a |
| Taxol+ E2-BSA 100 nm | 4.1 ± 0.3b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 6.1 ± 0.4a | — |
| Treatment | % Apoptosis (BrdU incorporation) | |
|---|---|---|
| MCF-7 cells | ZR-75-1 cells | |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| UV | 13.0 ± 1.0a | 12.1 ± 0.9a |
| UV+ E2 10 nm | 4.1 ± 0.5b | 3.0 ± 0.3b |
| UV+ E2+ ICI 1 μm | 10.1 ± 0.8a | 9.3 ± 0.7a |
| UV+ E2-BSA 100 nm | 5.9 ± 0.9b | — |
| UV+ E2-BSA+ ICI 1 μm | 11.3 ± 1.2a | — |
| Control | 1.8 ± 0.5 | 0.8 ± 0.2 |
| Taxol 20 μm | 7.6 ± 0.8a | 6.5 ± 0.5a |
| Taxol+ E2 10 nm | 3.4 ± 0.4b | 2.7 ± 0.3b |
| Taxol+ E2+ ICI 1 μm | 6.6 ± 0.5a | 5.3 ± 0.5a |
| Taxol+ E2-BSA 100 nm | 4.1 ± 0.3b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 6.1 ± 0.4a | — |
Apoptosis was determined by FACS analysis of cells stained with propidium iodide and BrdU. Data are the mean ± sem from three experiments combined.
P < 0.05 by ANOVA plus Scheffe’s for control vs. condition or UV or taxol versus UV or taxol + ICI.
P < 0.05 for UV or taxol vs. UV or taxol + E2. ICI alone was not different from control (data not shown).
To examine an early event in apoptosis, the cells were assessed for cell membrane binding of annexin V, again determined by FACS. In the cell undergoing programmed cell death, annexin V can bind phosphatidlyserine, which is expressed in the outer plasma membrane leaflet of dying cells. In both ER-positive breast cancer cell lines, UV or taxol induced 16-fold (MCF-7) and 8.5-fold (ZR-75) increases in annexin binding compared with control cells. E2 or E2-BSA significantly reversed this by 52–86% across all conditions and both cell lines (Table 2). ICI182,780 substantially reversed the effects of either E2 or E2-BSA.
Annexin-V Binding to the Membrane of Breast Cancer Cells Exposed to Ultraviolet (UV) Irradiation or Taxol, with or without Estrogen
| Treatment | % Apoptosis (Annexin V binding) | |
|---|---|---|
| MCF7 cells | ZR-75-1 cells | |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| UV | 12.8 ± 2.1a | 18.7 ± 2.1a |
| UV+ E2 10 nm | 2.5 ± 0.3b | 10.1 ± 1.2b |
| UV+ E2+ ICI 1 μm | 9.7 ± 1.2a | 15.5 ± 1.7a |
| UV+ E2-BSA 100 nm | 5.7 ± 0.6b | — |
| UV+ E2-BSA+ ICI 1 μm | 9.7 ± 1.3a | — |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| Taxol 20 μm | 8.3 ± 1.2a | 11.7 ± 2.1a |
| Taxol+ E2 10 nm | 2.7 ± 0.5b | 6.1 ± 1.1b |
| Taxol+ E2+ ICI 1 μm | 5.6 ± 0.3a | 9.1 ± 1.3a |
| Taxol+ E2-BSA 100 nm | 2.2 ± 0.6b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 5.4 ± 0.6a | — |
| Treatment | % Apoptosis (Annexin V binding) | |
|---|---|---|
| MCF7 cells | ZR-75-1 cells | |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| UV | 12.8 ± 2.1a | 18.7 ± 2.1a |
| UV+ E2 10 nm | 2.5 ± 0.3b | 10.1 ± 1.2b |
| UV+ E2+ ICI 1 μm | 9.7 ± 1.2a | 15.5 ± 1.7a |
| UV+ E2-BSA 100 nm | 5.7 ± 0.6b | — |
| UV+ E2-BSA+ ICI 1 μm | 9.7 ± 1.3a | — |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| Taxol 20 μm | 8.3 ± 1.2a | 11.7 ± 2.1a |
| Taxol+ E2 10 nm | 2.7 ± 0.5b | 6.1 ± 1.1b |
| Taxol+ E2+ ICI 1 μm | 5.6 ± 0.3a | 9.1 ± 1.3a |
| Taxol+ E2-BSA 100 nm | 2.2 ± 0.6b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 5.4 ± 0.6a | — |
Apoptosis was determined by FACS analysis of cells binding Annexin-V-FITC to phosphatidylserine on the membrane. Data are the mean ± sem from three experiments combined.
P < 0.05 by ANOVA plus Scheffe’s for control vs. condition, or UV or taxol vs. same + ICI.
P < 0.05 for UV or taxol vs. UV or taxol + E2. ICI was not different from control (data not shown).
Annexin-V Binding to the Membrane of Breast Cancer Cells Exposed to Ultraviolet (UV) Irradiation or Taxol, with or without Estrogen
| Treatment | % Apoptosis (Annexin V binding) | |
|---|---|---|
| MCF7 cells | ZR-75-1 cells | |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| UV | 12.8 ± 2.1a | 18.7 ± 2.1a |
| UV+ E2 10 nm | 2.5 ± 0.3b | 10.1 ± 1.2b |
| UV+ E2+ ICI 1 μm | 9.7 ± 1.2a | 15.5 ± 1.7a |
| UV+ E2-BSA 100 nm | 5.7 ± 0.6b | — |
| UV+ E2-BSA+ ICI 1 μm | 9.7 ± 1.3a | — |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| Taxol 20 μm | 8.3 ± 1.2a | 11.7 ± 2.1a |
| Taxol+ E2 10 nm | 2.7 ± 0.5b | 6.1 ± 1.1b |
| Taxol+ E2+ ICI 1 μm | 5.6 ± 0.3a | 9.1 ± 1.3a |
| Taxol+ E2-BSA 100 nm | 2.2 ± 0.6b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 5.4 ± 0.6a | — |
| Treatment | % Apoptosis (Annexin V binding) | |
|---|---|---|
| MCF7 cells | ZR-75-1 cells | |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| UV | 12.8 ± 2.1a | 18.7 ± 2.1a |
| UV+ E2 10 nm | 2.5 ± 0.3b | 10.1 ± 1.2b |
| UV+ E2+ ICI 1 μm | 9.7 ± 1.2a | 15.5 ± 1.7a |
| UV+ E2-BSA 100 nm | 5.7 ± 0.6b | — |
| UV+ E2-BSA+ ICI 1 μm | 9.7 ± 1.3a | — |
| Control | 0.8 ± 0.2 | 2.2 ± 0.5 |
| Taxol 20 μm | 8.3 ± 1.2a | 11.7 ± 2.1a |
| Taxol+ E2 10 nm | 2.7 ± 0.5b | 6.1 ± 1.1b |
| Taxol+ E2+ ICI 1 μm | 5.6 ± 0.3a | 9.1 ± 1.3a |
| Taxol+ E2-BSA 100 nm | 2.2 ± 0.6b | — |
| Taxol+ E2-BSA+ ICI 1 μm | 5.4 ± 0.6a | — |
Apoptosis was determined by FACS analysis of cells binding Annexin-V-FITC to phosphatidylserine on the membrane. Data are the mean ± sem from three experiments combined.
P < 0.05 by ANOVA plus Scheffe’s for control vs. condition, or UV or taxol vs. same + ICI.
P < 0.05 for UV or taxol vs. UV or taxol + E2. ICI was not different from control (data not shown).
Apoptosis Is Modulated through c-Jun N-terminal kinase (JNK) Activity
We then determined whether the induction of apoptosis by UV or taxol was dependent on JNK activity and could be modified by E2. JNK is known to mediate the activation of apoptosis in several cell types, in response to various stressors (20). We first found that either UV or taxol could significantly activate JNK activity by 7- and 2.5-fold, respectively, in MCF-7, and significantly in ZR-75–1 cells (Fig. 3A, upper and lower panels). We also found that either E2 or E2-BSA caused a 50–67% reduction in the stimulated JNK activity seen in response to either stressor in both cell lines. The comparable effects of the two estrogen compounds were blocked by ICI182,780, and when considered along with the rapidity of inhibition by E2 (determined after 15 min of cell exposure), these results support a membrane ER as mediating this action. The effects of E2 or E2-BSA were also concentration related, with significant inhibition seen at 1 and 10 nm E2 in both cell lines (Fig. 3B). In contrast, E2 did not reduce UV- or taxol stimulated-JNK activity in HCC-1569 cells (data not shown).
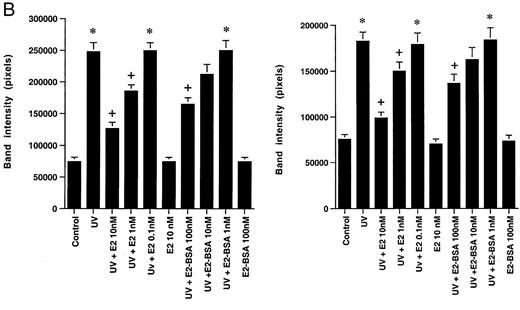
A, The c-Jun Kinase Activity in MCF-7 Cells (upper) and ZR-75–1 Cells (lower) after 15-Min Exposure to UV (left) or Taxol (right), in the Presence or Absence of E2, E2-BSA, or ICI182,780 JNK was immunoprecipitated from the treated MCF-7 cells, as described in Materials andMethods. A representative experiment of JNK activity directed against GST-c-Jun-(1–79) as substrate protein is shown, with the Jnk-1 immunoblots below each condition. The bargraph represents mean results ± SE of three experiments combined. *, P < 0.05 for control vs. UV or taxol; +, P < 0.05 for UV or taxol vs. UV or taxol plus E2 or E2-BSA; ++, P < 0.05 for UV or taxol plus E2 or E2-BSA vs. the former plus ICI. B, Concentration-related inhibition of UV- or taxol-stimulated JNK activity by E2 or E2-BSA in MCF-7 (left) and ZR-75–1 (right). The data reflect three separate experiments combined.
A critical issue is whether JNK activation is necessary and sufficient for the induction of apoptosis in this setting. As shown in Fig. 4A, MCF-7 cells transfected to express a dominant-negative Jnk-1 showed a significant reduction in the degree of apoptosis induced by UV [panel c vs. a (control)] or by taxol [panel d vs. b (control)]. Control MCF-7 cells were transfected with the empty plasmid vector and subjected to either of the two apoptotic stimuli. The reversal induced by dominant-negative Jnk-1 was substantial (63–67% for UV or taxol; Fig. 4B), which must be considered in the context that transfection efficiency does not allow complete inhibition of JNK activation. We had previously determined our transfection efficiency using this construct as approximately 75% (21). These results show the requirement of JNK for apoptosis induced by these two agents. Because UV- or taxol-induced JNK activation and apoptosis were blocked by this steroid, we believe that this is a novel target for the anti-apoptotic effects of estrogen.
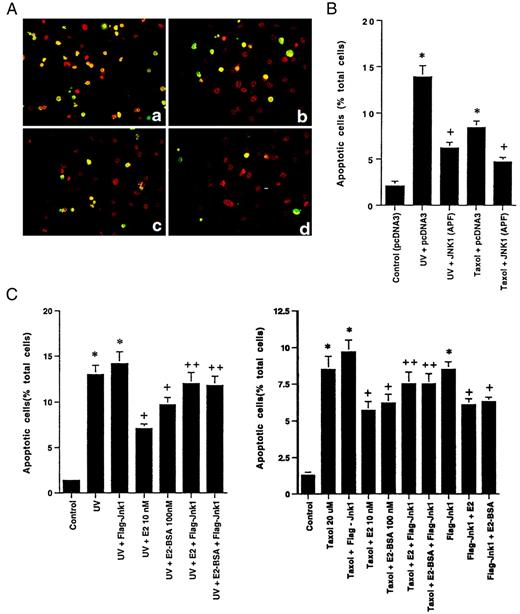
A, Apoptosis of pcDNA3-Transfected MCF-7 Cells in Response to UV (a) or Taxol (b) Is Substantially Greater Than That in Cells Transfected with Dominant-Negative Jnk-1 (pcDNA3 Flag-Jnk-1 APF), Then Exposed to UV (c) or Taxol (d) MCF-7 cells were transfected with dominant-negative (dom-neg) Jnk-1, recovered, then subjected to UV or taxol. B, Quantitation of three apoptosis experiments is shown in the bar graph. Control apoptosis (pcDNA3 transfected, but not subjected to UV or taxol) was 2% and is not shown. *, P < 0.05 for UV or taxol vs. UV or taxol plus dom-neg Jnk-1. C, Expression of constituitively active Jnk-1 (Flag-Jnk-1) reverses the E2 or E2-BSA inhibition of UV (left)- or taxol (right)-induced apoptosis. Data are from three experiments combined. *, P < 0.05 for UV or taxol vs. UV or taxol plus E2 or E2-BSA; +, P <0.05 for UV or taxol plus E2 or E2-BSA vs. the former in the presence of active Jnk-1
To further support this contention, we transfected MCF-7 cells with a mildly constituitively active Jnk-1 expression plasmid, a vector that we and others have previously characterized (20, 21). We then exposed the cells to UV or taxol in the presence or absence of E2 or E2-BSA. We found that active Jnk-1 resulted in a nearly complete reversal of the ability of E2 or E2-BSA to block UV- or taxol-induced apoptosis (Fig. 4C).
Bcl-2 and Bcl-xl Serve as Substrates for Jnk
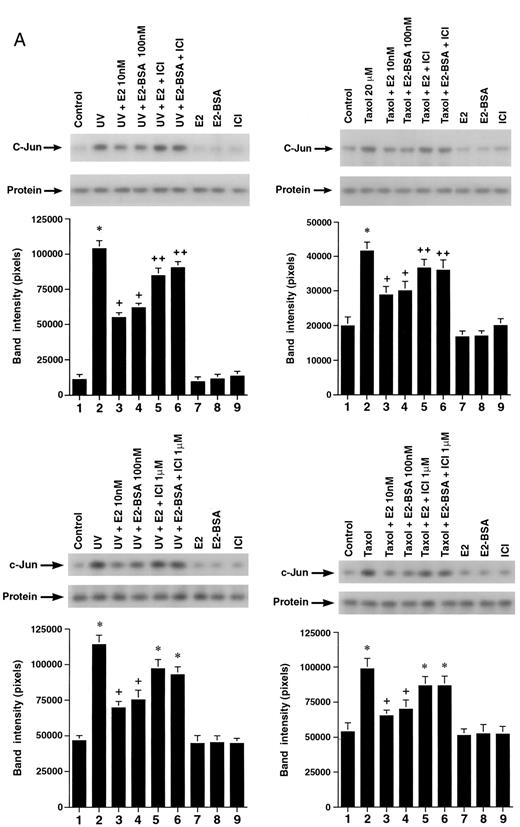
What JNK targets are critical for the apoptosis-inducing action of this enzyme? It has recently been shown that the anti-apoptotic protein Bcl-2 can be phosphorylated. In several cell types, phosphorylation by, for instance, protein kinase A or c-Jun kinase (JNK), down-regulates Bcl-2 actions to prevent cell death (22–24), although concomitant activation of phosphatases or phosphorylation of a different site may lead to activation of the protein in some contexts (25). However, recent data clearly indicate that JNK can inactivate Bcl-2 function by directly phosphorylating the loop domain of this protein (24). We determined that UV or taxol was capable of significantly stimulating the phosphorylation of Bcl-2 (Fig. 5A). Further, the phosphorylation was mainly attributable to JNK activation, as transfection of the MCF-7 cells with dominant-negative Jnk-1 almost completely reversed this effect of UV or taxol.
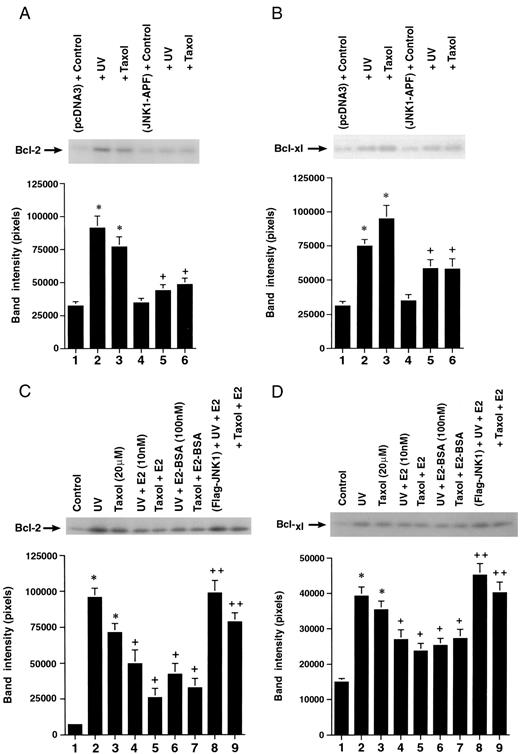
A, UV or Taxol Induces the Increased Phosphorylation of Bcl-2 and Bcl-xl Proteins in a JNK-Dependent Fashion after 15-Min Incubation JNK1-APF, Dominant-negative (dom-neg) Jnk-1. A representative study is shown, repeated three times to constitute the bar graph. *, P < 0.05 for control vs. UV or taxol; +, P < 0.05 for UV or taxol vs. UV or taxol plus dom-neg Jnk-1. B, UV or taxol induces the increased phosphorylation of Bcl-xl protein in a JNK-dependent manner. A representative study from three experiments is shown. C, E2 or E2-BSA inhibits UV- or taxol-induced phosphorylation of Bcl-2 (left) and Bcl-xl (right), which is reversed by expressing active Jnk-1 (Flag-JNK-1).
Another important anti-apoptotic protein in this family is Bcl-xl. We similarly found that UV and taxol induced JNK-dependent phosphorylation of this protein in MCF-7 cells (Fig. 5B). However, in this situation, another kinase may additionally contribute. As E2 or E2-BSA inhibits JNK activation, we hypothesized that they would block UV- or taxol-induced phosphorylation of Bcl-2 and Bcl-xl. We found this to be the case, in that either estrogen compound significantly inhibited the phosphorylation of these two proteins by 53–64% (Fig. 5C). To support a JNK-related mechanism of action, the blocking of either Bcl-2 or Bcl-xl phosphorylation by E2 was substantially reversed when a constituitively active Jnk-1 was expressed in the MCF-7 cells (Fig. 5C, lanes 8 and 9). This identifies a novel downstream target for the anti-apoptotic effects of the membrane ER, and the mechanism is probably mediated through the inhibition of JNK activation.
Caspase Activation Is JNK Dependent and Is Inhibited by E2 or E2-BSA
The inactivation of Bcl-2 or Bcl-xl induced by UV- or taxol-induced phosphorylation might lead to activation of caspase activity (22, 23). We determined this in whole cell lysates using a fluorogenic substrate that assesses caspase-4, -5, and -9 activities (26). UV- or taxol-treated cells showed 91% and 75% increases, respectively, in caspase activity, compared with nontreated, control MCF-7 cells (Table 3). Adding E2-BSA or E2 reduced the enhanced caspase activity by 41%–53% across both conditions. To determine a role for JNK, MCF-7 cells were transfected with the empty plasmid, pcDNA3, or the dominant negative Jnk-1 construct. In MCF-7 cells transfected with pcDNA3, the apoptosis was higher than that in nontransfected cells. However, UV and taxol still significantly enhanced caspase activity in this setting, by 68% and 51%, respectively (Table 3). Expression of Flag-Jnk-1 APF significantly reduced this stimulated caspase activity by 65% and 51%, respectively.
E2 inhibits UV or Taxol-Induced Activation of Caspase Activity
| Condition | Absorbance units/ (mg protein) |
|---|---|
| Control | 203 ± 11 |
| UV | 387 ± 14a |
| UV+ E2 10 nm | 299 ± 16b |
| UV+ E2-BSA 100 nm | 308 ± 12b |
| Taxol 20 μm | 355 ± 18a |
| Taxol+ E2 | 275 ± 13b |
| Taxol+ E2-BSA | 291 ± 11b |
| E2 | 208 ± 14 |
| E2-BSA | 198 ± 12 |
| pcDNA3 | 301 ± 21 |
| pcDNA3+ UV | 504 ± 24a |
| pcDNA3+ Taxol | 452 ± 18a |
| pcDNA3 Flag-JNK-1 APF+ UV | 372 ± 18c |
| pcDNA3 Flag-JNK-1 APF+ Taxol | 379 ± 20c |
| pcDNA3 Flag-JNK-1 APF | 278 ± 19 |
| Condition | Absorbance units/ (mg protein) |
|---|---|
| Control | 203 ± 11 |
| UV | 387 ± 14a |
| UV+ E2 10 nm | 299 ± 16b |
| UV+ E2-BSA 100 nm | 308 ± 12b |
| Taxol 20 μm | 355 ± 18a |
| Taxol+ E2 | 275 ± 13b |
| Taxol+ E2-BSA | 291 ± 11b |
| E2 | 208 ± 14 |
| E2-BSA | 198 ± 12 |
| pcDNA3 | 301 ± 21 |
| pcDNA3+ UV | 504 ± 24a |
| pcDNA3+ Taxol | 452 ± 18a |
| pcDNA3 Flag-JNK-1 APF+ UV | 372 ± 18c |
| pcDNA3 Flag-JNK-1 APF+ Taxol | 379 ± 20c |
| pcDNA3 Flag-JNK-1 APF | 278 ± 19 |
Data are the mean ± sem from three experiments combined. aP < 0.05 for control vs. condition by ANOVA plus Schefe’s test; bP < 0.05 for condition vs. condition plus E2 or E2-BSA; cP < 0.05 for condition vs. condition plus dominant negative c-jun-1 kinase (pcDNA3 Flag-JNK-1 APF). Caspase activity was determined as described in Materials and Methods using a fluorogenic substrate that identifies caspase 4, 5 and 9 activities.
E2 inhibits UV or Taxol-Induced Activation of Caspase Activity
| Condition | Absorbance units/ (mg protein) |
|---|---|
| Control | 203 ± 11 |
| UV | 387 ± 14a |
| UV+ E2 10 nm | 299 ± 16b |
| UV+ E2-BSA 100 nm | 308 ± 12b |
| Taxol 20 μm | 355 ± 18a |
| Taxol+ E2 | 275 ± 13b |
| Taxol+ E2-BSA | 291 ± 11b |
| E2 | 208 ± 14 |
| E2-BSA | 198 ± 12 |
| pcDNA3 | 301 ± 21 |
| pcDNA3+ UV | 504 ± 24a |
| pcDNA3+ Taxol | 452 ± 18a |
| pcDNA3 Flag-JNK-1 APF+ UV | 372 ± 18c |
| pcDNA3 Flag-JNK-1 APF+ Taxol | 379 ± 20c |
| pcDNA3 Flag-JNK-1 APF | 278 ± 19 |
| Condition | Absorbance units/ (mg protein) |
|---|---|
| Control | 203 ± 11 |
| UV | 387 ± 14a |
| UV+ E2 10 nm | 299 ± 16b |
| UV+ E2-BSA 100 nm | 308 ± 12b |
| Taxol 20 μm | 355 ± 18a |
| Taxol+ E2 | 275 ± 13b |
| Taxol+ E2-BSA | 291 ± 11b |
| E2 | 208 ± 14 |
| E2-BSA | 198 ± 12 |
| pcDNA3 | 301 ± 21 |
| pcDNA3+ UV | 504 ± 24a |
| pcDNA3+ Taxol | 452 ± 18a |
| pcDNA3 Flag-JNK-1 APF+ UV | 372 ± 18c |
| pcDNA3 Flag-JNK-1 APF+ Taxol | 379 ± 20c |
| pcDNA3 Flag-JNK-1 APF | 278 ± 19 |
Data are the mean ± sem from three experiments combined. aP < 0.05 for control vs. condition by ANOVA plus Schefe’s test; bP < 0.05 for condition vs. condition plus E2 or E2-BSA; cP < 0.05 for condition vs. condition plus dominant negative c-jun-1 kinase (pcDNA3 Flag-JNK-1 APF). Caspase activity was determined as described in Materials and Methods using a fluorogenic substrate that identifies caspase 4, 5 and 9 activities.
Active caspase-9 facilitates the cleavage and activation of the death effector caspases, resulting in DNA fragmentation and apoptosis. Caspase-9 activation mainly results from proteolytic processing of procaspase-9. This processing is indirectly restrained by Bcl-2 or Bcl-xl and is directly activated through association with the cytochrome c-Apaf-1 complex. We therefore determined whether UV and taxol cleaved procaspase-9 to caspase-9, yielding detectable active fragments of the zymogen. In control MCF-7 cells, only the 46-kDa zymogen was detected. Both taxol and UV induced cleavage of the procaspase, yielding smaller molecular mass bands at 34 kDa, which were detected after stressor exposure (Fig. 6). E2 or E2-BSA substantially prevented the cleavage of procaspase-9 in the setting of either stressor for the MCF-7 cells.
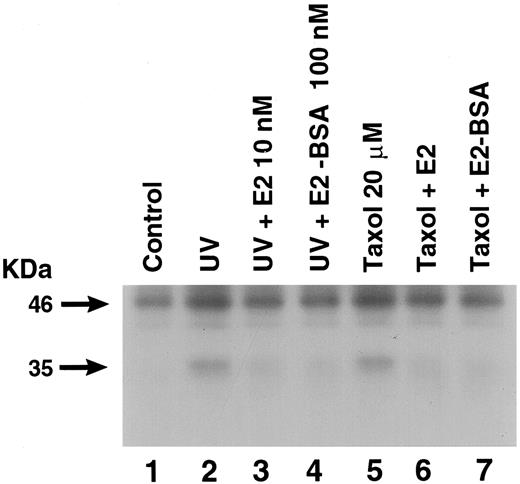
Procaspase-9 Is Cleaved by UV (1-Min Exposure) or Taxol (20 μm), Which Is Prevented by E2 or E2-BSA MCF-7 cells were exposed to UV or taxol in the presence or absence of E2 or E2-BSA, as described, then incubated for a total of 4 h. Cell lysates were then immunoprecipitated for caspase-9, using a monoclonal antibody that recognizes unprocessed and processed forms of the caspase-9 zymogen. After SDS-PAGE separation and membrane transfer, Western blot for caspase-9 was carried out. A representative study is shown, repeated three times.
Extracellular Signal-Regulated Protein Kinase (ERK) Activation Contributes to Antiapoptosis
Might there be a contribution of other signaling molecules to the protective actions of E2? We determined the possible role of the ERK member of the mitogen-activated protein kinase family. Both E2 and E2-BSA stimulated ERK activity by about 3-fold after 10-min exposure to the MCF-7 cells, and this was prevented by ICI182,780 (Fig. 7A). In the setting of UV or taxol, incubation of MCF-7 cells with PD 98059, a mitogen-activated extracellular protein kinase kinase (MEK) inhibitor, partially reversed the anti-apoptotic effects of E2 or E2-BSA; this reversal ranged from 33–48% (Fig. 7B). These results indicate that ERK activation by E2 occurs through the plasma membrane ER, and that this contributes to the anti-apoptotic effects of the sex steroid.
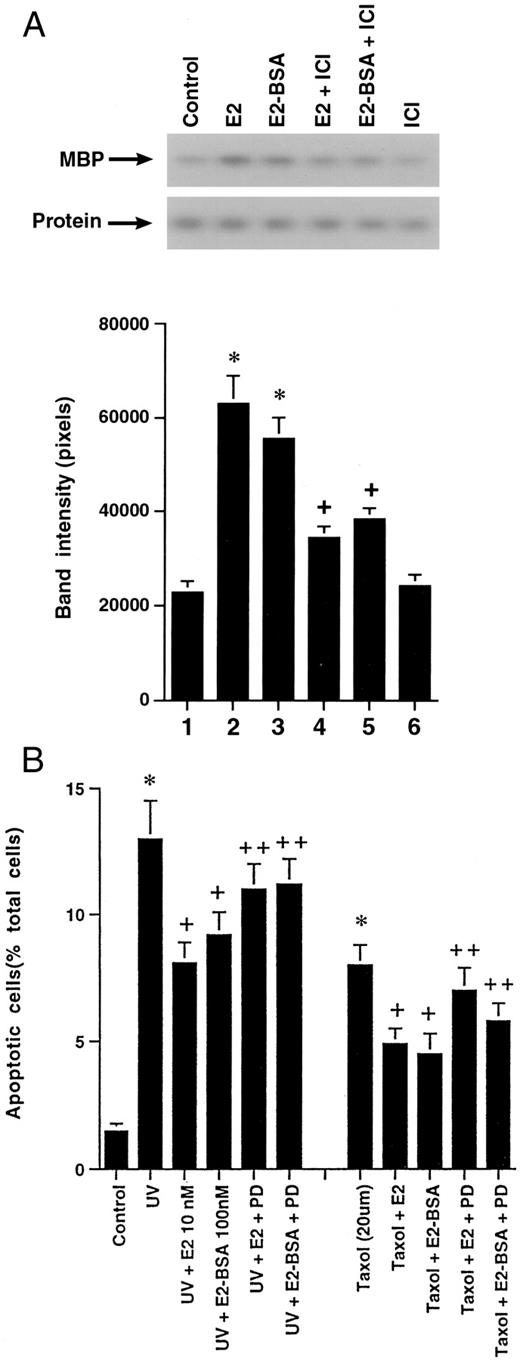
A, E2 and E2-BSA Stimulate ERK Activity MCF-7 cells were incubated with the steroids for 10 min, ERK activity was immunoprecipitated from cell lysate, and activity was determined against myelin basic protein (MBP). A representative study is shown, repeated two additional times. B, Bar graph of three experiments combined delineating the effects of the MEK inhibitor, PD 98059, to partially reverse the ability of E2 or E2-BSA to prevent apoptosis. Data are the mean ± se from three experiments. *, P < 0.05 for control vs. UV or taxol; +, P < 0.05 is UV or taxol vs. either treatment plus E2 or E2-BSA; ++, P < 0.05 is UV or taxol plus E2 or E2-BSA vs. the former plus PD 98059.
DISCUSSION
E2 is an acknowledged growth and/or survival factor for several cell types (1–3, 27, 28). Prenatally, E2 could limit the programmed cell death mechanism of remodeling, which is used as part of developmental plasticity in target organs (29). Postnatally, E2 protection of the ovarian follicle ensures fertility (30), whereas antiapoptosis in vascular cells probably contributes to the lowered incidence of arterial disease in estrogenreplaced postmenopausal women (31). However, the effect of E2 is cell and context specific. When advantageous, E2 can also induce apoptosis to protect against bone resorption by inducing the death of osteoclasts (32). A very important effect of E2 in this regard is the unfortunate ability of this sex steroid to prevent breast cancer cells from undergoing apoptosis in response to chemotherapy or radiation.
Here, we have shown that E2 prevents UV- or taxol-induced apoptosis of ER-positive breast cancer cell lines and have uncovered some novel mechanisms of action. E2 partially, but significantly, prevents UV- or taxol-induced JNK activation and separately stimulates the activation of ERK. Each mechanism contributes to the anti-apoptotic effects of this steroid (Fig. 8). E2 acts through a putative plasma membrane ER (13, 14), a receptor that has not yet been physically isolated, but for which strong functional evidence has now emerged (19, 33–35). We have recently shown that a single cDNA for ERα (or ERβ) can result in the expression of both nuclear and plasma membrane binding proteins. The membrane receptor is a G protein-linked receptor capable of signaling through multiple pathways after G protein activation (19). In the studies reported here, we show that E2 or E2-BSA inhibits the UV- or taxol-induced rapid activation of JNK. These effects are reversed by an ER antagonist.
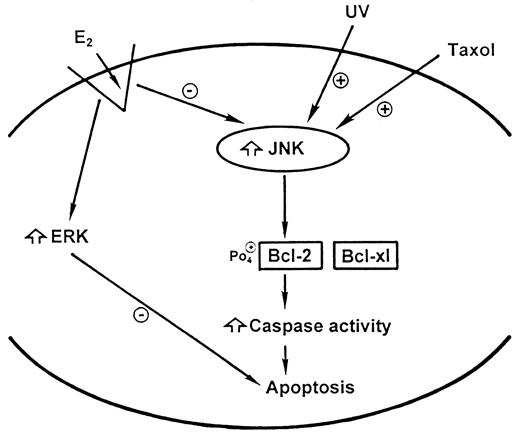
Schema of E2 Acting through the Plasma Membrane ER to Signal to Antiapoptosis
Several laboratories have previously shown that E2-BSA does not activate the nuclear ER (17, 19), and that this compound can be used as a membrane ER-specific ligand. However, this has recently been called into question. Stevis et al. proposed that commercially prepared E2-BSA substantially deconjugates to free E2 and BSA, and that the conjugated E2-BSA, but not free E2, has nonspecific effects to activate signal transduction, at least in neural cancer cells (36). They also did not find nuclear ER activation by intact E2-BSA. In the studies presented here, E2 and E2-BSA act similarly to inhibit signaling to apoptosis, and their effects are always significantly reversed by an ER antagonist. Additionally, E2, but not E2-BSA, activated an ERE-luciferase reporter gene; this indicates that E2-BSA did not bind the nuclear ER and did not dissociate substantially into free E2 and BSA. Furthermore, the focus should be not whether E2-BSA is a useful reagent to activate the membrane ER, but, rather, what are the nongenomic, rapid actions of E2? Importantly, the signal effects of E2 rapidly occur (within 5–15 min) and are unlikely to represent an unprecedented action of a nuclear receptor.
The nongenomic actions of E2 probably result from ERα ligation by the steroid. The latter conclusion derives from the fact that MCF-7 cells express almost exclusively the ERα receptor (37). Furthermore, we have previously shown in CHO cells transfected to singly express either ER that E2 ligation of ERα inhibits basal JNK activity (and stimulates ERK activation), whereas in CHO-ERβ cells, E2 activates JNK (22). It has been established here and previously that JNK is essential to apoptosis induced by UV or taxol in several cell types (24, 38, 39).
We have further defined downstream targets for E2-related antiapoptosis in breast cancer. UV and taxol were found to stimulate the phosphorylation, and hence the inactivation, of Bcl-2. These agents also stimulated phosphorylation of the Bcl-xl protein in MCF-7 cells. This occurs mainly through a JNK-mediated mechanism, which is also inhibited through the membrane ER. Recently, taxol-induced JNK activation has been shown to directly phosphorylate/inactivate Bcl-2 (24). Active Bcl-2 (and Bcl-xl) is proposed to prevent cytochrome c release from mitochondria (40), and therefore inhibit the complexing of Apaf-1, cytochrome c, and procaspase-9. In the presence of ATP, cytochrome c induces the oligomerization of Apaf-1, which then cleaves the procaspase-9 zymogen, yielding active caspase-9. We report here that taxol or UV induces the activation of caspase activity in a JNK-dependent fashion and also induces the cleavage of the procaspase-9 zymogen to caspase-9. These events are significantly prevented by E2 or E2-BSA.
Caspase-9 cleaves/activates the death effector caspases, such as procaspase-3 or -7, thereby effecting apoptosis (41, 42). Caspase-3 is not functional in MCF-7 cells (43), but other effector caspases, such as caspase-7, are present (confirmed by us). In addition to its actions in preventing the release of cytochrome c, Bcl-xl has been shown to form a ternary complex with Apaf-1 and caspase-9, perhaps inhibiting the ability of Apaf-1 to activate caspase-9 (44). Active Bcl-2 has many important functions in preventing cell death (40, 45), and each, theoretically, could be affected by and contribute to the effects of E2 shown here. It has previously been demonstrated that E2 can stimulate the transcription and protein synthesis of Bcl-2 (11, 46). Thus, E2 can prevent cell death by acute and more chronic modulation of both the activity and levels of this anti-apoptotic protein. This may be an example of coordinated cellular actions between the membrane and nuclear ERs, respectively.
The novel finding that UV and taxol induce Bcl-xl phosphorylation through a JNK-dependent mechanism deserves comment. This important anti-apo- ptotic protein can form homo- or heterodimers with other family members, including Bcl-2, Bad, Bax, etc., and it is believed that these interactions underlie the effects of Bcl-xl. However, it is not clear what the effect of phosphorylation is on Bcl-xl action. Analogous to Bcl-2, Bcl-xl contains a 60-amino acid loop domain that partially suppresses the anti-apoptotic functions of this protein (47). The identical domain in Bcl-2 can be phosphorylated by JNK, and this phosphorylation disables Bcl-2 cell survival function (24, 48). We would predict that this sequence is a target for Bcl-xl phosphorylation via JNK, causing inactivation of anti-apoptotic function. Very recently, Kufe and colleagues demonstrated that ionizing radiation causes the translocation of JNK to the mitochondria of leukemia cells, where JNK phosphorylates Bcl-xl (49). Expression of a phosphorylation mutant Bcl-xl in these cells prevents ionizing radiation-induced apoptosis, perhaps because the mutant protein cannot be inactivated by phosphorylation. Collectively, the data suggest that the ability of E2 to inhibit the JNK-induced, inactivating phosphorylation of Bcl-2 provides a partial understanding of the anti-apoptotic effects of this sex steroid and probably extends to Bcl-xl function as well.
It is likely that E2 stimulates other signal transduction mechanisms to inhibit apoptosis. In MCF-7, E2 stimulates the cascade that activates the ERK member of the mitogen-activated protein kinase family, and importantly contributes to DNA synthesis in this cell (50, 51). This kinase is recognized to mediate cell proliferation in response to a variety of growth factors targeting a myriad of cell types (52, 53) and has also been proposed to act as a survival protein (54, 55). We show here that E2 or E2-BSA activates ERK, and that inhibition of activated ERK with the soluble and specific MEK-1 inhibitor, PD 98059, partially reverses E2-induced antiapoptosis in MCF-7 cells. This indicates that ERK contributes to E2 effects in this regard. Singer et al. showed that excitotoxicity-induced necrosis of neurons can be prevented by E2 or nerve growth factor, mediated through ERK signaling to unknown downstream targets (56). Several mechanisms of ERK-induced protection against antiapoptosis have recently been elucidated. These include the phosphorylation of the pro-apoptotic BAD protein at serine 112 (57) and the activation of cAMP response element-binding protein-mediated transcription (58). Thus, it is likely that several pathways that originate from the plasma membrane ER lead to the preservation of target cells, and that it is the overall balance of pro- and anti-apoptotic signals that determines the fate of a cell.
Exactly where JNK (or ERK) fits in the apoptotic pathway is not clear, but we and others (48) have shown that JNK is upstream of Bcl-2. The complete effector pathways that prevent apoptosis will need to be defined in a situation-specific context (59). Furthermore, taxol is a microtubule-stabilizing agent, which theoretically acts through several mechanisms to induce apoptosis (60, 61). These conceivably could also be influenced by E2 in a JNK-independent fashion.
Based upon these studies, we speculate that the actions of the membrane ER in breast cancer could underlie the ability of E2 to effect cell survival in vivo (62). Understanding the mechanisms by which E2 induces antiapoptosis in cancer provides theoretical targets to prevent this undesirable action. Membrane ER-specific antagonists that are targeted to cancer cells might be a future means to enhance the response to therapy. This might also allow women to take hormone replacement to preserve desirable genomic effects of the nuclear ER. To support this strategy, further understanding of the discrete actions of E2 at both the cell membrane and the nucleus must occur.
MATERIALS AND METHODS
Cells and Apoptosis Determination
MCF-7 cells were grown on 18-mm coverslips in 12-well culture dishes in DMEM/F-12 medium without phenol red, but with added charcoal-stripped serum. For apoptosis studies, the cells were subjected to 1 min of UV irradiation (20 J/cm2) or paclitaxel (taxol; 20 μm) and incubated for 4 h at 37 C in the presence or absence of 10 nm E2 or 100 nm E2-BSA. At the end of incubation, the cells were washed with PBS and fixed with 1% freshly prepared paraformaldehyde in PBS, pH 7.4, at 4 C overnight. Apoptosis was then determined by the terminal deoxynucleotidyl transferase-stimulated incorporation of nucleotides into the 3-OH end of damaged DNA in the cell, detected by fluorescent antibodies to the nucleotides (terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling), using a kit from Intergen (Purchase, NY). From each experimental condition, 400 cells were visually scored for apoptosis, viewed by fluorescence microscopy using standard fluorescein excitation and emission filters. In addition, FACS-based cell counting for apoptosis was carried out after bromodeoxyuridine labeling, for MCF-7 and another ER-positive cell line, ZR-75–1. Apoptosis in both of these cell lines was also determined by FACS detection of annexin V binding using a kit (Becton Dickinson and Co., Mountain View, CA). In early apoptosis, the plasma membrane phospholipid, phosphatidylserine, translocates from the inner to the outer membrane leaflet. In cells undergoing apoptosis, phosphatidylserine is then available to bind phospholipid-binding proteins, such as annexin V. HCC1569 cells served as a control, ER-negative breast cancer cell line for these experiments.
Transient Transfections
Breast cancer cell lines were grown to 60–70% confluence in DMEM/F-12 (MCF-7) medium or RPMI-1640 (ZR-75–1 and HCC1569), without phenol red but with 10% FBS. The cells were then washed and transiently transfected with 5–10 μg of fusion plasmids after optimization depending on the plate size and the amount of cells. The plasmids included c-Jun wild type or dominant-negative mutant (see below), ERE-simian virus 40 luciferase (provided by Dr. B. Gehm), or respective backbone vectors. DNA was amplified and isolated using the QIAGEN maxi-prep kit (Chatsworth, CA), and care was taken to minimize carryover of salts, alcohol, or other confounding reagents. Transfections were performed with Lipofectamine reagent (Life Technologies, Inc., Grand Island, NY); cells were incubated with liposome-DNA complexes at 37 C for approximately 5 h, followed by overnight recovery in DMEM-F-12 medium containing 10% FBS. Then, before experimental treatment, cells were synchronized in serum-free DMEM-F-12 for 24 h and treated with 17β-E2 and/or related compounds. Cotransfections with a green fluorescent protein expression vector (Promega Corp., Madison, WI) indicated approximately 70–75% efficiency of transfection. Luciferase activity was determined as previously described (19, 63). The concentration of E2-BSA was calculated from the number of E2 molecules attached to each BSA molecule.
Kinase Assays
The c-Jun kinase activity was determined as previously described (21). Briefly, MCF-7 cells were incubated under various conditions for 15 min, then the cells were lysed, and lysate was immunoprecipitated for Jnk-1. JNK activity was determined against GST-c-Jun-(1–79) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) as substrate. Laser densitometry of the phosphorylated bands was used for quantitation, and three experiments were combined for statistical analysis. Immunoblot of Jnk-1 protein assured equal amounts of protein loaded in each condition. In some experiments, wild-type (pcDNA3Flag-Jnk-1) or dominant-negative Jnk-1 (pcDNA3 Flag-Jnk-1 APF) (58) was transfected into MCF-7 cells as previously described (21, 58). ERK activity was determined as previously described (18). MCF-7 cells were synchronized in serum- and growth factor-free medium for 24 h, then incubated with E2 or E2-BSA for 10 min, and immunoprecipitated ERK activity from cell lysates was determined against myelin basic protein as the substrate. For apoptosis experiments, cells were subjected to UV or taxol treatment with or without E2 or E2-BSA in the absence or presence of the specific ERK kinase (MEK-1) inhibitor, PD 98059 (10 μm).
Bcl-2 and Bcl-xl Phosphorylation
MCF-7 cells were incubated for 1 h at 37 C in phosphate-free medium containing 5% dialyzed FBS. At the end of the incubation, cells were washed and labeled with 32P (final concentration, 0.2 mCi/ml) for 2 h at 37 C in a CO2 incubator. Cells were further incubated with or without taxol, UV, and E2 or E2-BSA for 1 h. At the end of the labeling period, cells were washed with ice-cold PBS, then lysed with buffer for 30 min on ice. The lysates were microcentrifuged, and Bcl-2 and Bcl-xl immunoprecipitation from the supernatant was conducted using specific antibodies (Santa Cruz Biotechnology, Inc.) at 4 C. After centrifugation and washing, the immunoprecipitated Bcl-2 and Bcl-xl were resolved by SDS-PAGE on a 12% gel, followed by autoradiography.
Caspase Activity and Zymogen Proteolysis
Cultured MCF-7 cells in 100-mm dishes were exposed to UV or taxol as described for the previous experiments. The cytosolic extracts were repeatedly frozen in extraction buffer and thawed as previously described (26). Cell lysates were then diluted and incubated with 1 μm fluorescent substrate (caspase-4, -5, and -9 substrate) for 30 min at 30 C. At the end of the incubation, the fluorescence of the cleaved substrates was determined using a spectrofluorometer, set at an excitation wavelength of 400 nm and an emission wavelength of 505 nm.
Cleavage of the zymogen, procaspase-9, was assessed. MCF-7 cells were cultured as described, then treated with UV (1 min) or taxol (20μ m) for 4 h in the absence or presence of E2 or E2-BSA. At the end of the 4-h incubation, the cells were washed, then lysed in buffer for 30 min on ice. The lysates were microfuged, and the supernatants were immunoprecipitated with anti-caspase-9 polyclonal antibody, raised against a peptide corresponding to the unique amino acids 299–318 of human caspase-9 (Cayman Chemical, Ann Arbor, MI). The immunoprecipitants were then analyzed by Western blotting using the ECL kit from Amersham Pharmacia Biotech (Arlington Heights, IL).
Statistical Analysis
Pooled data from multiple experiments were compared by ANOVA and Scheffe’s test, using the StatView statistical program (P < 0.05 as significant). Bar graphs represent the mean ± sem from at least three experiments.
Acknowledgements
We thank Roger Davis for the JNK plasmids.
This work was supported by grants from the Research Service of the Department of Veteran’s Affairs, and the NIH (HL-59890; to E.R.L.).
References
Srivastava RK

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}