





Basic and translational research achievements over the past 2 decades have disclosed the molecular mechanisms underlying several genetic forms of hypopituitarism. Disorders that are limited to the hypothalamic, pituitary, GH axis are caused by mutations in individual components of that axis. Disorders involving GH and one or more additional pituitary hormones are caused by mutations in the homeodomain transcription factors that direct embryological development of the anterior pituitary gland. Pit-1 has a POU-specific and a POU-homeo DNA-binding domain. The phenotype produced by mutations in the PIT1 gene involves deficiencies of GH, PRL, and TSH. Pituitary glands are either small or normally sized. The PROP1 gene encodes a transcription factor with a single paired-like DNA-binding domain. Persons with inactivating mutations in PROP1 have deficiencies of LH and FSH, as well as GH, PRL, and TSH. Their pituitary glands may be small, normally sized, or extremely large and show suprasellar extension. Pituitary degeneration may produce acquired deficiency of ACTH. Expression of the HESX1 gene precedes expression of PROP1 and PIT1, and it is much more widespread. The protein has a paired-like domain, and it competes with the product of PROP1 for DNA-binding. Homozygosity for inactivating mutations of HESX1 produces a complex phenotype that resembles septo-optic dysplasia. Much more needs to be learned about the role of HESX1 mutations in other forms of hypopituitarism.
A GREAT DEAL has been learned about the genetic causes of hypopituitarism over the past 2 decades (1). By 1979, many families had been described with isolated deficiency of GH, resistance to the action of GH, or diminished production of GH and one or more additional pituitary hormones. The Little mouse provided a model of isolated GH deficiency, whereas Snell and Ames mice provided models of recessive multiple pituitary hormone deficiencies. The hormone contents and cellular composition of pituitary glands from the dwarf mouse strains was reasonably well understood. Nevertheless, in 1979, one could only make educated guesses about the genetic mechanisms responsible for hypopituitarism.
Hypothetically, disruption of the hypothalamic pituitary GH axis might be caused by mutations in any of the genes that encode critical components of that axis. Development of a complementary DNA (cDNA) probe for the pituitary GH gene permitted recognition of GH gene deletions in 1981 (2) and placental GH and chorionic somatotropin gene deletions in 1982 (3). The power of PCR amplification and DNA sequencing subsequently disclosed mutations and small deletions in GH-1 in other families with isolated GH deficiency (4). By 1989, discovery of the gene encoding the human GH receptor (5) facilitated recognition of GH-R deletions (6) and mutations (7) as the mechanism for the Laron syndrome of peripheral resistance to the action of GH. Description of the GHRH receptor gene in 1992 (8) was followed within a year by demonstration of a GHRH-R gene mutation in the Little mouse model of isolated GH deficiency (9). In 1996, a similar disorder was recognized in humans (10). The first, and to this point the only, example of homozygosity for deletion of portions of the insulin-like growth factor-I gene was described in 1996 (11).
The path to understanding the mechanisms underlying multiple pituitary hormone deficiency (MPHD) was less straightforward and could not have been predicted in 1979. How could mutation of a single gene cause deficiency of several hormones, the genes of which were widely dispersed through the genome? The combinations of missing hormones in affected individuals did not reflect the combinations of hormones influenced by hypothalamic releasing and release-inhibiting factors. Solutions to this riddle emerged with the discovery of transcriptional activation factors that direct the embryonic development of the anterior pituitary. This story began with the discovery in 1988 of a homeobox protein, termed Pit1 or GHF1, that bound to sequences in the promoter for the GH gene. It continued with the recognition of a legion of other pituitary and hypothalamic factors that orchestrate pituitary development. Together, they direct the formation of the anterior pituitary gland, the differentiation, the expansion and the definitive function of the five pituitary cell types that produce six pituitary hormones. This review will focus on three transcription factors that have been implicated as causes of multiple pituitary hormone deficiency in humans. In chronological order of their association with human disease, they are PIT1, PROP1, and HESX1.
PIT1
Discovery of PIT1 involved identification of a nuclear protein that bound to the GH promoter and partial sequencing of that protein, followed by detection of positive clones in cDNA expression libraries and characterization of genomic structure in mouse and man (12–14). PIT1 is also known as GHF1 (GH factor 1), and it has been officially designated as POU1F1 (POU domain, class 1, transcription factor 1). The mouse gene is located on chromosome 16, and the human is located on chromosome 3p11 (Table 1). Each consists of six exons that encode a protein of 290 amino acids (Fig. 1). There is an N-terminal transcriptional activation domain that is rich in hydroxylated amino acids, followed by two DNA-binding domains (15). The first is termed the POU-specific domain. Similar domains are found in the mammalian transcription factors Oct1 and Oct2, as well as in the Unc-86 protein of the flatworm Caenorhabditis elegans. POU (pronounced“ pow”) is an acronym for Pit, Oct, and Unc. The POU-homeo domain resembles DNA-binding domains found in a large family of homeobox proteins. Both domains contain three α helical regions, and both are required for specific binding. The POU-homeo domains contribute to recognition of AT-rich regions, and the POU-specific domain strengthens binding to regions containing a TATNCAT consensus sequence. There are two such regions in the GH and CS promoters, at least three in the PRL promoter, several more in a distal enhancer region preceding the PRL gene, one in the promoter of the gene encoding the β subunit of TSH and another in the promoter of the GHRH receptor gene. There are also binding sites in the PIT1 promoter and a complex Pit1 and retinoic acid receptor element some 10 kb upstream from PIT1 (16).
Pituitary homeobox genes associated with hypopituitarism
| Gene | Synonyms | Gene family | Location | Characteristics of protein | ||
|---|---|---|---|---|---|---|
| Human | Mouse | Size | Domains | |||
| PIT1 | GHF1, POU1F1 | POU homeo | 3p11 | 16 | 290 aa | TA, POUs, POUh |
| PROP1 | None | Paired-like | 5q | 11 | 223 aa | Paired-like, TA |
| HESX1 | RPX | Paired-like | 3p21 | 14 | 185 aa | Paired-like, TA |
| Gene | Synonyms | Gene family | Location | Characteristics of protein | ||
|---|---|---|---|---|---|---|
| Human | Mouse | Size | Domains | |||
| PIT1 | GHF1, POU1F1 | POU homeo | 3p11 | 16 | 290 aa | TA, POUs, POUh |
| PROP1 | None | Paired-like | 5q | 11 | 223 aa | Paired-like, TA |
| HESX1 | RPX | Paired-like | 3p21 | 14 | 185 aa | Paired-like, TA |
The gene designations are those of the human genes. Murine counterparts are PIT1, PROP1, and HESX1, respectively. The proteins are designated PIT1, PROP1, and HESX1.
Pituitary homeobox genes associated with hypopituitarism
| Gene | Synonyms | Gene family | Location | Characteristics of protein | ||
|---|---|---|---|---|---|---|
| Human | Mouse | Size | Domains | |||
| PIT1 | GHF1, POU1F1 | POU homeo | 3p11 | 16 | 290 aa | TA, POUs, POUh |
| PROP1 | None | Paired-like | 5q | 11 | 223 aa | Paired-like, TA |
| HESX1 | RPX | Paired-like | 3p21 | 14 | 185 aa | Paired-like, TA |
| Gene | Synonyms | Gene family | Location | Characteristics of protein | ||
|---|---|---|---|---|---|---|
| Human | Mouse | Size | Domains | |||
| PIT1 | GHF1, POU1F1 | POU homeo | 3p11 | 16 | 290 aa | TA, POUs, POUh |
| PROP1 | None | Paired-like | 5q | 11 | 223 aa | Paired-like, TA |
| HESX1 | RPX | Paired-like | 3p21 | 14 | 185 aa | Paired-like, TA |
The gene designations are those of the human genes. Murine counterparts are PIT1, PROP1, and HESX1, respectively. The proteins are designated PIT1, PROP1, and HESX1.
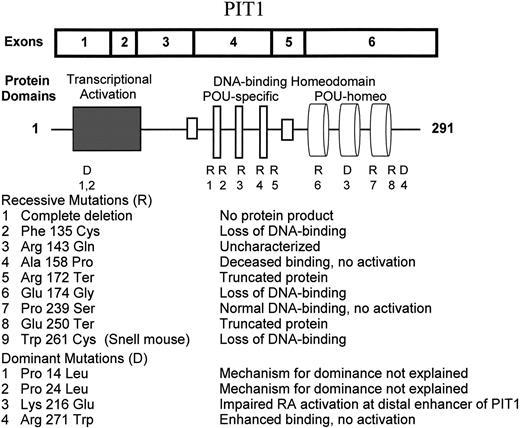
Pit1 cDNA, protein domains and mutations.
Expression of Pit-1 in the mouse pituitary begins at about 14 days of embryonic life and persists throughout life. The protein is restricted to the nuclei of somatotropic, lactotropic, and thyrotropic cells. Cotransfection experiments show that Pit-1 activates transcription from the GH, Prl, β-TSH, and PIT1 promoters. The Pit1 protein generally needs to bind as a dimer to activate transcription. It partners with different transcription factors, including Zn-15, HESX-1, Ptx-1, P-Lim, the thyroid hormone receptor, the estrogen receptor, and the retinoic acid receptor, in activating transcription from different target genes (17). The distribution and properties of Pit1 suggest that inactivating mutations would confer a phenotype of combined deficiencies of GH, PRL, and TSH. Detection of naturally occurring mutations in mouse and man has amply confirmed this expectation.
In 1990, Li et al. (18) found that Snell dwarfism in mice was caused by homozygosity for a missense mutation in Pit1. Snell mice lack GH, PRL, and TSH and have hypoplastic anterior pituitary glands. They are of normal size at birth, but their weights as adults are only one third those of their normal littermates. The Trp261Cys mutation in Snell mice eliminates specific binding of Pit1 to promoter sites. A phenotypically similar strain called dwarf Jackson carries a complex rearrangement of the Pit1 gene that precludes production of a functional Pit-1 protein.
The first human example of combined pituitary hormone deficiency due to PIT1 mutations was described by Tatsumi et al. (19) in 1992. Two sisters born to parents who were second cousins had profound neonatal hypothyroidism without elevation of TSH. One died of aspiration pneumonia at 2 months of age. The surviving sister also had deficiencies of GH and PRL. She was found to be homozygous for an Arg172Ter mutation in PIT1.
At least seven recessive and three dominant types of PIT1 abnormality have been recognized in sporadic cases and multiplex families with MPHD (20, 21). These mutations and their consequences are depicted in Fig. 1. Recessive mutations produce varying degrees of loss of DNA-binding and/or transcriptional activation functions. Dominant mutations require more elaborate explanations.
The ultimate example of a loss of function mutation is deletion of the entire coding sequence of PIT1. This constituted the maternal allele in two Dutch brothers with MPHD (22). Their paternal allele carried a recessive Ala158Pro missense mutation. The Arg172Ter (19) and Glu250Ter (23) nonsense mutations predict truncated proteins that are presumed to be incapable of binding DNA or activating transcription. The Phe135Cys (24) and Glu174Gly (25) mutations in the POU-specific domain eliminate DNA binding. Studies of the protein product of the Ala158Pro mutation show that it binds to the GH promoter with decreased affinity, does not form dimers, and does not activate transcription (22). The mutant Pro239Ser protein with an abnormality in the second α helix of the POU-homeo domain binds normally but is unable to stimulate transcription (26).
Radovick et al. (27) described the first autosomal dominant mutation of PIT1 in 1992. This mutation involves the substitution of tryptophan for arginine at position 271, near the end of the POU-homeo domain. The mutant protein has increased affinity for GH and PRL promoter sites. Dominance may reflect preferential occupancy of binding sites by Arg271Trp homodimers and Arg271Trp/wild type heterodimers that block, rather than stimulate, transcription. This mutation probably accounts for the majority of cases of PIT1-related MPHD. It involves a C to T transition at a CpG mutational hot spot. Most cases reflect new mutations, but there are several instances of vertical transmission. Okamoto et al. (28) reported a puzzling family in which the proband had typical features of MPHD, but the father, grandmother, and two aunts carried the Arg271Trp allele and were phenotypically normal. Lymphocytes from the proband expressed this allele as well as the normal allele, but lymphocytes from the father and aunt expressed only the normal allele. The only other suggestion of monoallelic expression through genomic imprinting of PIT1 is found in an article by Fofanova et al. (29). A girl with MPHD was heterozygous for a Pro14Leu missense mutation in the transcriptional activation domain. Her mother, maternal aunt, and maternal grandmother carried the same allele but were phenotypically normal. DNA-binding and transcriptional activation properties of the protein products of this mutation and those of the nearby Pro24Leu mutation described by Ohta et al. (30) have not been studied in transfection systems.
The most interesting example of a dominant mutation in PIT1 involves the Lys216Glu mutation described by Cohen et al. (31). The mutant protein behaves paradoxically as a superactivator of transcription from the GH and the proximal PRL promoters. In transfection systems, activation is approximately 4-fold and 1.5-fold, respectively, as great as that achieved with equivalent quantities of the normal Pit1 protein. Studies of activation of the distal enhancer of PIT1 tell a different story. The distal enhancer region is critical for autoregulation of Pit-1 regulation of Pit1. This region contains overlapping sites for Pit-1 and the retinoic acid receptor (RAR) binding. Both Pit-1 and RAR are required for transcriptional activation. The mutant protein is incapable of promoting activation, and it prevents activation by wild-type Pit-1. Thus, the mechanism responsible for dominance of the Lys216Glu mutation seems to be a quantitative defect in Pit-1 production caused by inhibition of the up-regulation of Pit-1 mRNA synthesis by Pit-1 and RAR at the distal enhancer site.
PROP1
Discovery of the Prop1 gene involved identification, by positional cloning, of the gene responsible for Ames dwarfism in mice (32). The Ames mouse, like the Snell and Jackson dwarf mice, has GH, PRL, and TSH deficiencies, and adults are roughly one third the size of normal littermates. The Ames gene is located on mouse chromosome 11, whereas the Snell and Jackson mutations involve the Pit1 locus on chromosome 16. Pituitary glands from Ames mice contain abnormally low quantities of Pit-1 mRNA and protein. The designation of Prop1 in mice and PROP1 in humans is an abbreviation for “prophet of Pit-1,” indicating a gene whose product normally precedes Pit-1 and is, in fact, a prerequisite for the expression of Pit-1.
The mouse Prop1 and human PROP1 genes are relatively compact, consisting of three exons over a distance of approximately 3 kb (Fig. 2). These genes encode proteins of 223 amino acids. There is a central DNA binding domain with three α helical regions that resembles the “paired” domains in Drosophila. A transcriptional activation domain follows the paired-like domain. There is divergence of amino acid sequence in the amino-terminal region of the protein, but very strong preservation of amino acid sequence between mouse and human versions in the middle and carboxy-terminal regions of the proteins. Prop-1 binds as a dimer to promoter elements containing palindromic TAAT ATTA sequences separated by 2- or 3-bp spacers. This type of element is recognized by other paired-like homeobox proteins including the pituitary transcriptional activation factor HESX1.
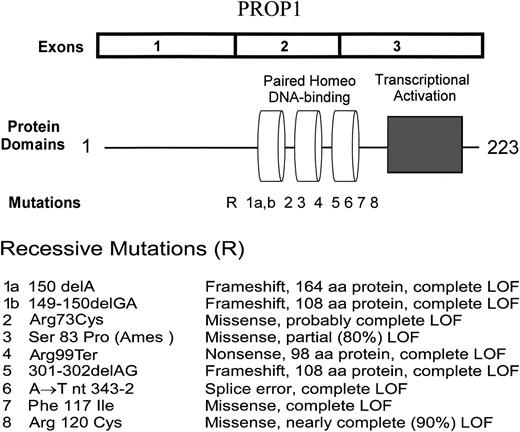
PROP1 cDNA, protein domains and mutations.
The Prop1 gene is first expressed at embryonic day 10.5 in the mouse, and expression is reduced by day 14, at the time of appearance of GH. Like Pit-1, Prop-1 is produced in somatotropic, lactotropic, and thyrotropic cells. Expression of Prop-1 seems to be required for extinction of HESX1 expression since Ames mice continue to express HESX1 beyond day 14. PROP1 mRNA continues to be expressed in normal human pituitary tissue and in pituitary tumors at levels that are readily detected by RT and PCR amplification of cDNA (33).
The Ames mouse mutation, described in 1996 (32), involves a substitution of proline for serine at position 83 in the paired-like homeodomain. The Ser83Pro protein retains about 20% of the DNA-binding and transcriptional activation activities of the wild type protein.
The first examples of PROP1 mutations in humans with MPHD were reported early in 1998 (34), some 15 months after publication of the report by Sornson et al. (32) of the Ames mutation. In humans, the hormonal phenotype involves deficiencies of LH and FSH, as well as GH, PRL, and TSH. At least seven human mutations have been recognized in the past 2 years (Fig. 2). All involve the paired-like DNA-binding domain encoded by exons 2 and 3, and all show recessive inheritance. The most common involves deletion of 2 bp from series of three GA repeats beginning at nucleotide 296 of the coding sequence. It is not possible to determine which two of the six nucleotides in the repeat are deleted. The deletion has been referred to as 301-302delAG and 296delGA in different publications. The consequence of this deletion is a shift in reading frame with divergence of amino acid sequence at codon 102 and termination of translation after residue 108. When the human mutation is introduced into mouse Prop1 cDNA, the expressed protein is devoid of DNA-binding and transcriptional activation properties (34). The 301-302delGA deletion introduces a new Bcg I restriction site so it is very easy to screen a large panel of MPHD patients for the presence of this deletion. The next most common mutations involve deletion of 1 or 2 bp from codon 50. In several families, the affected individuals are compound heterozygotes for 301-302delAG and a codon 50 deletion. Fofanova et al. (35) reported a 149-150delGA deletion in Russian cases of MPHD, and we detected a 150delA deletion in cases from Croatia (36), Poland (Brown, M. R., Lenartowska, I., Oltarzewski, M., et al., submitted for publication), and the United States (unpublished data). Pending exchange of specimens for analysis, it is not certain whether there are one or two classes of deletion in codon 50. The 1-bp deletion predicts divergence of amino acid sequence at residue 53 and termination after residue 163, whereas the 2-bp deletion predicts divergence at 52 and termination after residue 108. In either case, the mutant protein would lack functional DNA-binding and transcriptional activation domains. We recognized a C to T transition at nucleotide 295 that converts codon 99 to a translational stop codon (Brown, M. R., Lenartowska, I., Oltarzewski, M., et al., submitted for publication). The two affected siblings were compound heterozygotes for Arg95Ter and 301–302delAG, so neither allele would be expected to produce a functional Prop-1 protein. Deladoey et al. (37) have reported a T for A substitution a position-2 of the splice acceptor site preceding exon 3. This A to T transversion at nt 343–2 mutation predicts loss of much of the third α helix of the DNA-binding domain, as well as the transcriptional activation domain. It is very likely to be a complete loss of function mutation.
Missense mutations in PROP1 can result in partial or complete loss of function. The Arg120Cys mutation was first described in the study of Wu et al. (34) and the two families with homozygosity for this mutation were described in greater detail by Fluck et al. (38). When the Arg120Cys mutation is introduced into mouse Prop1 cDNA, the protein retains about 10% of normal DNA-binding and transcriptional activation. In contrast, the mutant Phe117Ile protein has no activity. Persons with partial loss of function mutations may have less severe hormone deficiency phenotypes, particularly with respect to LH and FSH production, than those with complete loss of function mutations. This issue will be discussed later in the review.
HESX1
The HESX1 gene plays an important role in the development of the optic nerves as well as the anterior pituitary gland. Its name denotes a “homeobox gene expressed in embryonic stem cells” (39). The same gene is also referred to as Rpx, denoting a “Rathke’s pouch homeo box” gene. The mouse gene is located on chromosome 14, and the human gene is located on chromosome 3p21.2 (32). The mouse and human genes contain four exons, span a distance of 1.7 kb and encode proteins of 185 amino acids (Figure 3). The proteins contain a putative repressor domain from amino acids 21 to 27 and a paired-like homeodomain extending from amino acid 110 to 167.
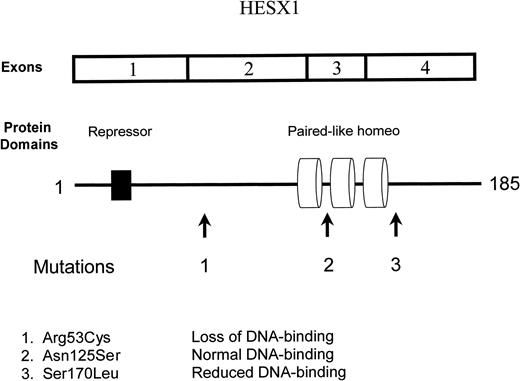
HESX1 cDNA, protein domains and mutations.
Expression of HESX1 begins before that of PROP1 and is more widespread. The mRNA and protein can be found in many organs, including the liver and brain. In the developing central nervous system, HESX-1 expression begins in a small patch of cells in the anterior midline visceral endoderm at the beginning of gastrulation, spreads to the adjacent ectoderm, then to the rostral neural folds, and subsequently the ventral diencephalon. At day 9.5 it is also expressed in a layer of oral ectoderm, which gives rise to Rathke’s pouch. It appears to be expressed in the precursors of all pituitary cell types, but expression declines after day 11.5 and is extinguished by day 15, following the appearance of PIT1.
Dattani et al. (40) produced a knockout of the HESX1 gene in transgenic mice. Homozygotes demonstrated reduction of prosencephalic tissue, anopthalmia, or micropthalmia and abnormalities of the corpus callosum and septum pellucidum. The anterior pituitaries were small and they were not connected with the posterior pituitary. In many ways, this phenotype resembles the syndrome of septo-optic dysplasia (SOD), also known as De Morsier syndrome, in humans.
Dattani et al. (40, 41) screened a total of 135 patients with pituitary disorders, including 35 with SOD, for mutations in the HESX1 gene. The brother and sister previously reported by Wales and Quarrell (42) were found to be homozygous for an Arg53Cys mutation in a conserved region between the repressor and paired-like domains. The mutant protein failed to bind to DNA-containing sequences that are normally recognized by HESX-1. No mutations were found in the HESX1 genes from patients with SOD. Heterozygosity for a Ser170Leu mutation was demonstrated in two siblings with isolated deficiency of GH. One of the siblings also had optic hypoplasia. The mutant protein had reduced affinity for HESX-1-binding elements, but did not prevent access of the normal protein. Another patient with isolated GH deficiency and pituitary hypoplasia was heterozygous for an Asn125Ser mutation. This mutation did not alter binding to DNA.
Although HESX1 mutations do not provide a general explanation for the syndrome of SOD, the findings of Dattani et al. (40, 41) suggest an important potential role for this gene in understanding recessive and dominant forms of hypopituitarism. There is a need for more complete characterization of the hormonal phenotypes of patients with HESX1 abnormalities There is no published information about GH, PRL, TSH, or gonadotropin responses to stimulation by releasing hormones. It will be interesting to see whether these and additional cases have defects at the level of the pituitary gland or involve defects in communication between the hypothalamus and pituitary.
Comparison of Phenotypes Produced by PIT1, PROP1, and HESX1 Mutations
There are important differences in the hormonal phenotypes produced by PIT1, PROP1, and HESX1 mutations in mice (44) and humans (Fig. 4). There are also some very interesting differences among persons with the same disease genotypes. The molecular basis for this type of variation has yet to be defined. It may well reflect polymorphic variability in other genes that contribute to pituitary development.
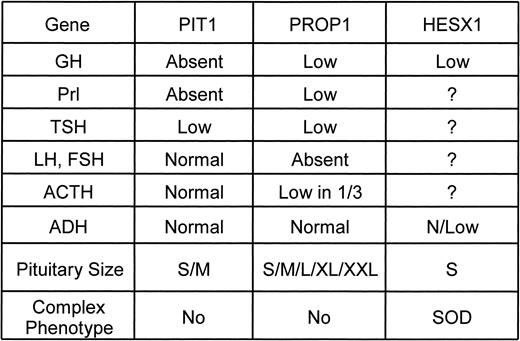
Comparison of phenotypes produced by PIT1, PROP1, and HESX1 mutations.
GH
Histological examination, immunohistochemistry, and in situ hybridization show that the pituitaries of Snell mice uniformly lack functional somatotropes (44). In contrast, the pituitary glands from some Ames mice have a very small number of functional somatotropes (45, 46). The cells tend to be located in clusters, suggesting a clonal origin. Estimates of somatotropes range from 10–20 cells per gland detectable in 20% of animals examined (45) to 100 cells per gland or about 0.05% of the normal number (46). Humans with PIT1 mutations generally do not release detectable amounts of GH after GHRH stimulation, and height sd scores during childhood are comparable with those of children with deletion of the GH-1 gene or complete GH insensitivity. Growth restriction and GH responses to GHRH stimulation are more variable in children with PROP1 mutations. This variability does not seem to be dependent on the type of PROP1 mutation because variability exists within sibships. For example, of the three siblings who were originally described as having hypopituitarism with large sella turcica (47), one was only 2 sd below mean height during childhood. She was later shown to have a peak GH level of 7 μg/L following GHRH stimulation. Her siblings were more than 4 sd below the mean before treatment and had no detectable response to GHRH. All three are compound heterozygotes for the 301-302delGA and 150delA mutations. Deladoey et al. (37) have suggested that GH deficiency in children with PROP1 mutations may be an acquired rather than congenital characteristic. Some patients show relatively normal growth during infancy, followed by profound growth failure in a manner similar to those with hypothalamic forms of GH deficiency. The nature of GH deficiency in patients with HESX1 mutations has yet to be defined.
PRL
Although both the Snell and Ames strains of dwarf mice seem to have total deficiency of pituitary PRL production throughout development (48), PRL-immunoreactive cells have been identified in the glands of Ames dwarfs (45, 49, 50) in a pattern similar to that described above for GH. Although PRL deficiency in humans has little clinical impact, measurement of PRL is a valuable step in recognizing those with pituitary rather than hypothalamic disorders. Persons with PIT1 and PROP1 mutations have subnormal PRL release following TRH stimulation, but peak responses may be somewhat higher in those with PROP1 abnormalities. Comparisons are difficult because of wide variation in the sensitivity of clinical assays for PRL. Patients with SOD tend to have high basal and stimulated PRL levels. It is not known whether this is true of those with HESX1 mutations.
TSH
Pituitary content of TSH is minimal in Snell mice and reduced in Ames mice (51, 52). There is wide variability in the severity of hypothyroidism among humans with PIT1 and PROP1 mutations. Some patients with PIT1 defects are born with features of cretinism (19). Others, including two of the patients described by Pfaffle et al. (22), have been euthyroid at diagnosis and developed mild hypothyroidism during GH treatment. The impact of severe prenatal hypothyroidism was illustrated in a family reported by de Zegher et al. (53). Both the mother and the child carried the Arg271Trp mutation in PIT1. The mother discontinued thyroid hormone replacement during pregnancy, and the infant demonstrated profound delay in respiratory and cardiovascular maturation, as well as impaired mental development. Cogan et al. (54) have shown that evaluation of the TSH response to TRH provides a valuable means of identifying sporadic cases of MPHD who might have abnormalities of PROP1. Twenty individuals were tested. Of the 17 who had normal responses, none were found to have PROP1 mutations. Of the three individuals with subnormal responses, two had PROP1 mutations. Thus, it is advisable to perform a TRH stimulation test with measurement of PRL and TSH before performing molecular genetic studies in patients with MPHD.
LH and FSH
Gonadotropes are present, but reduced in number, in the pituitaries of Snell and Ames mice (55). Fertility is reduced in both strains. Successful pregnancy and lactation can be established in female Ames mice (56) and fertility induced in male Ames mice (55) through injections of PRL or implantation of PRL-producing homografts. Patients with PIT1 abnormalities may experience delayed puberty because of delay in T4 or GH treatment. They eventually mature and have normal fertility. Hypogonadism is a striking and unexpected feature in patients with PROP1 mutations. It represents a major difference between the phenotypes produced by PIT1 and PROP1 mutations. There is variability in the clinical and hormonal expressions of hypogonadism among persons with PROP1 mutations. Most fail to enter puberty and show consistently low LH and FSH responses to LHRH stimulation. Fluck et al. (38) described two fascinating sibships with homozygosity for the Arg120Cys mutation. The affected children entered and then retreated from puberty. Three male and two females showed spontaneous pubertal development and had normal LH and FSH responses to LHRH. The two girls experienced menarche but developed secondary amennorhea. They were then found to have minimal gonadotropin responses to LHRH. The initial impression was that this phenomenon resulted from an incomplete loss of function mutation analogous to that in Ames mice. However, the same sequence of events was observed by Deladoey et al. (37) in several children with complete loss of function mutations. Variation in timing and severity of expression of hypogonadism is consistent with a model involving acquired, rather than congenital, gonadotropin deficiency. It would be fascinating to find out whether the observation of micropenis in newborn males is less common in PROP1-related than in other forms of hypopituitarism.
Puberty tends to occur on time, and sometimes ahead of the normal time, in persons with SOD. Little information regarding puberty is available for the limited number of subjects with HESX1 mutations.
ACTH
Corticotrope numbers and adrenal function are normal in Snell and Ames mice (57). Tests of the hypothalamic pituitary adrenal axis are uniformly normal in persons with PIT1 abnormalities. Most patients with PROP1 abnormalities do not have abnormalities of the adrenal axis. Some show diminished cortisol responses to ACTH late in the course of their disease. It is not known whether this reflects gradual attrition of corticotropes or whether it is a result of the sequence of pituitary hypertrophy followed by pituitary degeneration that is described in the next section.
Pituitary morphology
Snell and Ames mice have small anterior pituitary glands, reflecting absence of the somatotropes, lactotropes, and thyrotropes that normally account for about 80% of anterior pituitary cell content. Anterior pituitary size, as assessed by magnetic resonance imaging, is small or normal in persons with PIT1 mutations. There is no consistent relationship between the size of the pituitary and the age of the patient or the type of PIT1 mutation. Most persons with PROP1 mutations also have small or normal size anterior pituitary glands. The pituitary stalk is normal, and the posterior pituitary is not ectopic. There have been reports of families with PROP1 mutations and striking enlargement of the anterior pituitary (58). In some there has been progression from a large and full sella turcica to suprasellar extension of a pituitary mass, followed by areas of cystic change, loss of contrast enhancement of the mass, and eventual regression leaving a large and nearly empty sella with a rim of anterior pituitary tissue. The sequence of events is reminiscent of pituitary apoplexy. It is not consistent among affected siblings with the same PROP1 genotype. There is no information about the cell type that proliferates during the phase of active expansion. The phenomenon has not been observed in Ames mice. At the clinical level, it is important to recognize that PROP1 mutations may cause pituitary enlargement that can be mistaken for a pituitary adenoma or a craniopharyngioma or Rathke’s pouch cyst. The enlargement and degeneration sequence may also result in a transition to ACTH and cortisol deficiency.
Frequency of PIT1, PROP1, and HESX1 Mutations in Hypopituitary Patients
PROP1 mutations are turning out to be much more common than PIT1 mutations as a cause of MPHD. A survey from Moscow disclosed seven patients with PROP1, one with PIT1 and six with neither (35). Our study of a series of 52 patients from Poland identified 33 with PROP1, none with PIT1, and 19 with neither (37). A Swiss series found that 35 of 73 subjects with MPHD had PROP1 mutations (37). The numbers are even more impressive when one looks at multiplex families with more than one affected sibling. PROP1 mutations accounted for the disorder in 11 of 11 such families in the Polish series and 14 of 14 in the Swiss series. PROP1 mutations are also responsible for MPHD in some historically interesting isolates that were the subject of some of the initial reports of hypopituitarism. Hanhart’s dwarfs from the Isle of Krk in Croatia are homozygotes for 150delA (36), and the Hutterites with MPHD are homozygotes for 301-302delGA (Ref. 24 and personal observations).
Because PROP1 and PIT1 abnormalities account for such a large proportion of cases of familial hypopituitarism, it is appropriate to ask whether there is any point in searching for other genetic disturbances of pituitary development. Haploinsufficiency for the PTX2 or RIEG1 gene is known to account for the complex phenotype of Rieger syndrome, which includes variable degrees of multiple pituitary hormone deficiency along with coloboma and an increased risk of glaucoma (59). Many studies have shown a high frequency of the magnetic resonance imaging findings of hypoplastic pituitary stalk and ectopic posterior pituitary among patients with MPHD. These cases do not have PROP1 or PIT1 abnormalities, and it will be very interesting to see if this common condition can be matched to a candidate gene.
Clinical suspicion of PROP1 or PIT1 mutations should be high for any individual with MPHD, even those with features of organic hypopituitarism such an intrasellar or suprasellar mass. Familial cases and those with diminished hormonal responses to GHRH, TRH, or LHRH are appropriate candidates for genetic analysis. Molecular diagnosis will aid in developing appropriate strategies for hormone replacement, and it will permit presymptomatic diagnosis or exclusion of diagnosis in their younger siblings.
Supported by Grants 97-27 and 98-7R from the Genentech Foundation for Growth and Development and by a grant from Pharmacia & Upjohn (to J.S.P.), PHS Grant NS25987 (to C.J.P.), NSF Career Award IBN9600805 (to D.L.H.), and NIH Award DK02569 (to M.P.W.).
Mayo KE.
Dattani M, Brickman J, Tyrrell R, et al. 1999 Novel mutations of HESX1 associated with septo-optic dysplasia in man. Proc 81st Meeting of The Endocrine Society, San Diego, CA (abstract OR12–1).
de
Bartke A.

{kind=link}
{kind=link}
{kind=link}
{kind=link}