





Abstract
C-type natriuretic peptide (CNP), found in endothelial cells, chondrocytes, and neurons, binds its cognate transmembrane receptor, natriuretic peptide receptor-B (NPR-B/GC-B), and stimulates the synthesis of the intracellular signaling molecule, cGMP. The known physiologic consequences of this binding event are vasorelaxation, inhibition of cell proliferation, and the stimulation of long bone growth. Here we report that 10% fetal bovine serum markedly reduced CNP-dependent cGMP elevations in NIH3T3 fibroblast. The purified serum components platelet-derived growth factor and lysophosphatidic acid (LPA) mimicked the effect of serum on CNP-dependent cGMP elevations, but the latter factor resulted in the most dramatic reductions. The LPA-dependent inhibition was rapid and dose dependent, having t1/2 and IC50 values of approximately 5 min and 3.0 μm LPA, respectively. The decreased cGMP concentrations resulted from reduced CNP-dependent NPR-B guanylyl cyclase activity that did not require losses in receptor protein or activation of protein kinase C, indicating a previously undescribed desensitization pathway. These data suggest that NPR-B is repressed by LPA and that one mechanism by which LPA exerts its effects is through the heterologous desensitization of the CNP/NPR-B/cGMP pathway. We hypothesize that cross-talk between the LPA and CNP signaling pathway maximizes the response of fibroblasts in the wound-healing process.
C-TYPE NATRIURETIC PEPTIDE (CNP) is a member of a structurally and functionally related family of hormones called natriuretic peptides, which also includes atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) (1). In response to increased cardiac wall stretch, ANP and BNP are secreted from atrial and ventricular cardiomyocytes, respectively. Their primary function is to lower blood pressure by stimulating renal sodium and water excretion, vascular smooth muscle relaxation and inhibiting renin and aldosterone secretion. In contrast, CNP is not stored in granules; instead, it is regulated at the level of transcription. It is not found in significant quantities in blood but is highly concentrated in endothelial cells, chondrocytes, and the brain. With respect to endothelial cells, a vascular natriuretic peptide system has been suggested in which CNP regulates vascular function in a paracrine manner (2). In this scenario, vascular endothelial cells secrete CNP in response to specific cytokines or shear stress. Upon release, it binds natriuretic peptide receptor (NPR)-B found on adjacent vascular smooth muscle cells in which it relaxes them and inhibits their migration and proliferation. With respect to long bone growth, CNP stimulates the expansion of the proliferative and hypertrophic zones in cultured fetal mouse tibias, which is required for longitudinal bone growth (3). Consistent with these data, the targeted disruption of the CNP gene results in mice with impaired endochondral ossification and severe dwarfism (4).
Natriuretic peptides signal through the cell-surface family of guanylyl cyclase receptors, enzymes that catalyze the conversion of GTP into the intracellular second messenger cyclic GMP (5–8). Natriuretic peptide receptor-A (NPR-A) is activated by physiologic concentrations of ANP and BNP, whereas CNP activates NPR-B. In addition, all three natriuretic peptides bind NPR-C, which does not possess guanylyl cyclase activity. It has similar affinities for all three peptides and controls the local levels of these peptides that are available to bind NPR-A and NPR-B via receptor-mediated internalization and degradation. Consistent with this hypothesis, mice lacking NPR-C display a decreased ability to degrade circulating ANP and a phenotype indicative of the superactivation of NPR-A and NPR-B (9). On the other hand, evidence also suggests that NPR-C signals through the heterotrimeric GTP binding proteins Gi and/or Go, to inhibit adenylyl cyclase or activate phospholipase C (10–12).
The prodilatory/antiproliferative actions of natriuretic peptides are directly antagonistic to the vasoconstrictory effects of the pressor hormones angiotensin II, arginine-vasopressin, and endothelin as well as the mitogenic effects of platelet-derived growth factor (PDGF). Hence, it is not surprising that cross-talk exists between natriuretic peptides and these antagonistic signaling pathways (13–15). Recently, fetal bovine serum (FBS) was shown to inhibit CNP-dependent cGMP elevations in NIH3T3 cells stably overexpressing NPR-B (16). The serum factor responsible for the desensitization was suggested to be PDGF because it inhibited CNP-dependent cGMP elevations in untransfected Balb3T3 cells. In the process of confirming these data, we found that although PDGF clearly desensitized NPR-B in the overexpressing NIH3T3 cells, the effect of saturating concentrations of PDGF accounted for only a small portion of the desensitization elicited by 10% FBS. This suggested that an additional serum component or components also desensitizes NPR-B. Here we describe for the first time the inhibitory effect of lysophosphatidic acid (LPA) on the CNP/NPR-B/cGMP signal transduction pathway in fibroblasts.
Materials and Methods
Materials
Rat ANP and CNP were from Peninsula Laboratories, Inc. or Sigma-Aldrich, St., Louis, MO). Rat PDGF-BB was purchased from R&D Systems (Minneapolis, MN). LPA was from Avanti Polar Lipids (Alabaster, AL) as a powder or chloroform solution. The powder was dissolved in 95% ethanol by warming to 50 C and briefly sonicating in a water bath. Both forms of LPA gave equivalent results. The NIH3T3 cells (CRL-1658) were acquired from ATCC (Manassas, VA). The NIH3T3 cell line stably overexpressing NPR-B has been previously described (17). GF-109203X and the alumina resin used for cGMP purification were from Sigma. 32P-α-GTP (NEG-006H) was from PerkinElmer Life Sciences, Inc. (NEN Life Science Products, Boston, MA). The horseradish perioxidase-conjugated donkey antirabbit secondary antibody was purchased from Amersham Pharmacia Biotech (Piscataway, NJ).
Cell culture and preparation of crude membranes
NIH3T3 and NIH3T3-NPR-B cells were maintained in DMEM containing 10% FBS (Mediatech, Inc., Herndon, VA) in an atmosphere of 95% air and 5% CO2 at 37 C. To prepare crude membranes, 10-cm plates of approximately 85% confluent cells were washed and incubated overnight with DMEM in the absence of serum. After incubation with DMEM containing varying concentrations of LPA, FBS, or phorbol-12-myristate-13-acetate (PMA), cells were washed twice with PBS, scraped into 0.5 ml phosphatase inhibitor buffer (25 mm HEPES, 20% glycerol, 50 mm NaCl, 50 mm β-glycerol phosphate, 2 mm EDTA, 1 μm microcystin, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 μg/ml pepstatin), sonicated for 1–2 sec with a Misonix Sonicator XL2020, and centrifuged at 20,000 × g for 20 min at 2 C. Pellets were resuspended in phosphatase inhibitor buffer and assayed for guanylyl cyclase activity or stored at −80 C. Protein concentrations were estimated using a bicinchoninic acid assay or Coomassie dye binding protein assay kits (Pierce Chemical Co., Rockford, IL).
Whole-cell stimulations
Cells plated in 12-well dishes were grown to 90% confluency and incubated overnight without serum. The dishes were then placed on a slide warmer maintained at 37 C for 1 h. In some experiments the medium was aspirated and replaced with 0.5 ml DMEM containing 20 mm HEPES (to stabilize pH at room atmosphere), 0.5 mm 1-methyl-3-isobutylxanthine to inhibit phosphodiesterases, and various concentrations of LPA or vehicle. After a 30-min incubation, the media was aspirated and replaced with the same media containing various concentrations of CNP. The cells were then stimulated for 3 min and stopped by aspirating the media and adding 1 ml ice-cold 80% ethanol. The ethanol extract was transferred to 1.5-ml tubes and centrifuged for 10 min at 20,000 × g to remove any particulate matter. The supernatant was transferred to glass tubes, and the ethanol was evaporated to dryness in a speedvac apparatus. In experiments with the NIH3T3 cells overexpressing NPR-B, the assay was stopped with an equal volume of 1N perchloric acid, and 10 μl of this acidic extract was assayed for cGMP content. The amount of cGMP contained in each sample was estimated by RIA according to the manufacturer’s instructions (NEN Life Science Products).
Guanylyl cyclase assays
Guanylyl cyclase assays were performed in the presence of 25 mm HEPES, 50 mm NaCl, 0.1% BSA, 500 μm 1-methyl-3-isobutylxanthine, 1 mm EDTA, 5 mm creatine phosphate, and 0.1 mg/ml creatine kinase (as a nucleotide-regenerating system), 1 μm microcystin, and 1 or 0.1 mm GTP. Some assays included 0.1–0.2 μCi [α-32P]GTP. Activator cocktails consisted of 1 mm ATP, 0.1 or 1 μm CNP, and 5 mm MgCl2 or 1% Triton X-100, and 5 mm MnCl2. Between 25 and 50 μg crude membranes were assayed for 3 min at 37 C by the addition of a mixture containing the above reagents in a total volume of 100 μl. The reactions were initiated by the addition of a cocktail containing the substrate to the membranes. Reactions containing [α-32P]GTP were terminated with 500 μl of 110 mm zinc acetate. To purify the cGMP, 0.5 ml of 110 mm sodium carbonate was added to the mixture, vortexed, and centrifuged at 3000 × g for 10 min at 2 C. The supernatant was added to chromatography columns (model 731-1550, Bio-Rad Laboratories, Inc., Hercules, CA) containing 0.5 g dry neutral alumina resin (A9003, Sigma) acidified with 5 ml of 1 n perchloric acid. The columns were then washed with 10 ml of 1 n perchloric acid followed by 10 ml water. The purified [α-32P]GTP was then eluted with 5 ml freshly prepared 200 mm ammonium formate and quantitated using the Cerenkov method in a 3801 scintillation counter (Beckman). Some guanylyl cyclase assays were performed in the absence of [α-32P]GTP, and the amount of cGMP formed was estimated by RIA. These reactions were stopped by the addition of 400 μl of 50 mm sodium acetate buffer, pH 6.2, containing 5 mm EDTA followed by boiling for 5 min. The cGMP concentrations were estimated according to the manufacturer’s instructions (NEN Life Science Products).
Immunoblot analysis
NPR-B present in crude membranes was fractionated on an 8% SDS-polyacrylamide gel and blotted to polyvinylidene difluoride (Immobilon P) membrane using a Trans-Blot semidry transfer cell (Bio-Rad Laboratories, Inc.). The membrane was then incubated for 1 h in TBST [20 mm Tris (hydroxymethyl)aminomethane, 500 mm NaCl, and 0.05% polyoxyethylene sorbitan monolaurate, pH 7.5] containing 3% BSA, followed by three 5-min washes with TBST. The primary antiserum was diluted 1:2,500 in TBST and incubated with the membrane for 2 h, followed by 4 washes for 5 min with TBST. The specific antisera were raised against synthetic peptides corresponding the last 17 or 10 carboxyl-terminal amino acids of NPR-A or NPR-B, respectively, which were conjugated to keyhole limpet hemocyanin. These antisera are specific for each receptor and do not cross-react (see Fig. 2). The membrane was then incubated with donkey antirabbit horseradish peroxidase-conjugated secondary antibody (Amersham Pharmacia Biotech) diluted 1:10,000 in TBST for 45 min. After four washes for 5 min with TBST, the NPR-B antibody complex was visualized by chemiluminescence using the ECL Western blot detection system (Amersham Pharmacia Biotech).
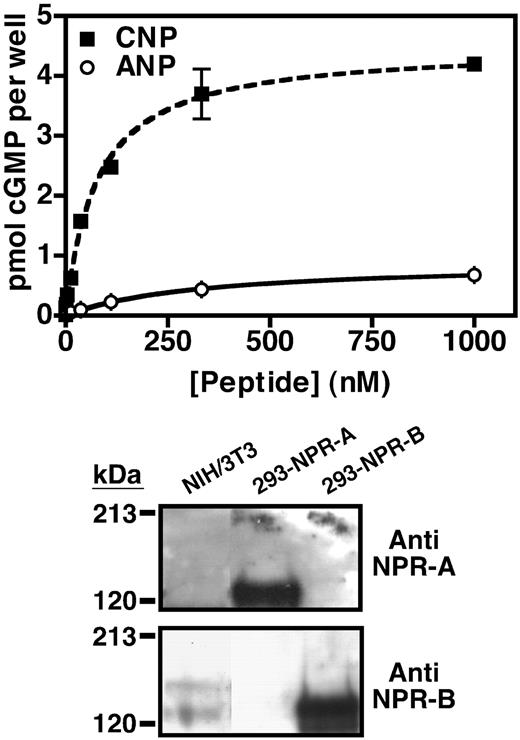
NIH3T3 cells express NPR-B. Top panel, NIH3T3 cells were serum starved and incubated with increasing concentrations of CNP (▪) or ANP (○) for 3 min. The reaction was terminated, and the cellular cyclic GMP concentration in each well was estimated by RIA. The wells contained between 50,000 and 200,000 cells. Values are the mean of three separate determinations (±sem) from one representative experiment. Where error bars are not visible, they are contained within the data point. Bottom panel, Twenty-five-microgram crude membranes of NIH3T3, 293-NPR-A and 293-NPR-B cells were separated by SDS-PAGE and blotted to polyvinylidene difluoride membrane. NPR-A (top panel) or NPR-B (lower panel) was detected by Western blot analysis using antiserum directed against NPR-A or NPR-B as indicated. This experiment was performed at least three times with similar results.
Data analysis and statistics
The data were graphed and IC50 estimated with GraphPad Software Prism for the MacIntosh (GraphPad Software, Inc., San Diego, CA). In Fig. 2, the dose-response curve was fit using the equation: Y = bottom + (top − bottom)/(1 + 10^((logEC50-X)*hill slope)). The top is the best-fit highest value, which the program determined to be 0.2381. The bottom is the best-fit lowest value, given by 0.05303. With these two values, the logEC50 was estimated at −5.540. The caveat to this method of estimation is its assumption that the lowest value is 0.05303, when it is unclear whether LPA is able to further inhibit cGMP elevations at higher concentrations. Hence, it is possible that this method may underestimate the IC50 value if LPA concentrations higher than 100 μm result in maximal desensitization of NPR-B. Unfortunately, we were unable to test concentrations of LPA higher than 100 μm because of nonspecific effects of the diluent. To determine the two-tailed P values as indicated in Fig. 3, the mean values of the cGMP concentrations from cells treated with or without LPA were analyzed using a t test.
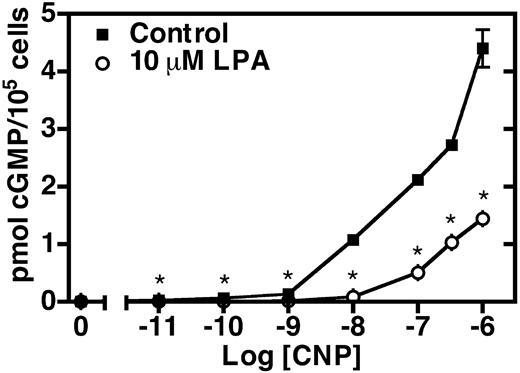
LPA inhibits CNP-dependent cGMP elevations in NIH3T3 cells. Serum-starved NIH3T3 cells were incubated in the presence (○) or absence (▪) of 10 μm LPA for 30 min. The media was aspirated, and the cells were stimulated with increasing concentrations of CNP for 3 min. The reaction was then terminated by aspirating the media and adding 1 ml 80% ethanol. Cellular cGMP levels were determined by RIA. Values are the mean of four separate determinations (±sem) of one representative experiment. Where error bars are not visible, the error is contained within the data point. The asterisk indicates that differences in cGMP levels from LPA-treated and control cells are statistically significant with a P value of < 0.01 as determined by a t test. This experiment was repeated three or more times with similar results.
Results
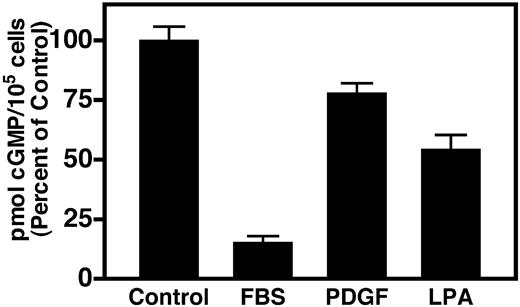
As a first step toward investigating the mechanism of the PDGF-dependent desensitization of NPR-B, we examined the ability of 10% FBS and 10 nm PDGF to inhibit CNP-dependent cGMP elevations in NIH3T3 cells stably expressing NPR-B. These cells are well characterized and have been used to study both homologous (17) and heterologous (16) desensitization of NPR-B. Similarly to a previous report (16), we found that prior exposure of these cells to FBS reduced CNP-dependent cGMP levels to less than 20% of the levels detected in untreated cells (Fig. 1). On the other hand, when these cells were incubated with saturating concentrations of PDGF, we found that the CNP-dependent cGMP levels were depressed to only approximately 80% of control values. The difference between the mean values of the cGMP concentrations detected in extracts from control and PDGF-treated cells was statistically significant at a P value of less than 0.01, which clearly indicates that PDGF desensitizes NPR-B in these cells. On the other hand, these results suggest that serum contains an additional desensitizing agent or agents because the majority of the serum-dependent desensitization was not accounted for by PDGF.
LPA inhibits CNP-dependent cGMP production in NIH3T3 cells overexpressing NPR-B. Serum-starved NIH3T3 cells stably transfected with NPR-B were treated with or without 10% FBS, 10 nm PDGF, or 10 μm LPA for 30 min. The media was aspirated, and the cells were stimulated with 20 nm CNP for 3 min. The reaction was terminated by the addition of 1 N perchloric acid. The total cyclic GMP concentration from each well was estimated by RIA. Values are the mean of at least four determinations from one representative experiment. This experiment was performed at least twice with similar results. The vertical lines centered within each column represent the sem. Control values are approximately 220 pmol/100,000 cells.
An important mitogenic factor found in high concentrations (1–5 μm) in mammalian serum is LPA (18). In fibroblasts, LPA stimulates cell proliferation, cell migration, DNA synthesis, and activation of protein kinase C (PKC) (19). The latter observation is particularly relevant to our study because activated PKC inhibits NPR-B (20). Therefore, because LPA is present in high concentrations in serum, the known actions of LPA are antagonistic to the actions of CNP, and LPA activates PKC, we investigated the ability of LPA to inhibit CNP signaling in these overexpressing NIH3T3 cells. We found that 10 μm LPA reduced CNP-dependent cGMP elevations to approximately 50% of control values, which is a substantially greater desensitization than was observed with PDGF in these cells (Fig. 1).
Because we were concerned about the physiological significance of studying the regulation of NPR-B in highly overexpressing cells, we asked whether NIH3T3 cells endogenously express NPR-B, and if so, whether it is also desensitized by LPA. To this end, we examined the sensitivity of untransfected NIH3T3 cells to ANP or CNP by measuring cellular cGMP production in response to increasing concentrations of each peptide. Exposure of whole cells to CNP resulted in a dose-dependent accumulation of intracellular cGMP (Fig. 2A, squares). Measurable increases were detected with CNP concentrations as low 0.45 nm, and the maximum concentration tested (1 μm) resulted in a 250-fold increase over cGMP levels detected in the absence of CNP (basal). In contrast, exposure of the NIH3T3 cells to similar concentrations of ANP only slightly stimulated cGMP production (Fig. 2A, circles). Significant cGMP elevations were first observed at 35 nm ANP, and exposure to 1 μm ANP resulted in a 34-fold stimulation. These dose-response curves are similar to those previously observed in NIH3T3 cells by others (21, 22). Furthermore, they are similar to those obtained using cells transfected with the rat or human version of NPR-B, which suggest that the ANP-dependent cGMP elevations in the NIH3T3 cells result from cross-activation of NPR-B (23, 24).
To confirm the expression of NPR-B in these cells, we performed Western blot analysis using antiserum specific for NPR-A or NPR-B (Fig. 2, bottom panel). When crude membranes from the NIH3T3 cells or 293 cells transfected with NPR-A (293-NPR-A) or NPR-B (293-NPR-B) were fractionated by SDS-PAGE, blotted to an Immobilon membrane support, and probed with antiserum specific for NPR-A, we detected a very strong signal in the 293-NPR-A membranes but no positive signal in membranes from the 293-NPR-B or NIH3T3 cells. These data indicate that the antiserum against NPR-A does not cross-react with NPR-B and that NPR-A is either not present or present at undetectable levels in the NIH3T3 cells. In contrast, when we probed a duplicate Immobilon membrane with antiserum specific for NPR-B, we observed a positive signal in membranes from the 293-NPR-B and the NIH3T3 cells but not the 293-NPR-A cells. Notably, a second, higher-molecular-weight band was also detected in the NIH3T3 membranes, which likely represents NPR-B with additional cell-specific glycosylation, similar to that observed with NPR-A expression in nervous tissue (25). These data indicate that the antiserum against NPR-B does not cross-react with NPR-A and provide strong support for the expression of NPR-B, but not NPR-A, in these NIH3T3 cells.
We then asked whether NPR-B expressed endogenously in these cells was inhibited by LPA. We found that the incubation of confluent, serum-starved NIH3T3 cells with 10 μm LPA for 30 min suppressed cGMP production at every CNP concentration tested (Fig. 3). The differences were significant at P values of 0.01 or less. Stimulation for 3 min with 100 nm CNP elevated cGMP levels to approximately 2.1 pmol/100,000 cells, whereas a 30-min exposure to 10 μm LPA decreased CNP-dependent cGMP formation to less than 25% of this value (0.5 pmol cGMP/100,000 cells). The inhibition was greater with lower CNP concentrations; at 10 nm CNP, LPA inhibited approximately 90% of cGMP production, whereas at 1000 nm CNP, only a 68% percent inhibition was observed. On the other hand, we did not detect any effect of LPA on basal (unstimulated) intracellular cGMP concentrations.
To determine whether the reduced cGMP levels were due to decreased NPR-B activity, we performed guanylyl cyclase assays on crude membranes isolated from NIH3T3 cells treated with or without 10 μm LPA for 30 min. Membranes were assayed for basal (5 mm MgCl2 and 1 μm ATP, open symbols) and CNP-stimulated (5 mm MgCl2, 1 μm ATP, and 100 nm CNP, closed symbols) guanylyl cyclase activities for 5 and 10 min (Fig. 4). LPA reduced hormone-dependent cGMP production to approximately 30% of the activity measured in membranes isolated from control cells, whereas no significant change in basal guanylyl cyclase activity was detected. These data mirror the effects of LPA on whole-cell cGMP concentrations (Fig. 3) and suggest that these reductions result solely from decreases in CNP-dependent guanylyl cyclase activity and not from increases in the degradation of cGMP by cyclic nucleotide phosphodiesterases. Additionally, the lack of an effect on basal activity suggests that the reduced hormone-dependent activity does not result from receptor degradation, which is consistent with the observation that LPA treatment does not reduce NPR-B protein levels as determined by Western blot analysis (Fig. 4, inset).
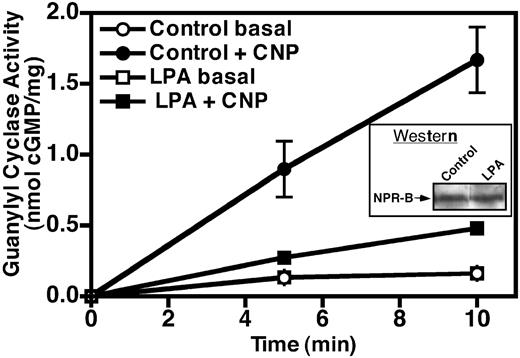
Incubation of NIH3T3 cells with LPA decreases NPR-B guanylyl cyclase activity. NIH3T3 cells were serum starved and treated with or without 10 μm LPA for 30 min. Then crude membranes were prepared and assayed for basal (1 mm ATP and 5 mm MgCl2) and hormone-dependent (1 mm ATP, 1 μm CNP, and 5 mm MgCl2) guanylyl cyclase activity. Cyclic GMP concentrations were estimated by RIA. Values are the mean of two separate determinations, which were assayed in duplicate (±sem). The data represent one of at least three similar experiments. Where error bars are not visible, they are contained within the data point. Inset, Fifty-microgram crude membranes prepared from NIH3T3 cells treated with or without 10 μm LPA were separated by SDS-PAGE and blotted to polyvinylidene difluoride membrane. The amount of NPR-B present was detected by Western blot analysis using antiserum directed against NPR-B, as described in Materials and Methods.
To further characterize the LPA-dependent desensitization of NPR-B, we examined the sensitivity of the NIH3T3 cells to increasing concentrations of LPA. In this experiment, we incubated the cells with various concentrations of LPA for 30 min and then measured cGMP production in the presence of 20 nm CNP. We found that LPA inhibited the CNPdependent elevations in these cells in a dose-dependent manner. We first detected an effect (28% inhibition) at 1 μm LPA, and the highest concentration tested (100 μm LPA) reduced cGMP concentrations to 25% of untreated levels. From these data we estimated an IC50 value for LPA of approximately 3 μm (Fig. 5).
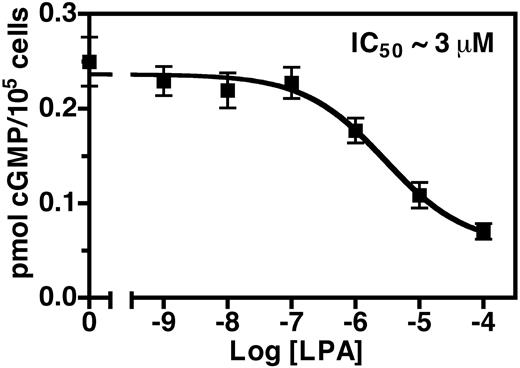
LPA decreases CNP-dependent cGMP production in a dose-dependent manner. Serum-starved NIH3T3 cells were treated with increasing concentrations of LPA for 30 min, the media were aspirated, and the cells were stimulated with 20 nm CNP for 3 min. The reaction was terminated by aspirating the media and adding 1 ml 80% ethanol. The cellular cGMP concentration from each well was estimated by RIA. Values are the mean of six separate determinations (±sem) from one of at least three representative experiments.
To determine the time course of the LPA-dependent desensitization, we serum starved the NIH3T3 cells overnight and then incubated them in the presence or absence of 10 μm LPA for 0, 10, 20, 40, and 960 min. Membranes were then prepared from these cells and assayed for hormone-dependent (5 mm MgCl2, 1 μm ATP, and 100 nm CNP, squares) or detergent-dependent (5 mm MnCl2, 1% Triton X-100, triangles) guanylyl cyclase activities (Fig. 6). The latter conditions maximally activate NPR-B independently of ligand and are an excellent indicator of total amount of guanylyl cyclase receptor present in any given preparation (17). The effect of LPA on CNP-dependent activity was rapid, having a t1/2 of approximately 5 min and was maximal by 10 min. The desensitization was maintained for about 20 min, but by 40 min activity was returning to control levels and by 960 min the effect was completely lost. The reason for the resensitization is not known, but because the addition of a fresh aliquot of LPA to cells exposed overnight to LPA restores the desensitization (data not shown), it likely results from the breakdown of LPA. These data indicate that NPR-B inhibition is an early and transient consequence of LPA receptor activation.
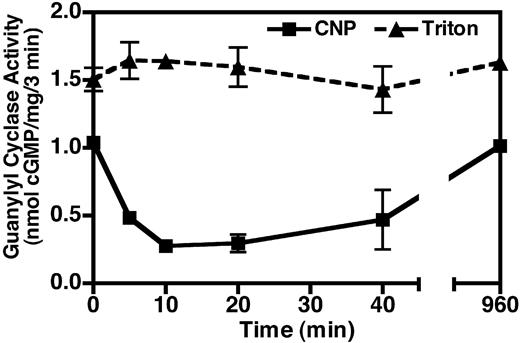
The LPA-dependent desensitization of NPR-B is rapid. Serum-starved NIH3T3 cells were incubated with 10 μm LPA for the indicated period of time, crude membranes were prepared from the treated cells, and assayed for guanylyl cyclase activity in the presence of 1 mm ATP, 100 nm CNP, and 5 mm MgCl2 (▪) or 1% Triton X-100 and 5 mm MnCl2 (▴) as described in Materials and Methods. cGMP concentrations were estimated by RIA. Values are the mean of two determinations, which were assayed in duplicate (±range). The data represent one of three experiments with similar results. Where error bars are not visible, they are contained within the data point.
The synthetic PKC activator, PMA, has been shown to desensitize NPR-B (15, 20, 26–28), and LPA has been shown to activate PKC in fibroblast (29). Therefore, we asked whether phorbol ester-sensitive PKC was required for the LPA-dependent desensitization of NPR-B in NIH3T3 cells (Fig. 7). We found that crude membranes isolated from NIH3T3 cells exposed to 100 nm PMA for 30 min contained only about 40% of the CNP-dependent guanylyl cyclase activity (black columns) obtained in membranes from untreated cells. Similarly, membranes from cells exposed to 10 μm LPA for 30 min contained only 25% of CNP-dependent activity, compared with control membranes. In contrast, neither PMA nor LPA treatment resulted in changes in detergent-dependent guanylyl cyclase activities (gray columns), indicating that the losses in hormone-dependent activity were not due to receptor degradation. As expected, preincubation of the cells for 1 h with 1 μm GF-109203X, a cell-permeable inhibitor of the α1, β1, β2, δ, ε, and γ PKC isoforms (30), abrogated the PMA-dependent desensitization. GF-109203X appears to be specific for PKC because it had no effect on cyclase activity measured in membranes from untreated cells. In surprising contrast to PMA, GF-109203X was unable to block the LPA-dependent desensitization of NPR-B. These data indicate that even though LPA has been shown to activate PKC in fibroblasts (29), LPA does not require PKC for the desensitization of NPR-B in NIH3T3 cells. Hence, these data provide evidence for the existence of a previously undescribed PKC-independent NPR-B heterologous desensitization pathway.
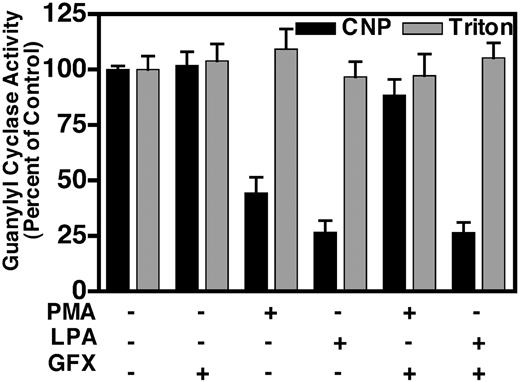
LPA-dependent desensitization does not require PKC. Serum starved, NIH3T3 cells were pretreated with 1 μm GF-109203X or mock treated with buffer for 1 h. The cells were then incubated with or without 100 nm PMA or 10 μm LPA for 30 min. Crude membranes were prepared and assayed for CNP-dependent (1 mm ATP, 1 μm CNP, and 5 mm MgCl2, black columns) and detergent-dependent (1% Triton X-100 and 5 mm MnCl2, gray columns) guanylyl cyclase activity for 3 min. These data are the average of two experiments. The vertical bar within each column represents the sem where n = 4. Control values for hormone and detergent-dependent guanylyl cyclase activities were approximately 0.4 and 1.0 nmol/mg per 3 min, respectively.
To further characterize the pathways involved in the inhibition of NPR-B, we treated NIH3T3 cells in the presence or absence of 50 μm LPA, 1 μm PMA, 10 nm PDGF, LPA and PMA, or LPA and PDGF for 30 min and then measured the guanylyl cyclase activity of crude membranes prepared from these cells (Fig. 8). Membranes isolated from cells incubated with LPA, PMA, and PDGF contained approximately 30%, 60%, and 33% of the CNP-dependent (black columns) activity obtained in membranes isolated from cells exposed to buffer alone. Interestingly, we found that PDGF desensitized NPR-B in membranes from the NIH3T3 cells to a much greater extent than that observed in the overexpressing line (Figs. 1 and 8). The detergent-dependent (gray columns) activity remained unchanged for all six treatments, indicating that there was no alteration in total amount of NPR-B. Given the results in Fig. 7, we expected that membranes prepared from cells treated with both LPA and PMA would contain less guanylyl cyclase activity than membranes from cells treated with either agent alone because the effects of the PKC-dependent and -independent pathways would be additive. Surprisingly, the combined treatment resulted in CNP-dependent activity close to 55% of the control, which was similar to the activity observed in membranes isolated from cells exposed to PMA alone. In contrast, when cells were incubated with both LPA and PDGF, the CNP-dependent activity of crude membranes was 20% of the control, which is less than that observed in membranes from cells treated with either agent alone.
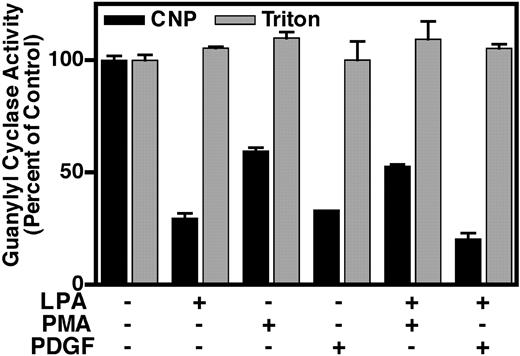
Combinatorial effects of LPA, PMA, and PDGF. Serum-starved NIH3T3 cells were exposed to 50 μm LPA, 1 μm PMA, 10 nm PDGF, or a combination of the treatments for 30 min. Crude membranes were prepared and assayed for CNP-dependent (1 mm ATP, 1 μm CNP, and 5 mm MgCl2, black columns) and detergent-dependent (1% Triton X-100 and 5 mm MnCl2, gray columns) guanylyl cyclase activity for 3 min. Values are the mean of two separate plate determinations (±range), and where the bar is not visible, the error is contained within the column. These data are representative of at least three experiments. Control values for hormone- and detergent- dependent guanylyl cyclase activities were approximately 0.2 and 0.6 nmol/mg per 3 min, respectively.
Discussion
To our knowledge, this is the first report of cross-talk between a bioactive lipid and natriuretic peptide signaling pathway. We show that 1) LPA decreases cGMP production in whole NIH3T3 cells; 2) the decreased cGMP levels are explained by reductions in the CNP-dependent, but not basal, guanylyl cyclase activity; 3) the LPA-dependent desensitization is rapid having a t1/2 of approximately 5 min, and 4) the reduced NPR-B guanylyl cyclase activity does not require reductions in receptor protein or the activation of PKC, which is indicative of a previously undescribed desensitization pathway. Together these data have important implications for both natriuretic and lipid signaling because they suggest that NPR-B is under the constant repression of LPA because of its high serum concentrations and that inhibition of NPR-B is an early consequence of LPA signaling. Based on these and other data, we suggest that the activity of NPR-B is regulated by not only local CNP concentrations but also the presence of various desensitizing agents in the extracellular milieu. Hence, the hormone responsiveness of NPR-B likely represents a steady state between the pathways that desensitize and resensitize the receptor, and subtle shifts in the balance of these pathways may significantly impact CNP-dependent responses.
LPA-dependent inhibition of NPR-B is dose dependent, which is consistent with a receptor-mediated process. Furthermore, directly adding 10 μm LPA to NIH3T3 membrane preparations does not affect NPR-B activity (data not shown), suggesting that LPA-dependent desensitization is not mediated by direct binding of LPA to NPR-B. The specific receptor responsible for this effect is unknown. LPA activates members of the endothelial differentiation gene (Edg) family of G protein-coupled receptors, a family consisting of eight receptors that are divided into two groups. The LPA receptor subfamily contains the receptors Edg2, Edg4, and Edg7, whereas the second subfamily binds a related biolipid called sphingosine-1-phosphate and includes Edg1, Edg3, Edg5, Edg6, and Edg8 (31–33). In NIH3T3 cells, Edg1 (34), and Edg2 but not Edg4 (35) mRNA transcripts were detected by RT-PCR and Northern analysis. Hence, Edg4 is not likely to be involved in LPA-dependent desensitization of NPR-B in these cells. In addition, because the EC50 value for LPA-dependent activation of Edg2 is approximately 100-fold lower than the concentrations necessary for the desensitization of NPR-B, it is unlikely to be involved in this process. Recently, LPA was shown to be a low-affinity agonist for Edg1, having an apparent dissociation constant of approximately 2.3 μm, which is similar to the 3 μm IC50 observed for the desensitization of NPR-B (36). Thus, Edg1 is a reasonable candidate for the receptor that mediates the desensitization of NPR-B in NIH3T3 fibroblasts. Additional studies will be required to unequivocally identify the specific LPA receptor involved in this response.
Although we have shown that LPA can inhibit NPR-B in cultured fibroblasts, the physiologic significance of this interaction remains to be determined. One scenario in which LPA may inhibit CNP signaling in fibroblasts is during wound healing. As an early response to injury, fibroblasts proliferate and then migrate to the wound insult (37). This process is stimulated in part by LPA, which is released from platelets during blood clotting (38). In contrast, CNP inhibits cell proliferation and migration (39, 40). Therefore, LPA may block the inhibitory effects of CNP on these processes to maximize the ability of the fibroblast to participate in the healing process. Consistent with this idea, ANP has been shown to inhibit epidermal growth factor-dependent wound closure in corneal epithelial cells via a cGMP-dependent mechanism (41). Additional experiments will be required to determine whether CNP can similarly inhibit LPA-dependent wound healing in fibroblasts.
The molecular mechanism employed by LPA as well as other antagonistic hormones that desensitized NPR-B is currently undefined. In this study, we show that LPA mediated desensitization does not require NPR-B degradation or GF-109203X-sensitive forms of PKC. Therefore, LPA appears to employ a previously undescribed desensitization mechanism. The lack of involvement of PKC is particularly surprising because phorbol ester-dependent activation of PKC has been shown by many groups to desensitize NPR-B. In fact, we have recently defined the molecular basis of this inhibition by demonstrating that PMA causes the selective dephosphorylation of NPR-B at Ser-523 (20). Importantly, this single dephosphorylation event appears to be the mechanism for the PMA-dependent desensitization because the mutation of Ser-523 to alanine or glutamate completely abrogates the effect of PMA on NPR-B.
The results in Figs. 7 and 8 suggest that the molecular mechanisms involved in NPR-B heterologous desensitization are more complex than previously appreciated. For example, treatment of NIH3T3 cells with PMA and LPA resulted in the more moderate desensitization than was observed with PMA exposure, not an increased desensitization that would have been anticipated if the desensitory effects of PMA and LPA had been additive. One explanation for these data are that PMA-dependent dephosphorylation of NPR-B at Ser-523 alters the structure of the enzyme such that it is no longer susceptible to inhibition by LPA signaling. In contrast, PDGF treatment slightly increases the NPR-B inhibition beyond that observed when cells are exposed to LPA alone. Although this is consistent with LPA and PDGF using separate pathways to inhibit NPR-B, we cannot rule out the possibility that the concentration of LPA used in these experiments did not maximally desensitize NPR-B. In fact, this is likely because we were unable to achieve a saturating dose of LPA in our experiments because of the relative insolubility of LPA in our solvent (Fig. 5).
Lastly, future experiments will focus on determining the role of receptor dephosphorylation in LPA-induced inhibition of NPR-B activity and identifying the signaling pathways that are required for this process. Recently we demonstrated that arginine-vasopressin inhibits NPR-B activity in a calcium-dependent manner in vascular smooth muscle cells (42). Whether the LPA-dependent desensitization requires elevations in calcium as well remains to be determined.
Acknowledgments
This work was supported by a National American Heart Association Scientist Development Grant (013938N), an equipment grant from the Minnesota Medical Foundation, and a grant-in-aid from the University of Minnesota Graduate School.
We thank Alyssa Cody for excellent technical assistance.
Abbreviations
- ANP
Atrial natriuretic peptide
- BNP
brain natriuretic peptide
- CNP
C-type natriuretic peptide
- Edg
endothelial differentiation gene
- FBS
fetal bovine serum
- LPA
lysophosphatidic acid
- NPR
natriuretic peptide receptor
- PDGF
platelet-derived growth factor
- PKC
protein kinase C
- PMA
phorbol 12-myristate-13-acetate

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}