





Human progesterone receptors (PR) rapidly activate cytosolic signaling pathways, in addition to their classical function as ligand-activated transcription factors. Using ER+/PR-B+ T47D breast cancer cells, we probed the role of progestin-stimulated rapid PR signaling in the transcriptional regulation of target genes involved in breast cancer cell proliferation. Epidermal growth factor receptor (EGFR) was rapidly activated after a 10-min treatment with R5020. Progestin induced EGFR-, c-Src-, and MAPK-dependent phosphorylation of PR-B on the MAPK consensus site, Ser345. Ser345-phosphorylated PR-B receptors strongly associated with specificity protein 1 (Sp1) transcription factors to regulate PR cell cycle (p21) and growth-promoting (EGFR) target genes whose promoters lack canonical progesterone response element sequences. Inhibitors of EGFR, c-Src, or MAPK activities blocked PR tethering to Sp1 and progestin-stimulated S-phase entry. Mutant PR-B receptors defective for c-Src binding (mPro) were not phosphorylated on Ser345 in response to progestin and failed to interact with Sp1. Hormone-induced complexes containing Sp1 and wild-type PR-B, but not S345A or mPro PR-B, were recruited to Sp1 sites within the endogenous p21 promoter. Progestin-induced S-phase entry was attenuated in T47D cells containing wild-type PR-B and treated with EGFR, c-Src, or MAPK kinase inhibitors or in T47D cells stably expressing mPro or mutant DNA-binding domain PR-B. In sum, rapid progestin-activated PR signaling leads to PR Ser345 phosphorylation and tethering to Sp1. These events are critical for progestin-stimulated regulation of Sp1 target genes and breast cancer cell proliferation. Our data demonstrate the therapeutic potential for PR-targeted breast cancer treatment by exploiting multiple nodes along the PR signaling pathway, including PR-B, EGFR, c-Src, MAPK, or Sp1.
PROGESTERONE IS AN ovarian steroid hormone that orchestrates key aspects of female reproduction and mammary gland development. Progesterone and its synthetic progestin analogs are employed clinically as methods of contraception and hormone replacement therapy. It has been shown that inclusion of progestins in combined hormone replacement therapy confers an increased breast cancer risk (1). The health risks and benefits associated with progesterone and synthetic progestins call for a better understanding of their function in normal breast cell biology and health and, in turn, the deregulation that occurs in breast tumor malignancy.
A cholesterol derivative, progesterone diffuses across the lipid bilayer of a target cell to bind and activate its receptor. The progesterone receptor (PR) is expressed as three isoforms, PR-A, -B, and -C, all of which are members of the large superfamily of nuclear receptor transcription factors. Classically, progesterone binding to PR induces a change in receptor conformation, followed by dimerization and localization to progesterone response elements (PRE) in the promoter or enhancer regions of target genes. In addition to directly contacting DNA, PR can tether to other transcription factors, including specificity protein 1 (Sp1), signal transducer and activator of transcription 5 (Stat5), and activator protein 1 (AP1), to regulate gene promoters that lack canonical PRE sequences, including p21, β-casein, and cyclin D1 (2–4). To activate target gene transcription, ligand-activated PR recruits chromatin-modifying enzymes and the RNA polymerase II transcription apparatus. PR may also mediate ligand-independent gene transcription and/or repression (5, 6).
The intriguing ability of progestin and PR to rapidly initiate signaling through activation of multiple cytoplasmic kinases (7–10) could theoretically serve to potentiate several aspects of PR function, including changes in subcellular location, phosphorylation state, transcriptional activity, and degradation rate. Steroid receptor coactivators (SRC-1 or SRC-3) and histones are also potential substrates of kinase activity initiated by progestin and PR (11–13). Ligand binding to membrane-proximal or cytosolic PR results in the rapid interaction of PR with the soluble protein tyrosine kinase c-Src. This association occurs directly through a PR polyproline motif that is a ligand for the SH3 domain of c-Src family kinases (8) or indirectly through PR association with estrogen receptors (ER) and subsequent ER interaction with SH2 domains of c-Src molecules (7). After PR or ER ligand binding, protein-protein interaction with c-Src relieves an intermolecular inhibitory conformation, allowing for the autoactivation of c-Src. Active c-Src initiates the Ras/Raf/MAPK cascade (7, 8). Although it can be quite modest, progestin-induced activation of c-Src kinase and Erk1/2 MAPK have been shown to be of similar intensity to that elicited by EGF stimulation in T47D breast cancer cells (7).
The addition of progesterone or progestins to cultured, quiescent breast cancer cells stimulates a swift acceleration through one or more rounds of the cell cycle, followed by G1 arrest (14–16). Progestins are mitogenic to breast cancer cells growing in soft agar (17) and protect breast cancer cells from apoptosis (18). Although these studies provide insight into the proliferative advantage conferred by progestins to human breast tumors, the mechanism by which progestins mediate proliferation remains unclear. Paradoxically, many of the cell cycle or proliferative gene targets of PR do not contain a canonical PRE in their promoter regions and may thus be regulated by nonclassical PR tethering and/or rapid signaling independent of direct PR DNA binding. PR-B, but not PR-A, rapidly activates cytosolic signaling (8), mediates the proliferative actions of progestins (17), and is the key isoform required for mammary gland development (19). Herein, we hypothesized that the dual functions of PR-B as activators of both membrane-proximal signaling pathways and transcription converge upon the transcription factor Sp1 to stimulate nonclassical gene transcription and breast cancer cell proliferation. We show that epidermal growth factor receptors (EGFR) are rapidly activated by liganded PR-B upstream of the MAPK module. These membrane-proximal signaling events induce specific PR-B phosphorylation on Ser345 to mediate interaction with Sp1 and subsequent regulation of noncanonical target gene transcription. As such, Sp1 is a node integrating the distinct membrane-proximal signaling and transcriptional properties of PR-B and is a central mediator of the proliferative action of progestin.
RESULTS
EGFR Is Rapidly Activated by Agonist-Bound PR and Is Required for Progestin-Stimulated Cell Cycle Progression
Rapid estrogen signaling to receptor tyrosine kinases, including IGF-I receptors and EGFR, occurs via c-Src/Shc/ER complexes (20, 21). This prompted us to test whether EGFR is activated at early time points (10 min) after progestin treatment of T47D breast cancer cells stably expressing PR-B (T47D-YB) (22). Cells were treated with or without the synthetic progestin R5020 for 10 min and then lysed and EGFR was immunoprecipitated. Because Tyr1173 is a known site of autophosphorylation indicative of EGFR activation (23), active and total EGFR proteins were visualized using phospho-Tyr1173- and total-EGFR-specific antibodies (Ab), respectively (Fig. 1A). Surprisingly, EGFR is significantly more phosphorylated when cells are treated with R5020 for 10 min relative to vehicle control.
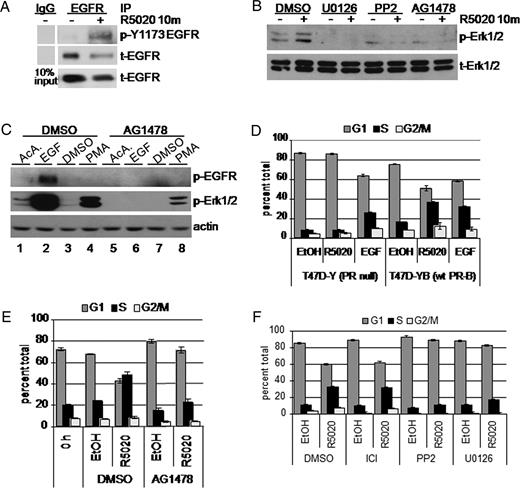
Progestins Induce Rapid Activation of EGFR Upstream of Erk1/2 MAPK A, Serum-starved T47D cells stably expressing wt PR-B (T47D-YB) were treated with R5020 (10 min). EGFR in whole-cell lysates was immunoprecipitated (IP). Immune complexes were subjected to Western blotting with phospho-Tyr 1173 Ab. Membranes were stripped and reprobed to detect total EGFR in each IP and lysates (10% input). IgG was included as a specificity control for IP-specific Ab. B, T47D-YB cells were pretreated with DMSO vehicle control, U0126, PP2 (10 μm each), or AG1478 (1 μm) for 30 min before addition of R5020 or EtOH for 10 min. Western blotting of whole-cell lysates was performed to detect phospho (p-) and total (t-) Erk1/2 MAPK. C, Serum-starved T47D-YB cells were incubated with DMSO or AG1478 for 30 min, followed by EGF (10 min) or PMA (30 min). Cell lysates were subjected to Western blotting as above; actin blots indicate equal protein loading. D, Triplicate cultures of PR-null (T47D-Y) or wt PR-B (T47D-YB) expressing cells were grown in the absence of steroid hormones for 48 h. After 18 h treatment with EtOH vehicle control, R5020, or EGF, cells were fixed and stained with propidium iodide. DNA content was measured by flow cytometry. Bars indicate percentage of cells in the indicated phase of the cell cycle: G1 (dark gray), S (black), G2/M (light gray); error bars represent sd (n = 3). E, Steroid-starved T47D-YB cells were pretreated for 30 min with DMSO vehicle control or AG1478 (10 μm), followed by 18 h of treatment with R5020 or EtOH vehicle control and analyzed as above. F, Steroid-starved T47D-YB cells were pretreated with vehicle control and the ER antagonist ICI 182,780 (0.10 μm) or PP2 or U0126 (10 μm each), treated with or without R5020 for 18 h, fixed with EtOH, and stained with propidium iodide. Bars indicate percentage of cells in the indicated phase of the cell cycle: G1 (dark gray), S (black), G2/M (light gray): error bars represent sd (n =3). All experiments were repeated at least three times with similar results.
We therefore tested whether EGFR kinase activity is required for rapid (10 min) activation of Erk1/2 MAPK by progestins. T47D-YB cells were pretreated with small molecule inhibitors of MAPK kinase (MEK)-1/2 (U0126), c-Src (PP2), or EGFR (AG1478) for 30 min, followed by treatment with ethanol (EtOH) vehicle control or R5020 for 10 min. Western blotting confirmed that U0126, PP2, or AG1478 blocked the rapid activation of Erk1/2 MAPK induced by progestin binding to PR-B (Fig. 1B). To confirm the specificity of the EGFR inhibitor AG1478, we conducted control experiments using the peptide growth factor EGF and the phorbol ester, phorbol 12-myristate 13-acetate (PMA), a strong activator of protein kinase C. EGF, but not PMA treatment, resulted in EGFR tyrosine phosphorylation, whereas both agents strongly activated Erk1/2 MAPK at 10 min compared with their respective vehicle controls (Fig. 1C). AG1478 completely blocked EGF-induced activation of EGFR, as measured by EGFR tyrosine phosphorylation and downstream Erk1/2 MAPK activation. The fold activation of Erk1/2 MAPK by PMA was unchanged in the presence of AG1478, given the corresponding reduction in basal Erk1/2 MAPK phosphorylation (Fig. 1C, compare lane 3 to lane 7). Although PMA has been shown to transactivate EGFR (24), we did not detect this in our assay (lane 4).
We have shown previously that rapid signaling upon PR-B ligand binding is a critical component of the progestin-induced proliferative response of cultured MCF-7 breast cancer cells (25). To elucidate the pathway involved in progestin-induced proliferation of T47D cells, we examined whether EGFR is required for the mitogenic effects of progestins in our assay of progestin-stimulated S-phase entry. Steroid-starved, ER-positive, and PR-null T47D-Y cells failed to enter S-phase upon treatment with R5020 compared with vehicle control (Fig. 1D), whereas treatment with EGF induced a 32% increase in S-phase cells. Stably expressed PR-B in T47D-YB cells restored the R5020 proliferative response to a level comparable to EGF, with 36% of cells in S-phase (Fig. 1D). Inhibition of EGFR tyrosine kinase using the tyrphostin, AG1478, resulted in complete blockade of the mitogenic effects of R5020 observed at 18 h, with a comparable percentage of cells entering S-phase as seen for control cells treated with dimethylsulfoxide (DMSO) and EtOH (Fig. 1E). In similar experiments, the antiestrogen (ICI 182,780) had no effect on progestin-induced S-phase entry, whereas both the c-Src inhibitor (PP2) and the MEK-1/2 inhibitor (U0126), included as a control for MAPK inhibition, significantly dampened the R5020 response (Fig. 1F). These data suggest that PR-B mediates the mitogenic effects of progestins via rapid, MAPK-dependent signaling events downstream of activated EGFR.
Phosphorylation of PR-B Serine 345 Is EGFR, c-Src, and Erk1/2 MAPK Dependent
One prospective role of membrane-proximal signaling pathway activation may be to provide a feed-forward mechanism for rapid, direct phosphorylation of PR coincident with ligand binding. We searched for MAPK consensus phosphorylation sites (PXXSP) in PR-B that were also sensitive to MAPK or c-Src kinase inhibition. Ser294 and Ser345 are the only reported ligand-inducible Ser-Pro motifs (26) that are MAPK consensus sites; Ser400 is a basally phosphorylated Pro-directed kinase (cdk2) site (27) that is also sensitive to both progestins and mitogens (28).
We observed modest PR-B Ser294 phosphorylation after 30 min R5020 treatment that increased at 60 min of R5020 in the presence of DMSO vehicle control (Fig. 2A). Pretreatment of cells with U0126 did not significantly reduce PR-B Ser294 phosphorylation after treatment with R5020 at 30 or 60 min, whereas PP2 slightly diminished PR-B Ser294 phosphorylation after 60 min R5020 treatment relative to DMSO vehicle control. Using custom-designed Ab recognizing phosphorylated Ser345 and Ser400 PR (28), we observed robust phosphorylation of PR-B Ser345 and Ser400 after 30 and 60 min of R5020 treatment (Fig. 2A). Phosphorylation of PR-B Ser400 at 30 and 60 min was relatively insensitive to both kinase inhibitors. In contrast, PR-B Ser345 phosphorylation was completely blocked by U0126 and PP2 at the 30-min time point, with some recovery by 60 min (Fig. 2A). In separate experiments, duplicate cultures of T47D-YB breast cancer cells treated with R5020 demonstrate the specificity of complete inhibition of PR Ser345 in the presence of PP2 relative to DMSO vehicle control and the p38 MAPK inhibitor, SB203580 (Fig. 2B). Consistent with EGFR activation upstream of Erk1/2 MAPK (Fig. 1), the EGFR inhibitor AG1478 also completely blocked PR-B Ser345 phosphorylation (Fig. 2B).
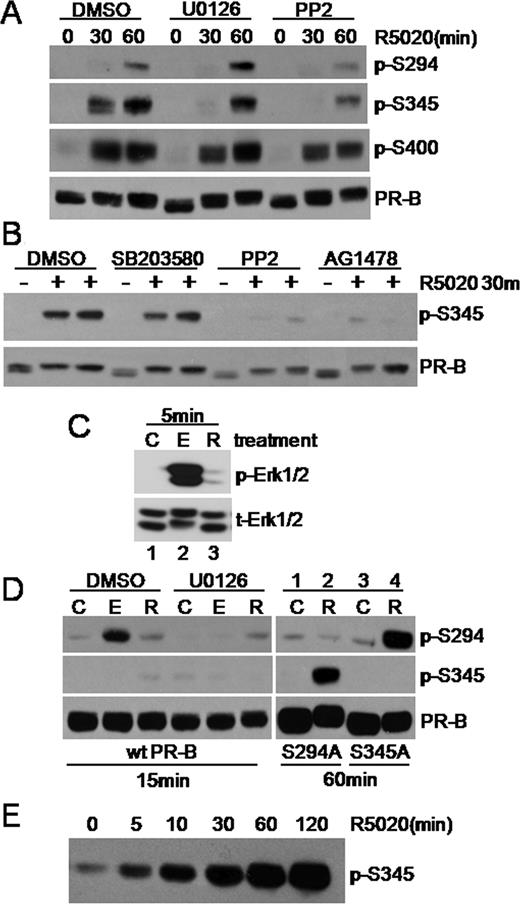
Rapid Progestin-Induced PR Signaling Induces Ser345 PR-B Phosphorylation A, T47D-YB cells were pretreated with DMSO, U0126, or PP2, followed by 0, 30, or 60 min of treatment with R5020. Cell lysates were subjected to Western blotting using phospho-Ser294, -Ser345, or -Ser400 or total PR Ab. Care was taken with exposure times, Ab concentrations, and negative control mutant S294A PR-B (50 ) to avoid cross-reactivity with purchased monoclonal phospho-Ser294 Ab. B, T47D-YB cells were pretreated with inhibitors as above followed by a 30-min treatment with R5020. Whole-cell lysates were subjected to Western blotting using phosphospecific and total PR Ab. C, Western blots detected total and active Erk1/2 MAPKs in whole-cell lysates from T47D-YB cells treated with control (C), EGF (E), or R5020 (R) for 5 min. D, T47D cells stably expressing wt PR-B (left panel) or either S294A or S345A mutant PR-B (right panel) were treated as indicated. Whole-cell lysates were subjected to Western blotting with phospho-Ser294, phospho-Ser345, or total-PR Ab. Note the weak nonspecific band in lysates from cells stably expressing the S294A mutant PR-B probed with the phospho-Ser294 Ab (lanes 1 and 2). E, COS cells overexpressing wt PR-B were treated with R5020 for 0–120 min, and whole-cell lysates were Western blotted using PR Ser345 phosphospecific Ab. Experiments in A–D were conducted at least three times with similar results.
To further examine the specific regulation of PR-B MAPK sites, Ser294 and Ser345, we compared the phosphorylation of these sites by EGF and progestin. EGF is a potent mitogen (Fig. 1D) that also induced robust activation of MAPK at early time points (Fig. 2C, lanes 1 and 2). R5020-induced MAPK activity is modest relative to induction of MAPK activity by EGF at 5 min (Fig. 2C). Interestingly, EGF treatment stimulated PR-B Ser294 but not Ser345 phosphorylation in a U0126-sensitive manner (Fig. 2D, left panels). PR-B Ser345 phosphorylation is faintly visible in whole-cell lysates after 15 min of R5020 treatment (Fig. 2D) relative to the higher level of phosphorylation seen at 30–60 min (Fig. 2A). These results mark an interesting distinction between MAPK activity stimulated by progestin relative to EGF. Several factors may explain this divergence including the formation of a PR-c-Src complex in response to progestins relative to EGF (8), subcellular localization, or differential signaling strength and duration (Fig. 2C). Notably, similar to our results with EGF (Fig. 2D), overexpression of constitutively active MEK-1 induced phosphorylation of PR Ser294 but not Ser354 (not shown), suggesting that ligand-induced PR-protein kinase complex formation is required for phosphorylation of Ser345. Consistent with this idea, a time course conducted in COS cells overexpressing PR demonstrated that PR Ser345 phosphorylation occurs rapidly, within 5 min of progestin treatment (Fig. 2E), but remains completely insensitive to EGF treatment (not shown). Both PR-B phospho-mutants, S294A and S345A (Ser344/345Ala), were phosphorylated on Ser345 and Ser294, respectively, in response to 60 min of R5020 treatment (Fig. 2D, right panel, lanes 1–4), demonstrating the specificity of the phosphospecific Ab used in this study. In sum, these results show that PR ligand binding is required for phosphorylation of Ser345 but not Ser294. Interestingly, PR Ser345 is phosphorylated in a MAPK-dependent manner after R5020 treatment but is insensitive to stimulation of cells with EGF or overexpression of activated MAPK pathway members (29–31). The divergence of PR Ser294 and Ser345 phosphorylation after MAPK activation by EGF relative to progestins suggests the formation of a progestin-dependent PR complex with rapidly activated protein kinases that coordinate the phosphorylation of Ser345.
Ser345Ala (S345A) Phospho-Mutant PR Displays Specific Defects in Nonclassical PR Target Gene Regulation
To further investigate the function of rapid, progestin- and MAPK-dependent PR-B Ser345 phosphorylation, we engineered PR-null T47D-Y cells stably expressing the Ser344/345Ala (S345A) phospho-mutant PR-B. Stable S345A PR-B expression levels were slightly greater than that of wild-type (wt) PR-B stably expressed in the same parental cells (Fig. 3A). S345A PR-B was not recognized by the phospho-Ser345 Ab in cells treated with R5020 but did undergo an up-shift in gel mobility indicative of ligand binding and subsequent receptor phosphorylation at sites other than Ser345 (Fig. 3A). These results do not support the previously reported role of Ser345 phosphorylation in retardation of PR gel migration (26). We next tested the rapid signaling and transcriptional activation function of S345A PR-B relative to wt PR-B. S345A PR-B and wt PR-B stimulated Erk1/2 MAPK phosphorylation, after a 10-min R5020 treatment (Fig. 3B). Importantly, both transiently and stably expressed S345A and wt PR-B induced comparable levels of transcription from a PRE-luciferase reporter plasmid (Fig. 3C and data not shown). Stably expressed S345A PR-B is thus functionally similar to wt PR-B in assays of progestin-induced rapid MAPK phosphorylation and PRE-driven transcription; both PR-dependent events occurred in response to equal concentrations (≥10 nm) of progestin.
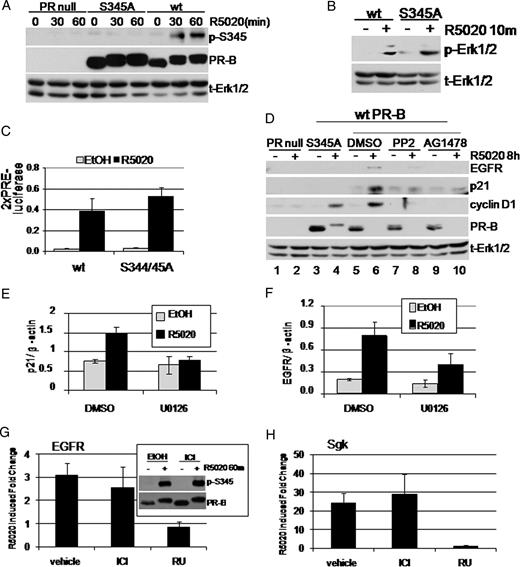
Functional Analysis of S345A Phospho-Mutant PR-B and MAPK Dependence of Nonclassical PR Gene Targets A, T47D PR-null cells or T47D cells stably expressing either wt or S345A PR-B were treated with R5020 for the indicated times. Phospho-Ser345 and total PR were visualized using phospho-PR and total PR Ab. Phospho-Ser345 blotting demonstrates the specificity of phosphospecific Ab. Total Erk1/2 blots were included as protein loading controls. B, T47D cells expressing wt or mutant S345A PR-B were treated for 10 min with R5020. Whole-cell lysates were subjected to phospho-Erk1/2 and total Erk1/2 Western blotting. C, PR-null HeLa cells were transiently transfected with either wt or S345A PR-B and PRE-luciferase and renilla-luciferase (control) reporter constructs. Luciferase activity was measured in whole-cell lysates after EtOH or R5020 treatment (24 h) and represented as firefly/renilla. Similar results were obtained with T47D cell lines stably expressing wt or S345A PR-B. Error bars represent sd (n = 3). D, T47D PR-null cells or T47D cells stably expressing S345A or wt PR-B were treated for 8 h with R5020. Cells expressing wt PR-B were pretreated with DMSO, PP2, or AG1478. Cell lysates were subjected to Western blotting to detect EGFR, p21, and cyclin D1. Total PR blotting indicates PR expression levels and ligand-dependent PR down-regulation. Total Erk1/2 was included as a loading control. E and F, T47D-YB cells were treated with or without R5020 (18 h) in the presence of vehicle control (DMSO) or the MEK inhibitor U0126. Real-time quantitative PCR was then used to measure p21 (E) or EGFR (F) mRNA. Values were normalized to β-actin. Error bars represent sd of triplicate cultures. G and H, T47D-YB cells were treated with or without R5020 (18 h) in the presence of vehicle control (DMSO), antiestrogen ICI 182,780 (ICI; 1.0 μm), or antiprogestin RU486 (RU; 10 μm). Real-time quantitative PCR was then used to measure EGFR (G) or Sgk (H) mRNA. Values were normalized to β-actin. Error bars represent sd of triplicate cultures. G, Inset, Western blot showing progestin-induced PR Ser345 phosphorylation in the presence or absence of ICI 182,780 (1.0 μm). All experiments were repeated at least three times with similar results.
In addition to classical PRE-driven target genes, PR-B stimulates transcription of target genes that do not contain canonical PREs in their promoter regions. Progestin-induced up-regulation of the cell cycle molecule p21 occurs via a PR-Sp1 interaction at Sp1 sites located in the proximal p21 promoter (2). The EGFR promoter additionally does not encode a canonical PRE; however, the presence of numerous Sp1 sites may provide for a similar mechanism of PR regulation of EGFR expression via Sp1 tethering (32, 33). We measured p21 and EGFR regulation by progestin as a model of nonclassically regulated PR target genes and included cyclin D1 as a known MAPK-dependent PR target molecule (25). Endogenous p21 and EGFR protein expression was increased by R5020 treatment of wt PR-B-expressing cells but not in negative control PR-null cells (Fig. 3D, compare lanes 5 and 6 and lanes 1 and 2). In contrast, neither EGFR nor p21 protein levels were increased by R5020 treatment in cells containing S345A phospho-mutant PR-B (Fig. 3D, lanes 3 and 4). This phenotype was specific to S345A PR-B, because R5020-treated cells expressing the S294A phospho-mutant PR-B up-regulated p21 and EGFR at levels similar to wt PR (data not shown). As a control for MAPK-dependent gene regulation, we also probed for cyclin D1. Both wt and S345A PR-B mediated an increase in cyclin D1. We then evaluated the role of PR-induced rapid signaling in the up-regulation of EGFR, p21, and cyclin D1 protein levels. Inhibition of c-Src and EGFR kinase activity with PP2 and AG1478 blocked the up-regulation of EGFR, p21, and cyclin D1 protein in R5020-treated cells stably expressing wt PR-B, compared with DMSO vehicle control (Fig. 3D, lanes 5–10). Total PR Western blotting indicated that ligand-dependent PR down-regulation was unaffected by inhibitors. These results suggest that a specific subset of nonclassical PR target genes, including EGFR and p21, are regulated by MAPK-dependent Ser345 PR-B phosphorylation.
If our model is correct, PR-initiated rapid c-Src→MAPK signaling should be reflected in changes in the transcription of p21 and EGFR. Indeed, we found an up-regulation in p21 and EGFR mRNA using real-time PCR (Fig. 3, E and F), which was inhibited by U0126 as compared with DMSO vehicle control. Similar results were obtained using c-Src (PP2) and EGFR (AG1478) inhibitors (data not shown). Importantly, the expression of progestin-induced endogenous EGFR and SGK (a control gene with a PRE-containing promoter) mRNA was sensitive to the antiprogestin RU486 but not the antiestrogen ICI 182,780 (Fig. 3, G and H). Additionally, ICI 182,780 did not alter progestin-induced PR Ser345 phosphorylation (Fig. 3G, inset). Together, these data link rapid MAPK-dependent phosphorylation of PR-B Ser345 to nonclassical PR target gene transcription and demonstrate that these PR-induced events occur independently of ERα.
PR-B Tethering to Sp1 Is Regulated by Rapid c-Src→MAPK Activation and Ser345 Phosphorylation
After establishing that progestin-bound PR stimulates rapid activation of EGFR/c-Src/MAPK signaling and subsequent PR-B Ser345 phosphorylation, we next sought to determine whether these events regulate transcriptional machinery through Sp1. Immunoprecipitation (IP) of PR from R5020-treated cells (60 min) stably expressing wt PR-B revealed the ligand-dependent formation of a PR-Sp1 protein complex (Fig. 4A, left panels). The reverse IP of Sp1 also demonstrated PR-B interaction, albeit in a ligand-independent fashion (Fig. 4A, middle panels). Sp3 was also weakly detected in PR IP, but this was not regulated in response to progestin (not shown), as was Sp1. We next investigated whether PR-B Ser345 phosphorylation is critical for regulating the interaction of PR-B with Sp1. T47D cells stably expressing either wt or S345A PR-B were treated with progestin. Ser345-phosphorylated PR were immunoprecipitated using phosphospecific Ab (Fig. 4B). Sp1 was strongly enriched in immunoprecipitates of Ser345-phosphorylated PR from cells expressing wt but not S345A PR-B (Fig. 4B, compare Sp1 in PR IP to 10% input). In further support of the requirement for PR-B Ser345 phosphorylation in PR tethering to Sp1, ligand-activated wt PR, which is Ser345 phosphorylated, pulled down significantly more Sp1 relative to EtOH control in IP performed using total PR Ab, whereas S345A PR had no such effect (Fig. 4C). Despite some basal interaction with Sp1, S345A PR-B displayed no ligand-dependent increase in association with Sp1 after R5020 treatment for 30 or 60 min relative to wt PR-B. These data indicate that PR-B must be phosphorylated at Ser345 for ligand-induced PR tethering to Sp1 to occur.
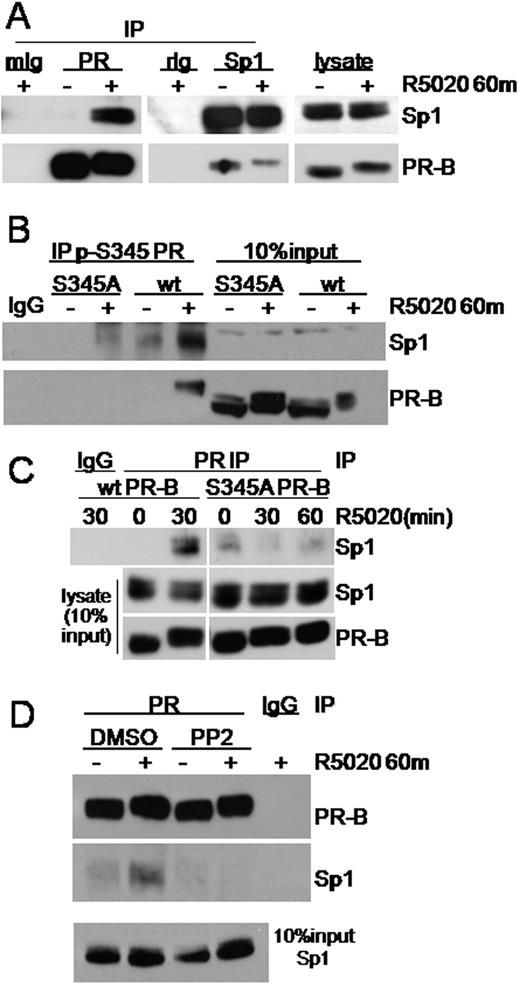
PR-B Interacts with Sp1 after c-Src/MAPK-Mediated PR Ser345 Phosphorylation A, PR or Sp1 was immunoprecipitated from whole-cell lysates of T47D-YB cells treated with EtOH or R5020 for 60 min. Immunoprecipitates were subjected to Western blotting using PR- or Sp1-specific Ab. IgG was included as a control for Ab specificity. Each lysate represents 10% of input. B, T47D cells stably expressing either wt PR-B or S345A PR-B were treated with R5020 for 60 min. Ser345 phosphorylated PR was immunoprecipitated from whole-cell lysates using phospho-Ser345-specific PR Ab. Immune complexes were subjected to Western blotting for Sp1 and total PR. Lysates represent 10% of IP input. IgG was included as a control for IP Ab specificity. C, PR was immunoprecipitated from whole-cell lysates of T47D cells stably expressing either wt or S345A PR-B as in B but using total PR Ab. Immunoprecipitations were subjected to Western blotting using specific Ab. D, T47D-YB cells were pretreated with DMSO or PP2 and then treated without or with R5020 for 60 min. PR was immunoprecipitated and subjected to Western blotting for Sp1 or PR. IgG was included to control IP Ab specificity. Lysate representing 10% IP input is shown. Experiments were repeated three to four times with similar results.
We next investigated the requirement for c-Src activation in PR tethering to Sp1. Again, PR was immunoprecipitated from T47D breast cancer cells stably expressing wt PR-B treated with or without R5020 for 60 min (Fig. 4D). PP2 (or vehicle control) was included to inhibit progestin-stimulated c-Src activation. As above, progestin treatment stimulated the coprecipitation of Sp1 with PR-B compared with vehicle control (Fig. 4D). In contrast, no Sp1 was present in ligand-activated PR-B immune complexes under conditions of c-Src inhibition with PP2. These data suggest that c-Src activation and subsequent PR Ser345 phosphorylation are critical for PR-B interaction with Sp1.
Activation of c-Src can either proceed directly through a proline-rich domain of PR (8) or through an indirect interaction of PR with unliganded ER (7, 34). To test whether the membrane-proximal direct interaction of PR-B with c-Src is required for PR-B, Ser345 phosphorylation and Sp1 tethering, we employed a mutant PR-B (mPro) that fails to directly interact with c-Src. PR-B mPro was created by disrupting the proline-rich domain of PR, thereby precluding formation of the c-Src-PR complex and subsequent activation of c-Src and downstream Erk1/2 (8). Stable expression of PR-B mPro in T47D-Y cells was obtained at a level slightly less than stably expressed wt PR-B (Fig. 5A) (17). As above (Fig. 2A), wt PR-B phosphorylation on Ser345 and Ser294 was stimulated after 30 and 60 min of R5020 treatment (Fig. 5A). Interestingly, Ser294 phosphorylation on mPro PR-B was greatly reduced relative to wt, but clearly increased along a similar time course. In contrast, Ser345 phosphorylation was completely undetectable in mPro PR-B-expressing cells at all time points examined (Fig. 5A and data not shown). These data support the notion that PR-B Ser345 phosphorylation is dependent on the direct, membrane-proximal interaction of progestin-bound PR with c-Src and confirms our results that c-Src inhibition with PP2 blocks Ser345 phosphorylation (Fig. 2).
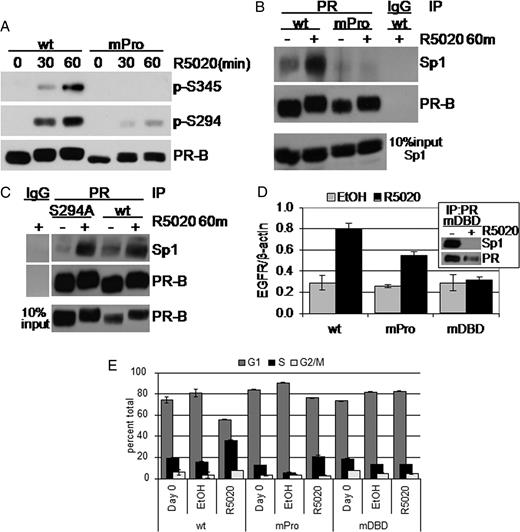
PR-B Interaction with c-Src Is Required for Sp1 Tethering A, T47D cells stably expressing either wt or mPro PR-B were treated for 30–60 min with R5020 or EtOH vehicle control, and whole-cell lysates were Western blotted using phosphospecific and total PR Ab. B, T47D cells stably expressing either wt or mPro were treated with or without R5020 for 60 min. Cell lysates were subjected to immunoprecipitation using PR-specific Ab; immune complexes were Western blotted for Sp1 or PR-B. IgG was included as a specificity control for the IP Ab; lysate representing 10% IP input is shown. C, The experiment shown in B was repeated in T47D cells stably expressing wt or S294A PR-B. D, Progestin-induced (R5020, 18 h) EGFR mRNA up-regulation was measured by real-time quantitative PCR in T47D cells stably expressing wt, mPro, or mDBD PR-B. Values were normalized to β-actin. Error bars represent sd of triplicate cultures. Inset, PR-Sp1 co-IP showing the mDBD PR basal interaction with Sp1 is disrupted upon progestin treatment. E, Cell cycle analysis of EtOH-fixed and propidium iodide-stained nuclei in T47D cells expressing wt, mPro, or mDBD PR-B and treated for 18 h with or without R5020. Similar results were obtained in three independent experiments.
Ser345-phosphorylated PR-B strongly associated with Sp1 (Fig. 4, B and C). Given the absence of Ser345 phosphorylation on mPro PR-B, we predicted that mPro PR-B would not interact with Sp1 in similar co-IP of PR from progestin-treated T47D cells. Indeed, no Sp1 was detected in mPro PR-B immune complexes, as compared with wt PR-B (Fig. 5B). S294A phospho-mutant PR-B efficiently copurified with Sp1 in a ligand-dependent manner (Fig. 5C), even though the mPro mutation reduces phosphorylation at this site (Fig. 5A), suggesting that Ser294 phosphorylation is dispensable for PR-Sp1 tethering. Thus, the loss of mPro PR-B’s ability to interact with Sp1 is most likely due to its inability to directly interact with c-Src, which in turn disrupts subsequent rapid c-Src and MAPK activation and phosphorylation of PR Ser345.
To confirm that mPro PR-B was also functionally impaired on Sp1-regulated genes, we measured the induction of EGFR mRNA in response to progestin treatment of T47D cells stably expressing either wt or mPro PR-B. A PR-B DNA-binding mutant [mutant DNA-binding domain (mDBD, C587A)] was included as a negative control for activation of transcription via PR zinc-finger-mediated DNA binding (35). EGFR mRNA was clearly induced in R5020-treated cells expressing wt PR-B, whereas by comparison, EGFR mRNA was nearly halved in mPro PR-B-expressing cells treated with R5020 (Fig. 5D). Surprisingly, R5020 treatment of cells expressing the mDBD mutant PR-B showed no increase in EGFR mRNA. We speculated that mDBD PR-B is unable to tether to Sp1 because it may undergo significant conformational changes due to a loss of the first zinc finger of PR. Indeed, we detected a high basal interaction of mDBD PR-B with Sp1 that was completely disrupted upon progestin treatment (Fig. 5D, inset). These data support a model whereby direct binding of PR-B to c-Src activates downstream kinase pathways to regulate PR-Sp1 tethering and important aspects of nonclassical gene transcription. Interestingly, mPro PR-B retained the partial ability to induce EGFR gene transcription despite its inability to efficiently tether to Sp1 (Fig. 5D), indicating that PR-B may regulate this gene in part via mechanisms independent of direct c-Src interaction (34), perhaps via distant sites (36), or that a greatly weakened PR-Sp1 interaction (Fig. 5B) is able to sustain some level of EGFR regulation over the 18-h time course (Fig. 5D).
To validate whether direct interaction between c-Src and PR-B and/or the presence of an intact DBD are required for progestin-induced S-phase entry, we tested the proliferative response of ER+ T47D cells stably expressing mutant mPro or mDBD PR-B. In previous work, stable expression of wt, but not mPro, PR-B induced a significant proliferative response to R5020 in MCF-7 breast cancer cells stably expressing low levels of PR (25). Similar results have been reported in ER-null breast cancer cell lines stably expressing mutant (mPro) PR (T47D C42 and MCF-7C412) (37). In the present study, R5020 treatment of wt PR-B-expressing cells stimulated a 44% increase in S-phase compared with EtOH vehicle control (Fig. 5E). Stable expression of mPro PR-B in ER+ T47D cells resulted in an attenuated, but measurable increase (29%) in S-phase entry compared with EtOH vehicle control (Fig. 5E); this effect was repeated in several independent mPro PR-B stable T47D clones (not shown). In contrast, stable expression of the mDBD PR-B mutant failed to stimulate a proliferative response to cells treated with R5020 (Fig. 5E). These results are consistent with the attenuation of EGFR up-regulation by these mutants (Fig. 5D). PR-null cells failed to respond to progestins (Fig. 1D), and the PR antagonist RU486 completely blocked progestin-induced S-phase entry in PR-B-expressing T47D cells (not shown). These data suggest that although PR rapid signaling to c-Src is clearly important for the proliferative response of breast cancer cells to progestins, PR nuclear action is still required (Fig. 5E).
PR Tethering to Sp1 Regulates Growth-Promoting PR Target Genes and Progestin-Stimulated Proliferation
PR regulation of p21 expression was localized to Sp1 sites in the proximal promoter region (2). To confirm that phosphorylated PR is recruited to Sp1-binding sites in the endogenous p21 promoter, we performed chromatin IP (ChIP) assays (Fig. 6). Coadministration of RU486 with R5020 was used to inhibit PR transcriptional activity in T47D cells stably expressing wt PR-B. PR or Sp1 molecules were immunoprecipitated from cross-linked chromatin preparations. After reverse cross-linking and DNA isolation, PCR was performed using p21 promoter-specific primers that flank the proximal GC-rich Sp1 sites (38); primers that flank a distal element located outside the promoter were used as specificity controls (Fig. 6A). ChIP assays demonstrated ligand-dependent recruitment of PR-B and Sp1 to the GC-rich p21 proximal promoter. These events were blocked by the PR antagonist RU486, but not by ICI 182,780 (not shown). In contrast to wt PR-B in the presence of progestin, mutant S345A PR-B and mPro PR-B interacted very weakly with the p21 promoter region encompassing the Sp1 binding site and remained unresponsive to ligand in multiple experimental repeats. Notably, Sp1 was also not associated with the p21 promoter in cells containing PR mutants (Fig. 6A). These data demonstrate the specific recruitment of wt PR and Sp1 to the Sp1-responsive element in the p21 promoter and further implicate the importance of PR Ser345 phosphorylation in Sp1 tethering. Similar results as those seen in T47D cells (Fig. 6A) were observed in HeLa cells transiently expressing PR (Fig. 6B), wherein p21 promoter (Sp1 site specific) recruitment of wt PR-B occurred in response to progestin. Basal interaction of S345A PR-B was high in transient assays but remained unresponsive to progestin.
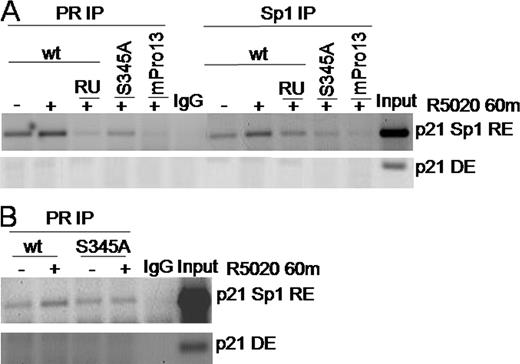
Wild-Type But Not Phospho-Mutant S345A PR-B Is Recruited to the p21 Promoter A, T47D cells stably expressing wt, S345A, or mPro PR-B were treated for 60 min with vehicle, R5020, or R5020 and RU486 before ChIP with Ab for PR, Sp1, or rabbit IgG for a specificity control. PCR were carried out using primers spanning the p21 promoter proximal GC-rich Sp1 response element (RE) or a control distal element (DE) and visualized by ethidium bromide staining of agarose gels. B, HeLa cells transiently transfected with either wt or S345A PR-B were treated for 60 min with vehicle or R5020 and subjected to ChIP with PR Ab or rabbit IgG. PCR using primers specific for the p21 promoter Sp1 (RE) or (DE) were visualized as in A. Results were repeated at least three times with similar results.
We next investigated whether other properties regulate PR tethering to Sp1, in addition to PR-B Ser345 phosphorylation. Although Sp1 is also a substrate of Erk1/2 MAPK (39), we were unable to detect changes in Sp1 phosphorylation in response to progestin treatment (data not shown). Sp1, however, was clearly recruited to the p21 proximal promoter in response to progestins (Fig. 6). Therefore, to test whether the interaction of PR with Sp1 is dependent upon aspects of Sp1 binding to DNA, we blocked Sp1 DNA binding using mithramycin A, an aurelic acid that prevents transcription factor interactions with GC box consensus sequences in chromatin (40). PR was again immunoprecipitated from R5020-treated T47D cells that stably express wt PR-B. As above (Fig. 4), R5020 stimulated an increased association of Sp1 with PR immune complexes (Fig. 7A). Interestingly, inhibition of Sp1 binding to DNA using mithramycin A completely blocked the ligand-dependent interaction of PR with Sp1 relative to methanol (MeOH) vehicle control (Fig. 7A). These data suggest that progestin-stimulated PR tethering to Sp1 requires Sp1 interaction with DNA. Consistent with this finding, EGFR expression was blocked in T47D cells expressing wt PR-B treated with R5020 in the presence of mithramycin A relative to MeOH vehicle control (Fig. 7B). Sp1-DNA binding is therefore a critical component of the mechanism of EGFR up-regulation by progestins, as has been reported for progestin-induced regulation of p21 (2).
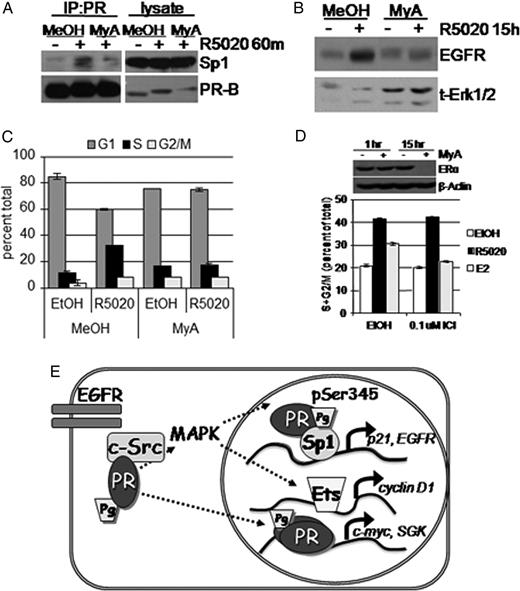
Sp1 DNA Binding Is Required for Progestin-Stimulated PR Tethering to Sp1, EGFR Up-Regulation, and S-Phase Entry A, PR was immunoprecipitated from whole-cell lysates of T47D-YB cells pretreated with MeOH vehicle control or mithramycin A (MyA, 0.4 μm) followed by EtOH or R5020 for 60 min. Immunoprecipitates were subjected to Western blotting with Sp1 or PR Ab. Lysate represents 10% IP input shown for Sp1 and PR. B, T47D-YB cells were treated without or with R5020 in the presence of vehicle control or R5020 for 15 h. EGFR was detected in whole-cell lysates using specific Ab. Total Erk1/2 was included as a loading control. C, Cell cycle analysis of T47D-YB cells treated for 18 h with R5020 in the presence of Sp1 inhibitor mithramycin A (MyA) or MeOH vehicle control. Bars indicate percentage of cells in G1 (dark gray), S (black), or or G2/M (light gray) phase of the cell cycle; error bars represent sd (n = 3). D, T47D-YB cells were treated with vehicle or mythramycin A (0.4 μm) for 1 or 15 h. Whole-cell lysates were subjected to Western blotting (upper panel) using total ERα Ab and β-actin as a loading control. Cell cycle analysis (lower panel) was conducted as in C except T47D-YB cells were pretreated for 45 min without (EtOH) or with ICI 182,780 (0.10 μm), followed by 18 h EtOH vehicle (dark gray bars), R5020 (black bars), or E2 (light gray bars). Bars indicate combined percentage of S+G2/M population of proliferating cells; error bars represent sd (n = 3). Results shown in A–D were repeated in at least three independent experiments. E, Integration of rapid PR signaling and nuclear transcription activity. Classically, ligand-activated PR dimers contact PRE in the promoter regions of target genes such as c-myc and SGK to initiate transcription. Progestin binding to PR-B also mediates nonclassical gene transcription through extranuclear rapid activation of the EGFR, c-Src, and Erk1/2 MAPK cascade to stimulate feed-forward phosphorylation of PR-B Ser345 phosphorylation. Ser345-phosphorylated PR tether to Sp1 to regulate EGFR and p21 transcription. Additionally, rapid Erk1/2 MAPK activation may regulate cyclin D1 transcription independently of PR transcriptional activity (25 ).
Finally, to confirm the importance of PR tethering to Sp1 in progestin-stimulated cell cycle progression, T47D-YB cells were treated with or without R5020 in the presence of mithramycin A or MeOH vehicle control. S-phase entry was measured by flow cytometry. Again, R5020 stimulated 32% of the cell population to enter S-phase compared with 11% in the EtOH control (Fig. 7C). Inhibition of Sp1 with mithramycin A completely blocked R5020-mediated S-phase entry, with only 17% of cells in S-phase relative to 16% in vehicle control. Although mithramycin A clearly blocked progestin-induced EGFR expression (Fig. 7B) and S-phase entry (Fig. 7C), it is possible that this effect occurred due to loss of ERα gene expression, which is also Sp1 dependent (41, 42). T47D cells express low levels of ERα that were reduced by mithramycin A at 15 h but not at 1 h (Fig. 7D). However, both progestin-stimulated EGFR expression (Fig. 3G) and S-phase entry (Fig. 7D) in T47D cells expressing wt PR were insensitive to ICI 182,780 (0.1–1.0 μm), indicating that loss of ERα could not account for the inhibition of progestin-induced EGFR expression (Fig. 7B) or S-phase entry (Fig. 7C) after 15–18 h of mithramycin A treatment. Taken together, our data support a model whereby progestins mediate breast cancer cell proliferation by first binding to PR. Ligand-activated PR then initiates rapid activation of membrane-proximal EGFR, c-Src, and MAPK, leading to PR-B Ser345 phosphorylation and PR tethering to Sp1 (Fig. 7E). Subsequent nonclassical PR target gene regulation by the PR-Sp1 complex facilitates the induction of S-phase entry.
DISCUSSION
Herein we present evidence in support of our hypothesis that progestin mediates breast cancer cell proliferation through a rapid PR signaling pathway. In this model, PR has a dual function in one pathway mediating the proliferative response of breast cancer cells, first as a rapid signaler to membrane-proximal kinases and then as a critical component of Sp1-regulated transcriptional activity (Fig. 7E). Expanding upon the mechanism of PR signaling, we found that EGFR was rapidly activated after a 10-min treatment with progestin. Rapid EGFR, c-Src, and Erk1/2 MAPK signaling was required for ligand-inducible PR-B Ser345 phosphorylation. Ser345-phosphorylated PR-B strongly associated with the transcription factor Sp1. This c-Src- and MAPK-dependent PR-Sp1 complex regulated PR cell cycle (p21) and growth-promoting (EGFR) target genes whose promoters lack canonical PRE sequences. Stimulation of S-phase entry by progestin was blocked by inhibitors of EGFR, c-Src, Erk1/2 MAPK, and Sp1. Taken together, our results demonstrate the critical link between rapid PR signaling and nonclassical modes of PR transcriptional activation in progestin-stimulated breast cancer cell proliferation.
Originally referred to as nongenomic signaling, it is now recognized that significant nuclear consequences are regulated by membrane-proximal, extranuclear PR signaling. Transcriptional up-regulation of several PR target genes have been reported to be MAPK dependent, including cyclin D1 (25, 37), DUSP-1, EGF, EGFR, and the MMTV promoter (12). Rapid signaling has been proposed to regulate PR-B nuclear accumulation (37) and/or targeting to PRE sequences and subsequent histone modifications (12). Notably, most PR target genes, including those listed above (except MMTV), do not encode canonical PREs in their promoters. We investigated progestin-bound PR rapid signaling pathway regulation of p21 and EGFR, which are two examples of nonclassical PR target genes with known cell cycle and growth-promoting functions. Low levels of p21 expression are hypothesized to be required in the initial functional assembly of G1 cyclin/cdk complexes (reviewed in Ref. 43). EGFR and ErbB family members are overexpressed in 25–30% of breast tumors (44), and EGFR kinase is required for progestin-stimulated anchorage-independent growth (17). Both p21 and EGFR promoters are regulated by the transcription factor Sp1 (2, 45). ChIP experiments have previously demonstrated PR interaction with Sp1 at the p21 promoter (38). Our data confirm the recruitment of PR and Sp1 to the p21 promoter and further implicate the importance of ligand-dependent PR Ser345 phosphorylation in this process (Fig. 6). In addition, progestin-bound PR up-regulation of EGFR and p21 mRNA and protein was also sensitive to inhibition of c-Src, Erk1/2, and Sp1 DNA binding. Ligand-activated S345A and mPro PR-Bs did not interact with Sp1 and did not support increased p21 and EGFR expression compared with wt PR-B (Figs. 3–5). We believe that our data support a model in which progestin binding to cytosolic and membrane-proximal PR initiates the kinase-dependent interaction of phospho-Ser345 PR with Sp1 (Fig. 7E).
Our data using small-molecule kinase inhibitors are well supported by our results with PR mutant receptors that are either not phosphorylated on Ser345 (S345A) or incapable of binding c-Src or activating MAPK (mPro). Of note is that PR complex formation with c-Src requires PR ligand binding (8). Our data suggest that phosphorylation of PR Ser345 also requires ligand binding (Fig. 2), complex formation with c-Src (Fig. 5A), and c-Src activity (Fig. 5B). EGF treatment activates both c-Src and MAPK but does not lead to PR Ser345 phosphorylation (Fig. 2D). Because the PR/c-Src complex is ligand dependent, overexpression of active kinases upstream of MAPK is predicted to have no effect, similar to EGF treatment (Fig. 2D). Indeed, overexpression of constitutively active protein kinases (MEK-1) fails to alter PR Ser345 phosphorylation.
We recently showed that knockdown of the PR-target gene, wnt-1, prevented progestin/PR-dependent sustained activation of EGFR, sustained MAPK activity, cyclin D1 expression, and soft-agar growth of T47D cells (17). An important conclusion of that study was that progesterone-induced breast cancer cell growth is sensitive to PR rapid signaling. The work is well supported by the findings of multiple groups showing that either PR−/ER+ or PR−/ER− breast cancer cells stably expressing mPro PR-B exhibit significantly reduced progestin-induced proliferation (17, 25, 37). However, the input of rapid signaling to PR nuclear actions remained unclear in previous studies. Our results described herein provide a link between the rapid and transcriptional actions of PR, in that we have now mapped the site of rapid c-Src and MAPK activities to PR Ser345 and Sp1 binding. In addition, we found that EGFR is activated upstream of MAPK after only 10 min of R5020 treatment and required for PR rapid signaling to MAPK (Fig. 1). Taken together, our data suggest the formation of persistent and constitutive signaling complexes that may initially contain liganded PR and activated c-Src in addition to activated EGFR. After their down-regulation, liganded PR are perhaps replaced by additional components, including signaling molecules that may be up-regulated by nonclassical but nuclear PR-dependent mechanisms. Such molecules may include wnt-1 (17), EGFR ligands (46), MAPK scaffolds, or other adaptor molecules.
Increasingly, rapid membrane proximal or extranuclear events are acknowledged to be important factors in steroid hormone receptor action and in breast cancer. Work by Santen and colleagues (47) has revealed that adaptive hypersensitivity, displayed by breast cancer cell culture and xenograft models of estrogen deprivation, is not primarily associated with an increased sensitivity at the level of ER transcription, but rather the augmentation of extranuclear signaling by ER and an up-regulation of growth factor pathways, including MAPK, phosphatidylinositol 3-kinase (PI3K), and mammalian target of rapamycin (mTOR) (reviewed in Ref. 48). The finding that PR-B-rich tumors are more likely to become tamoxifen resistant relative to PR-A-rich tumors (49) suggests a link to PR-B-dependent MAPK activation described herein. Recurrence of endocrine-resistant tumors is a critical issue in the treatment of steroid receptor-positive tumors, and breast cancer patients may benefit from combination treatment options that include inhibitors of estrogen signaling, c-Src, EGFR, ERK1/2 MAPKs, and/or PR, aimed at blocking hypersensitive mechanisms of cell proliferation.
MATERIALS AND METHODS
Cell Lines
The wt and mutant PR-B receptors (S294A, S345A, mPro, and mDBD) were reintroduced into ERα-positive, PR-null T47D-Y cells as described previously (17, 22, 50). T47D-Y and HeLa cervical carcinoma cell lines were grown at 37 C in a 5% CO2 humidified environment in T75 flasks with MEM (GIBCO, Rockville, MD) supplemented with 5% fetal bovine serum (FBS), 1% penicillin-streptomycin, 1% nonessential amino acids (Invitrogen, Carlsbad, CA), and 6 ng/ml insulin. T47D-YB cells lines stably expressing PR-B mPro, S294A, S345A, and mDBD were additionally maintained in 200 μg/ml G418 (Calbiochem, La Jolla, CA). For experiments involving the synthetic progestin R5020, growth medium was replaced 24 h after plating with starvation media, specifically phenol-red-free Iscove’s MEM (Invitrogen), supplemented with 5% dextran-coated charcoal-stripped serum (HyClone, Logan, UT) for 48 h. EGF treatments were conducted in phenol-red-free Iscove’s MEM in the absence of serum.
Reagents
U0126 (10–20 μm; Calbiochem), SB203580 (10 μm; Calbiochem), AG1478 (1–10 μm; Calbiochem), PP2 (10 μm; Calbiochem), mithramycin A (0.4 μm; Sigma-Aldrich, St. Louis, MO) and ICI 182,780 (0.1–10 μm; Tocris Cookson Inc., Ellisville, MO) were added to cell culture media 30–45 min before addition of R5020 (10 nm; NEN, Boston, MA), EGF (30 ng/ml; Sigma-Aldrich), or PMA (20 nm; Calbiochem). A polyclonal Ab that recognized the phosphorylated form of human PR Ser345 was commissioned from Invitrogen and was produced using the peptide sequence KAPPRS(pS)PCASS (phosphorylated at Ser345) as an antigen. A polyclonal Ab that recognized phosphorylated PR Ser400 was described previously (28). Total (Ab-8 for immunoblotting) and phospho-Ser294 PR Abs, total ERα (Ab-15), and monoclonal EGFR (Ab-13 for IP) were obtained from NeoMarkers (Fremont, CA). Phospho-Erk1/2 and total Erk1/2 MAPK Abs were purchased from Cell Signaling (Beverly, MA). Cyclin D1, phospho-Y1173 EGFR, and 4G10 phosphotyrosine-specific Abs were purchased from Upstate (Lake Placid, NY). Total EGFR and actin Abs were purchased from Sigma-Aldrich. Polyclonal PR Ab (H190 for IP), Sp1, and goat antisheep IgG horseradish peroxidase-conjugated secondary Ab were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Goat antimouse and goat antirabbit IgG horseradish peroxidase-conjugated secondary Ab were obtained from Bio-Rad Laboratories (Hercules, CA).
Immunoblotting and IP
Whole-cell lysates were collected on ice by scraping in RIPA buffer [10 nm sodium phosphate (pH 7.0), 150 mm NaCl, 2 mm NaCl, 2 mm EDTA, 1% (wt/vol) Nonidet P-40 (NP-40), 0.1% (wt/vol) SDS, 1% sodium deoxycholate, 0.1% (vol/vol) β-mercaptoethanol, supplemented with 20 μg/ml aprotinin, 0.1 m sodium fluoride, 1 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride], followed by sonication for 10 sec (1/2Dx5“L, setting 9, XL2000-Microson; Misonix, Farmingdale, IL). Lysates were clarified by centrifugation for 10 min at 14,000 rpm at 4 C. Soluble proteins were quantified by the Bradford method using Bio-Rad reagent, and equal amounts of protein were resolved by SDS-PAGE. The proteins were electrotransferred to polyvinylidene difluoride membrane (Millipore, Billerica, MA), immunoblotted with specific Ab, and developed using SuperSignal West Pico Chemiluminescent Substrate according to the manufacturer’s protocol (Pierce, Rockford, IL). For IP experiments, cell lysates were collected in NP-40 lysis buffer [0.5% (vol/vol) NP-40, 50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, and supplemented as above] and incubated 45 min on ice with periodic vortexing. Cell lysates containing equivalent protein concentrations were incubated for 2 h at 4 C with 1 μg appropriate Ab or control IgG, followed by overnight incubation with protein G agarose (Roche Diagnostics, Indianapolis, IN). Immune complexes were washed three times with NETN buffer [0.1% (vol/vol) NP-40, 150 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, and supplemented with protease and phosphatase inhibitors as above], resuspended in Laemmli sample buffer containing dithiothreitol, boiled for 5 min, and subjected to Western blotting analysis.
Flow Cytometry
For propidium iodide analysis of cell cycle distribution, cells were plated in six-well plates at a concentration of 220,000 cells per well. After indicated treatment, cells were washed once with 0.25% trypsin, detached from the plate using 0.25% trypsin, and collected with 1% FBS (1× PBS) in a 15-ml conical tube along with previously saved culture media and trypsin wash. Cells were spun at 1600 rpm for 4 min at 4 C, washed once with ice-cold 1% FBS (PBS), and spun again. A single-cell suspension was made in 300 μl 1% FBS (PBS), fixed with 4.2 ml 80% ice-cold EtOH, and stored at −20 C. Fixed cells were washed once with 1% FBS (PBS) and then resuspended in 400 μl fluorescence-activated cell sorting staining buffer [1× PBS, 1% FBS, 0.1% Triton X-100, 0.5 μm EDTA, 50 μg/ml propidium iodide (Sigma-Aldrich)]. DNA content of the nuclei, as reflected by the fluorescence signal of the propidium iodide, was measured by flow cytometry, and cell cycle phase distributions were determined using Modfit software program (Becton Dickinson, Franklin Lanes, NJ).
Real-Time Quantitative PCR
Two micrograms of RNA were reverse transcribed with random hexamers and SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer’s protocol. Each PCR contained 5 μl cDNA, and 0.5–1 μmol/liter of each primer. Real-time quantitative PCR analysis was performed with a LightCycler (Roche Diagnostics) or a Realplex Mastercycler EP (Eppendorf, Hamburg, Germany) according to the manufacturer’s instructions as described previously (17). Melting-curve and agarose gel electrophoresis analysis confirmed product-specific amplification; the results were normalized to β-actin.
Transcription Assay
PR transcriptional activity in cells was measured using the Dual-Luciferase Reporter Assay (Promega, Madison WI) according to manufacturer’s protocol. Twenty-four hours after plating, T47D-YB or S345A PR-B cells were transiently transfected using FuGene HD (Roche Diagnostics) according to manufacturer’s instructions with 500 ng of a PRE-2X-TATA luciferase reporter constructs and 30 ng of a control Renilla luciferase construct (pRL-TK) for normalizing transfection efficiency. For transient PR expression, Hela cells were additionally transfected with 20 ng wt or S345A hPR-1 (in pSG5). After overnight incubation with DNA complexes, the medium was replaced with starvation medium and incubated for an additional 24 h. Cells were treated overnight with or without R5020, and 20–30 μl lysate were analyzed for firefly/Renilla luciferase.
ChIP
Two million cells were plated in 100-mm dishes, starved for 48 h, and treated with R5020 (10 nm), R5020 and ICI 182,780 (10 μm), or R5020 and RU486 (10 nm) for 1 h. Cells were then fixed using 1% formaldehyde for 5 min at 37 C followed by treatment with glycine for 2 min at room temperature. Cells were scraped in cold PBS (supplemented with Complete protease cocktail inhibitor tablets from Roche Diagnostics, 20 μg/ml aprotinin, 0.5 m β-gylcerol phosphate, 0.5 m sodium fluoride, 1 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin), washed twice, lysed with supplemented SDS lysis buffer, and diluted with supplemented ChIP dilution buffer from Upstate (Temecula, CA). Lysates were sonicated in cycles of 30 sec on/30 sec off for 15 min at 4 C using a Bioruptor from Diagenode (Liege, Belgium), and input controls were then collected. PR Ab H190, Sp1 Ab PEP-2, and control normal rabbit IgG (Santa Cruz Biotechnology) were used for IP along with protein G agarose/salmon sperm DNA beads (Upstate). Complexes were washed, eluted form the agarose beads, reverse cross-linked overnight at 65 C, and treated with proteinase K. DNA was recovered using phenol/chloroform extraction and ethanol precipitation. PCR were carried out using primers specific to the p21 promoter, to amplify either the proximal region containing the Sp1 response element or a distal element (38). Amplified products were run on a 2% agarose gel.
Acknowledgments
We gratefully acknowledge Dr. Kathryn Horwitz (University of Colorado Health Sciences) for the kind gift of the PR-null T47D cell line and T47D variant stably expressing wt PR-B (22 ) and Mike Franklin (University of Minnesota Division of Hematology, Oncology, and Transplantation) for helpful editorial comments.
This work was supported by National Institutes of Health Grants R01 CA123763 and R21 CA116790 (to C.A.L.), Susan G. Komen Breast Cancer Postdoctoral Fellowship PDF0201956 (to E.J.F.), and Department of Defense Predoctoral Fellowship DOD BC043761 (to A.R.D.).
Disclosure Statement: The authors have nothing to disclose.
Abbreviations
- Ab,
Antibody;
- ChIP,
chromatin immunoprecipitation;
- DMSO,
dimethylsulfoxide;
- EGFR,
epidermal growth factor receptor;
- ER,
estrogen receptor;
- EtOH,
ethanol;
- FBS,
fetal bovine serum;
- IP,
immunoprecipitation;
- mDBD,
mutant DNA-binding domain;
- MeOH,
methanol;
- MEK,
MAPK kinase;
- mPro,
mutant PR-B receptors defective for c-Src binding;
- NP-40,
Nonidet P-40;
- PMA,
phorbol 12-myristate 13-acetate;
- PR,
progesterone receptor;
- PRE,
progesterone response element;
- Sp1,
specificity protein 1;
- SRC,
steroid receptor coactivator;
- wt,
wild type.
Pierson-Mullany LK, Lange CA 2004 Phosphorylation of progesterone receptor serine 400 mediates ligand-independent transcriptional activity in response to activation of cyclin-dependent protein kinase 2. Mol Cell Biol 24 10542–10557
Hynes N 2002 Mechanisms of resistance to endocrine therapy. In: Miller WR, Ingle JN, eds. Endocrine therapy in breast cancer. New York: Marcel Dekker
Santen RJ, Song RX, Zhang Z, Kumar R, Jeng MH, Masamura A, Lawrence Jr J, Berstein L, Yue W 2005 Long-term estradiol deprivation in breast cancer cells up-regulates growth factor signaling and enhances estrogen sensitivity. Endocr Relat Cancer 12(Suppl 1):S61–S73

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}