
Abstract
Helicobacter pylori infection remains challenging as it mainly colonized beneath the deep gastric mucosa and adheres to epithelial cells of the stomach. Concanavalin-A (Con-A)-conjugated gastro-retentive poly (lactic-co-glycolic acid) (PLGA) nanoparticles of acetohydroxamic acid (AHA) and clarithromycin (CLR) were prepared and evaluated under in vitro conditions. Solvent evaporation method was employed for preparation of nanoparticles and characterized for particle size distribution, surface morphology, percent drug entrapment, and in vitro drug release in simulated gastric fluid. Optimized nanoparticles were conjugated with Con-A and further characterized for Con-A conjugation efficiency and mucoadhesion and tested for in vitro anti-H. pylori activity. The conjugation with Con-A further sustained the drug release over a period of 8 h when compared to non-conjugated nanoparticles of AHA and CLR. In vitro anti H. pylori study confirmed that Con-A-conjugated nanoparticles containing both drugs, i.e., CLR and AHA, had shown maximum zone of inhibition compared to other formulations. In a nut shell, results suggest that the developed systems could be used for better therapeutic activity against H. pylori infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Helicobacter pylori colonizes on the surface of the epithelium of the stomach within the mucus layer and causes the inflammation or active chronic gastritis, gastric ulcer, and gastric carcinoma (1,2). It generally attaches to the inner lining of the stomach and establishes a favorable environment to grow up (3). In the course of H. pylori infection, urease hydrolyzes the urea (present in gastric epithelium) into the ammonia and carbamate, which increases the pH of the stomach (4). Neutralization of acidic environment of the stomach serves as defense mechanism for the survival of H. pylori (5).
Acetohydroxamic acid (AHA) is similar in structure to urea which acts as an antagonist of bacterial enzyme (urease) and used as urease inhibitor (6). Clarithromycin (CLR) is a broad-spectrum macrolide antibiotic and inhibits the bacterial protein synthesis. It is more effective against gram-negative bacteria, e.g., H. pylori. It is stable in acidic gastric environment, is readily absorbed, and provides an oral bioavailability of ∼50% with a prolonged t 1/2 of 3–4 h (7). The compromised effectiveness of AHA alone for such infections is reported in some literatures but its combination with other antibiotics such as CLR results into better performance against H. Pylori (8). AHA maintains the acidic environment of the stomach and creates the unfavorable condition for growth of H. pylori and enhances the anti-microbial activity of CLR. Thus, for the effective treatment of H. pylori, AHA and CLR were chosen as lead drugs.
Concanavalin-A (Con-A), a lectin isolated from jack-bean (Canavalia ensiformis), is useful as mucoadhesive carrier. Its specificity to bind mono-, oligo-, and poly-saccharides with terminal non-reducing R-D-mannopyranosyl, R-D-glucopyranosyl, or D-fructofuranosyl residues makes it suitable to fulfill the bioadhesive objectives for drug delivery purpose (9,10). Con-A is also stable in acidic gastric environment which perfectly complements the physico-chemical stability of both drugs, i.e., AHA and CLR.
Receptor-mediated targeting of lipobeads bearing AHA was prepared for the treatment of H. pylori infection (11). Tripathi et al. (12) reported the pH-sensitive gastro-retentive polymeric buoyant beads of AHA for clearance of H. pylori. Rajinikanth and Mishra (13) reported stomach-specific in situ gelling system for the treatment of H. pylori containing CLR as a drug. Oral carbopol-loaded amoxicillin nanospheres were developed which binds with the mucosa after delivery to the stomach and increase the efficacy of the drug, providing both an immediate and a sustained action (14). Tripathi (15) prepared pH stimuli-sensitive microspheres of CLR based on the principle of cation-induced gelification.
The different attempts mentioned above lack the effective active targeting of incorporated therapeutic agents due to several reasons, i.e., degradation in gastric environment, lower retention time of dosage form which results in insufficient drug availability at the site, and failure to cope with normal physiological gastric emptying process. In addition to the present attempt for the development of an effective delivery system against H. pylori, some previous research endeavors were also focused on the development of lectin-conjugated gastro-retentive microspheres containing different antibiotic agents (16,17). To achieve the objectives of interest, lectin-conjugated nanoparticles containing CLR and AHA were developed for effective eradication of H. pylori infection.
MATERIALS AND METHODS
Clarithromycin (CLR) and concanavalin-A (Con-A) were procured as gift samples from M/s Matrix Laboratories Ltd., India, and Bio-Research Products, Inc., 323 W. Cherry St., North Library (IA 5231), respectively. Acetohydroxamic acid (AHA), poly (lactic-co-glycolic acid) (PLGA) 50:50, N-hydroxysuccinimide (NHS), 1-ethyl-3,3z-(dimethylaminopropyl) carbodiimide (EDC), Pluronic F-68, nutrient broth (NB), nutrient agar (NA), Sabouraud dextrose agar (SDA), Sabouraud dextrose broth (SDB), and fluorescein isothiocyanate (FITC) were purchased from HiMedia Laboratory Pvt. Ltd., Mumbai, India. Dimethyl sulfoxide (DMSO) was procured from E. Merck Ltd., Mumbai, India. Acetone was purchased from CDH, Mumbai. The H. pylori strain 1101 was supplied by Radiant Research Services Pvt. Ltd., Bangalore, India. All other chemicals used were of analytical reagent grade.
The ex vivo study was performed at Guru Ghasidas Vishwavidyalaya, according to the protocols approved by the Committee for Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Social Justice and Empowerment, Government of India, on the recommendations of Institutional Animal Ethical Committee of Guru Ghasidas Vishwavidyalaya (Bilaspur, India).
Preparation of PLGA Nanoparticles
The PLGA nanoparticles were prepared by solvent evaporation method (18). In brief, PLGA (500 mg) was dissolved in 10 mL acetone followed by dispersion of individual drugs (CLR and AHA) and their combination with continuous stirring (Table I). This resultant dispersion was added drop wise via a needle (24 G) to 1% w/v Pluronic F-68 solution (100 mL) with continuous stirring. Polymeric dispersion was sonicated for 10 min by a probe sonicator (Frontline, Unitech Instrument, Vadodara) and stirred continuously with a magnetic stirrer at room temperature for 3 h. The hardened nanoparticles were washed twice with distilled water and processed for ultracentrifugation (Remi, Mumbai, India) at 16,000–20,000 rpm for 15 min at 4°C. Nanoparticles were lyophilized (Macro Scientific Work Pvt. Ltd., New Delhi) and stored at 4°C.
The prepared nanoparticles were optimized on the basis of process variables, i.e., drug polymer ratio and stirring time (Table I). Formulation properties, i.e., particles size and drug entrapment efficiency, were used to decide the optimized formulation.
Conjugation of Con-A with Nanoparticles
Conjugation process generally depends on the availability of functional groups over the carrier surface. Covalent attachment is the preferred phenomenon, involved for the conjugation of lectins to polymeric carriers (19). The coupling of lectin with polymeric carriers was carried out using carbodiimide technique (20). Optimized nanoparticle formulations CF7, AF7, and CF7-AF7 were selected for the conjugation with Con-A. Activation of carboxyl group of 500 mg nanoparticles was carried out by addition of 10 mL of 0.1 M 1-ethyl-3, 3-(dimethylaminopropyl) carbodiimide (EDC) and 10 mL of 0.1 M N-hydroxysuccinimide (NHS) in phosphate buffer (pH 5.8). After 3 h of incubation at room temperature, excess coupling agent was removed by washing with phosphate buffer pH 5.8. After activation, 10 mL of Con-A solution (1% w/v in phosphate buffer pH 5.8) was added and incubated overnight. Conjugated nanoparticles were collected by centrifugation at 20,000 rpm, washed three to four times with distilled water, lyophilized, and stored at 4°C until further use.
Conjugation Efficiency
The amount of Con-A bound to PLGA nanoparticles containing AHA and CLR was calculated as the difference between Con-A added initially and Con-A recovered after incubation. The amount of Con-A in the supernatant was estimated using Folin-Ciocalteu reagent. Absorbance was measured against blank using UV-visible spectrophotometer (Shimadzu 1800, Japan) at 750 nm.
Drug Entrapment
Accurately weighed quantity of nanoparticles (50 mg) from each batch were taken separately and dissolved in 5 mL of dimethyl sulfoxide (DMSO). Ten milliliters of 0.05 M NaOH solution containing 0.5% SDS was added and stirred on a magnetic stirrer for 1 h to extract out the drug completely. Extracted solution was analyzed by a UV spectrophotometer at 545 and 502 nm for CLR and AHA, respectively. Positive control solution of CLR and AHA was taken as blank while estimating CLR and AHA simultaneously in order to avoid the interference of absorbance of CLR and AHA to each other, respectively.
Particles Size and Surface Morphology
For the determination of particle size of developed formulations, Zetasizer instrument (Nano ZS, Malvern Instruments, UK) was used. A suspension of developed nanoparticles in de-ionized water was used for the analysis. The morphology of optimized formulations was studied by transmission electron microscope (TEM) (Morgagni 268D, Fei Electron Optics, USA). Samples (placebo and drug loaded nanoparticles) were fixed with 2% phospho-tungustanic acid and examined under TEM.
Infrared Spectroscopy
IR spectroscopy of drugs, polymer, and formulation was performed on FT-IR spectrophotometer (Shimadzu, Japan). The sample was mixed in the ratio of 95:5 with KBr for compression as disk and scanned over 4000–400 cm−1.
In Vitro Drug Release and Data Analysis
The in vitro release study of CLR and AHA containing nanoparticles were carried out in 20 mL of simulated gastric fluid (SGF, pH 1.2), using Franz diffusion cell at rotation speed of 100 rpm. Different formulations, equivalent to 50 mg drug, were taken in donor chamber. Aliquots (1 mL) were removed at predetermined intervals and replenished immediately with the same volume of fresh media followed by passing through Whatman filter paper (no. 41) and analyzed spectrophotometrically at 547 and 502 nm for CLR (21) and AHA (22), respectively.
Five kinetic models, including zero-order, first-order, Higuchi matrix, Peppas-Korsmeyer, and Hixson-Crowell release equations were applied to interpret the in vitro release data.
In Vitro Anti H. pylori Study and Determination of Zone of Inhibition
For these studies, the H. pylori strain 1101 was sub-cultured from the refrigerated mother cultures using modified standard procedures in nutrient broth with 50% fetal bovine serum (FBS) (23). The sub-cultured organism was standardized using nutrient broth with a spectrophotometer by adjusting 0.3 optical density at 650 nm. Samples of each formulation (10 mg), i.e., LCF7, LAF7, and L(CF7-AF7), were weighed and suspended in 1 mL of dimethyl sulfoxide (DMSO). From this stock, different dose levels were prepared in broth/DMSO (Table II).
Standardized culture of test organism (0.1 mL) was spread uniformly over the previously prepared nutrient agar (NA) plates. Wells were prepared using a sterile borer of diameter 10 mm and capacity of 100 μL to get the final concentration of test substance, i.e., 500 and 1000 μg/mL. Ciprofloxacin was added at equivalent concentration as a standard. The plates were kept at 40°C for 1 h to allow diffusion of test solution into the medium and maintained in anaerobic environment (desiccator) at room temperature for 24 h for sufficient growth of at least 10 to 15 generations. The zone of inhibition of microbial growth around the well was measured.
Determination of Minimum Inhibitory Concentration
The experiment was performed as per the previously reported procedure with minor modifications (12). CLR, AHA, and their formulations were suspended in DMSO separately. One hundred microliters volume of each suspension was added into wells of a 96-well plate. Antibiotics and their formulations were added at the fourfold concentration of the reported minimum inhibitory concentration (MIC) of each antibiotic for H. Pylori. Serial dilutions were made using a multichannel pipette. A set of positive, negative, and a standard (ciprofloxacin) control were also added to the wells. Fifty microliters of nutrient broth medium was added to each of the wells. Resazurin indicator solution (10 μL) and fetal bovine serum (FBS) (30 μL) were added to each well. Finally, 10 μL of standardized culture of H. pylori was added to each well and the plate was wrapped loosely with cling film to ensure that cultures did not become dehydrated. Wells were observed for color change from purple to pink or colorless after 24 h of anaerobic incubation at room temperature, and MIC was calculated. Each experiment was performed in triplicate.
Ex Vivo Mucoadhesive Study
Con-A-conjugated nanoparticles (10 mg) were incubated with 1 mL of 1% w/v solution of fluorescein isothiocyanate (FITC) in ethanol for 24 h prior to ex vivo study. Overnight fasted Wistar rats (220 ± 20 g) with ad libitum access to water were used for ex vivo study and handled as per the guidelines of CPCSEA. Animals were sacrificed, and the freshly collected stomach was rinsed with warm Tyrode solution (37°C) to remove any lumenal contents. Four equal segments were cut from the stomach of each animal. Three segments were dipped into a suspension of 10 mg of each fluorescent formulation, i.e., LCF7, LAF7, and L(CF7-AF7), respectively, in 5 mL SGF (pH 1.2), and the control segment was dipped in fluorescent marker only. All segments were immediately incubated in dark at 37°C in Tyrode solution gassed with oxygen. After 2 h, the stomach segments were washed thrice in ice-cold SGF to remove residual nanoparticles and observed under fluorescent microscope (Carl Ziess, Germany) (24).
RESULTS AND DISCUSSION
Nanoparticles were prepared by solvent evaporation technique and optimized on the basis of drug-polymer ratio and stirring time. Particle size and entrapment efficiency were considered as dependent variables for the selection of optimized formulation. The different stirring times, i.e., 2, 3, and 4 h, and different drugs, polymer ratios, i.e., 1:1, 1:2, and 1:3, were chosen for optimization experiments. The formulation with a combination of minimum particle size and maximum entrapment efficiency was scrutinized. A chemical process was adopted for the conjugation of PLGA with Con-A (Fig. 1). EDC is not particularly efficient itself in cross-linking because it fails to react rapidly with an amine. Its slow reaction with amine causes hydrolysis which results in regeneration of carboxyl moiety. EDC reacts with a carboxyl group of PLGA, forming an amine-reactive O-acylisourea intermediate. This intermediate may react with an amine of Con-A, producing a conjugate molecule. A stable amide bond forms during conjugation between two molecules. However, due to the risk of hydrolysis, this intermediate posed the problem concerned with stability. To overcome the issue of stability, NHS was added to stabilize the amine-reactive intermediate which converts it to an amine-reactive NHS ester, and hence, it results into more efficient coupling reactions between polymer and Con-A (25,26). The overall process allows easy and smooth two-step cross-linking procedure with no alteration to the carboxyl groups present on PLGA (17).

Schematic diagram of chemical reaction for lectin conjugation with PLGA nanoparticles
Particle Size and Surface Morphology
The particle size of non-conjugated formulations ranged between 400 and 600 nm whereas conjugated formulations ranged between 550 and 700 nm. Greater size of conjugated formulations resulted due to the formation of conjugated bulky network between PLGA and Con-A. In case of non-conjugated formulations, anionic COO− groups of PLGA cause repulsion between particles to restrict aggregation and give rise to particles with smaller size (27). Experimental observation clearly suggests that particle size of nanoparticles was directly proportional to PLGA concentration. Smaller particles were produced with lower polymer concentration which may be owed to higher surface area per unit volume (28). The data set for the stirring time of different batches indicates that an increase in the stirring time results into reduction in particle size. The TEM analysis showed that conjugated and non-conjugated formulations were almost spherical in shape and also confirms their size range within the nano-limits (Fig. 2). While both drugs, i.e., CLR and AHA, were incorporated simultaneously in the formulations, no significant change was observed on the particle size front (Tables III and IV).

TEM images of drug-loaded nanoparticles (AF7 and CF7), lectin-conjugated nanoparticles (LAF7 and LCF7), and combination batch (AF7-CF7)
Drug Entrapment
CLR formulations showed better drug entrapment efficiency compared to AHA formulations. The values for entrapment efficiency were ranged between 40.06 ± 1.21 to 55.02 ± 0.91 and 80.16 ± 0.23 to 90.12 ± 0.14% for AHA and CLR nanoparticles, respectively. These differences in entrapment efficiency of both drugs may be possibly due to the higher aqueous solubility of AHA in comparison to CLR (27,29). The incorporation of both drugs simultaneously (AF7-CF7 formulation) did not affect the entrapment efficiency of each drug significantly (Tables III and IV).
Characterization of Con-A-Conjugated Nanoparticles
IR spectrum of CLR showed the characteristic peaks, i.e., 1170.79, 1377.13, 1420, 1691, 1732, 2922.16, and 3410.15 cm−1, for different functional groups, i.e., –C–O–C stretching, CH2 group, amine group, carbonyl stretching, lactone ring, alkane stretching, and hydroxyl group, respectively (Fig. 3). Characteristic IR peaks of AHA were found to be, i.e., 1650–1550, 1680–1630, 1750, 2850–2950, and 3650–3550 cm−1 for amine group, amide group, saturated ketone, alkane, and –OH group, respectively (Fig. 4). The obtained IR spectrum of PLGA showed various peaks, i.e., 1313.32, 1710.71, and 2925.51 cm−1 for ether, ketone, and C–H stretching, respectively. In Con-A, IR showed their characteristic peaks, i.e., 3001.1 cm−1 for carboxylic acid and 1629.4 and 1510.3 cm−1 for amine. The comparison of IR spectra of LCF7 with CF7 and LAF7 with AF7 confirmed the attachment of Con-A with respective nanoparticles formulations. IR spectrum of Con-A-conjugated nanoparticles, represented by peaks (1707, 3498.87 cm−1) for the amide groups suggested the presence of amide group in AF7 and CF7 and absence in LAF7 and LCF7.

IR spectra of CLR, PLGA, Con-A, CF7, and LCF7

IR spectra of AHA, PLGA, Con-A, AF7, and LAF7
In Vitro Drug Release Study
The in vitro drug release study of different batches of AHA nanoparticles and CLR nanoparticles was performed in simulated gastric fluid. Drug release from the formulation with a drug polymer ratio 1:3 (AF7, AF8, and AF9) was found to be lower in comparison to formulation with drug polymer ratio 1:2 (AF4, AF5, and AF6) and 1:1 (AF1, AF2, and AF3) (Fig. 5). Reduced release from the formulations AF7, AF8, and AF9 may be due to increase in polymer concentration, resulting into the formation of dense polymer network which hinders the drug release. The cumulative percent drug release from different batches of AHA nanoparticles was in the following order: AF3 > AF2 > AF1 > AF6 > AF5 > AF4 > AF9 > AF8 > AF7.

In vitro drug release of acetohydroxamic acid (AHA) from different nanoparticles in pH 1.2 at 37°C. Values are mean ± SD (n = 3). AF 1–9 = formulation containing AHA as a drug
Similarly, in case of CLR nanoparticles, the batches with drug polymer ratio 1:3 (CF7, CF8, and CF9) showed least drug release when compared to the formulation with drug polymer ratio 1:1 (CF1, CF2, and CF3) and 1:2 (CF4, CF5, and CF6). Cumulative percent drug released of different formulation of CLR nanoparticles, followed the order CF3 > CF2 > CF1 > CF6 > CF5 > CF4 > CF9 > CF8 > CF7 for different formulations of CLR (Fig. 6).

In vitro drug release of clarithromycin (CLR) from different nanoparticles in pH 1.2 at 37°C. Values are mean ± SD (n = 3). CF 1–9 = formulation containing CLR as a drug
Formulations, AF7 and CF7, had shown best results in terms of drug entrapment with an extent of 55.02 ± 0.91 and 90.04 ± 0.23%, respectively, and mean particle size of 427.7 ± 3.1 and 401.0 ± 4.4 nm, respectively. Therefore, these two formulations, i.e., AF7 and CF7, were further processed for conjugation with Con-A and coded as LAF7 and LCF7, respectively. All the four formulations were subjected to in vitro drug release in simulated gastric fluid.
Over a period of 8 h, AF7 and CF7 showed cumulative percent drug release up to the extent of 37.69 ± 0.10 and 30.80 ± 0.43%, respectively, whereas LAF7 and LCF7 showed a cumulative percent drug release of 34.90 ± 0.17 and 29.09 ± 0.25%, respectively (Fig. 7). There was a very slight decrement in the drug release from conjugated formulations in comparison to respective non-conjugated formulations, which may be due to hindrance arisen by conjugated network. The release data of all formulations was subjected to different kinetic models, i.e., zero-order, first-order, Higuchi kinetics, Korsemeyer-Peppas kinetics, and Hixson-Crowell model in order to determine the type of diffusion responsible for the drug release from nanoparticles. AF7 follows the zero-order kinetics with R 2 value of 0.987 and CF7 best fitted to Hixson-Crowell model with R 2 value of 0.986. In CF7, the drug released is rate-limited; it may due to particle erosion not by the diffusion (30) whereas AF7 follow zero-order kinetics which indicates more efficient drug release control in comparison to CF7 nanoparticles (Tables V and VI).

Comparative cumulative percent drug release profile of AF7, LAF7, CF7, and LCF7. CF7 = formulation containing CLR as a drug. AF7 = formulation containing AHA as a drug. LCF7 = lectin-conjugated formulation containing CLR as a drug. LAF7 = lectin-conjugated formulation containing AHA as a drug. Values are mean ± SD (n = 3)
The effect of particle size on release of drug was clearly evidenced from the release pattern. Smaller particles release drug in greater extent due to the higher surface exposure to the biological environment. This increased exposure allows higher water inclusion within the polymeric matrix network which facilitated the drug diffusion process from nanoparticles (27).
In Vitro Growth Inhibition Study
All the formulations exhibited inhibitory properties against H. pylori. L(CF7-AF7) exhibited potent inhibitory properties with zone of inhibition (ZOI) values 28.3 and 26.4 mm at 1000 and 500 μg/mL, respectively. LCF7 exhibited potent inhibitory properties alone with ZOI values 24.0 and 21.6 mm at 1000 and 500 μg/mL, respectively. LAF7 have least inhibitory properties with ZOI values 12.4 and 16.3 mm at 500 and 1000 μg/mL, respectively (Table VI). In MIC method, CLR exhibited potent activity with MIC value of 3.9 μg/mL. AHA exhibited the low potency (MIC value 75 μg/mL) to inhibit the H. pylori, but along with the other antibiotics increases the inhibitory action of antibiotics. Incorporation of these drugs into formulation significantly increases their potency. Formulation containing both drugs, i.e., AHA and CLR showed the highest activity, i.e., MIC 1.1 μg/mL (Table VII).
Ex Vivo Mucoadhesive Study
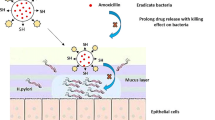
The bioadhesion of lectin-conjugated nanoparticles with gastric mucosa is due to the interaction between the sugar residue present on the epithelial tissues and Con-A (31). It was possible that Con-A would recognize the mannose and glucose residue present in gastric mucosa and bind to the competing sugar. The interaction between Con-A-conjugated nanoparticles and gastric mucosa was confirmed by fluorescence microscopy which elucidates the binding sites of Con-A-conjugated nanoparticles everywhere. These findings suggests that the bioadhesive property of Con-A-conjugated nanoparticles on rat gastric mucosa may be attributed to the true lectin-specific binding (Fig. 8).

Mucoadhesive fluorescent images of nanoparticle formulation containing clarithromycin (LCF7) acetohydroxamic acid (LAF7), and both L(CF7-AF7)
CONCLUSION AND FUTURE PERSPECTIVES
Con-A-conjugated PLGA nanoparticles of AHA and CLR were successfully prepared by solvent evaporation method. The conjugation of lectin (Con-A) to PLGA nanoparticles was performed through carbodiimide technique. Release of drug, i.e., AHA and CLR, from these nanoparticles in a controlled manner along with mucoadhesive property could be advantageous against H. pylori infection in gastric environment. The preliminary results of the present study suggests that these Con-A-conjugated PLGA nanoparticles offers a potential approach to be employed for the incorporation of other antibiotics along with urease inhibitor for the effective treatment of H. pylori infection. Such formulations could be further subjected to in vivo anti H. pylori and in vivo mucoadhesive studies to assess their capability for complete eradication of H. pylori infection.
References
Tanih NF, Ndip LM, Clarke AM, et al. An overview of pathogenesis and epidemiology of Helicobacter pylori infection. Afr J Microbiol Res. 2010;4:426–36.
Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;8390:1311–15.
Prasanthi CH, Prasanthi NL, Manikiran SS, et al. Focus on current trends in the treatment of Helicobacter pylori infection: an update. Int J Pharm Sci Rev Res. 2011;9:42–51.
Burne RA, Chen YM. Bacterial ureases in infectious diseases. Micro Infect. 2000;2:533–42.
Rektorschek M, Weeks D, Sachs G, et al. Influence of pH on metabolism and urease activity of Helicobacter pylori. Gastroenterology. 1998;115:628–41.
Phillips K, Munster DJ, Allardyce RA, Bagshaw PF. Antibacterial action of the urease inhibitor acetohydroxamic acid on Helicobacter pylori. J Clin Pathol. 1993;46:372–3.
Tripathy KD. Essential of medical pharmacology. 5th ed. New Delhi: Jaypee brother’s medical publishers (P) LTD; 2008.
Goldie J, van Veldhuyzen Zanten SJ, Jalali S, et al. Inhibition of urease activity but not growth of Helicobacter pylori by acetohydroxamic acid. J Clin Pathol. 1991;44:695–97.
Borrebaeck C, Mattiasson B. A study of structurally related binding properties of concanavalin A using differential scanning calorimetry. Eur J Biochem. 1980;107:67–71.
Jain SK, Gupta M, Sahoo AK, et al. Lectin conjugated gastro-retentive microspheres of amoxicillin for effective treatment of H. pylori. Curr Sci. 2014;106:267–76.
Umamaheshwari RB, Jain NK. Receptor-mediated targeting of lipobeads bearing acetohydroxamic acid for eradication of Helicobacter pylori. J Control Rel. 2004;99:27–40.
Tripathi GK, Singh S, Nath G, et al. Evaluation of pH sensitive gastroretentive polymeric blend buoyant beads of acetohydroxamic acid for clearance of Helicobacter pylori. Asian J Pharm Sc. 2011;6:132–40.
Rajinikanth PS, Mishra B. Preparation and in vitro characterization of gellan based floating beads of acetohydroxamic acid for eradication of H. pylori. Acta Pharm. 2007;57:413–27.
Harsha S. Dual drug delivery system for targeting H. pylori in the stomach: preparation and in vitro characterization of amoxicillin-loaded Carbopol nanospheres. Int J Nanomed. 2012;7:4787–96.
Tripathi GK. Formulation and evaluation of pH stimuli sensitive multiparticulate delivery system of clarithromycin for the treatment of Helicobacter pylori. Int J PharmTech Res. 2013;5:7–16.
Jain SK, Jangdey MS. Lectin conjugated gastro-retentive multiparticulate delivery system of clarithromycin for the effective treatment of Helicobacter pylori. Mol Pharm. 2009;6:295–304.
Jain SK, Gupta M, Sahoo AK, et al. Lectin conjugated gastro-retentive microspheres of amoxicillin for effective treatment of Helicobacter pylori. Curr Sci. 2014;106:267–76.
Zou W, Liu C, Chen Z, et al. Studies on bioadhesive PLGA nanoparticles: a promising gene delivery system for efficient gene therapy to lung cancer. Int J Pharm. 2009;370:187–95.
Ponchel G, Irache J. Specific and non-specific bioadhesive particulate systems for oral delivery to the gastrointestinal tract. Adv Drug Del Rev. 1998;34:191–219.
Olde Damink LHH, Dijkstra PJ, van Luyn MJA, et al. Cross-linking of dermal sheep collagen using a water-soluble carbodiimide. Biomaterials. 1996;17:765–73.
Kanfer I, Skinner MF, Walker RB. Analysis of macrolide antibiotics. J Chromatogr A. 1998;812:255–86.
Fukuda M, Peppas NA, McGinity JW. Floating hot-melt extruded tablets for gastroretentive controlled drug release system. J Control Rel. 2006;115:121–29.
Kojima N, Seino K, Sato Y, et al. Carbohydrate carriers affect adhesion of H. pylori to immobilized Leb–oligosaccharide. FEBS Lett. 2002;517:32–6.
Yin YS, Chen D, Qiao M, et al. Lectin conjugated PLGA nanoparticles loaded with thymopentin: Ex vivo bioadhesion and in vivo biodistribution. J Control Release. 2007;123:27–38.
Staros JV, Wright RW, Swingle DM. Enhancement by N-hydroxysulfosuccinimide of water-soluble carbodiimide mediated coupling reactions. Anal Biochem. 1986;156:220–22.
Grabarek Z, Gergely J. Zero-length crosslinking procedure with the use of active esters. Anal Biochem. 1990;185:131–35.
Mohammadi G, Nokhodchib A, Barzegar-Jalali M, et al. Physicochemical and anti-bacterial performance characterization of clarithromycin nanoparticles as colloidal drug delivery system. Colloids Surf B: Biointerfaces. 2011;88:39–44.
Jain SK, Kumar A, Kumar A, et al. Development and in vitro characterization of a multiparticulate delivery system for acyclovir-resinate complex. Artif Cells Nanomed Biotechnol. 2015; Early Online:1-10.
Salem II, Duzgunes N. Efficacies of cyclodextrin-complexed and liposome encapsulated clarithromycin against Mycobacterium avium complex infection in human macrophages. Int J Pharm. 2003;250:403–14.
Kalam MA, Humayun M, Parvez N, et al. Release kinetics of modified pharmaceutical dosage forms: a review. Continental J Pharm Sci. 2007;1:30–5.
Khin MM, Hua JS, Cong HN, et al. Agglutination of Helicobacter pylori coccoids by lectins. World J Gastroentero. 2000;6:202–9.
Acknowledgments
One of the authors, Sunil K. Jain, gratefully acknowledges All India Council for Technical Education, New Delhi, India, for providing financial support (F.No. 20/AICTE/RIFD/ RPS (POLICY-III) 13 (Govt)/2012-13).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The ex vivo study was performed at Guru Ghasidas Vishwavidyalaya, according to the protocols approved by the Committee for Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Social Justice and Empowerment, Government of India, on the recommendations of the Institutional Animal Ethical Committee of Guru Ghasidas Vishwavidyalaya (Bilaspur, India).
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Jain, S.K., Haider, T., Kumar, A. et al. Lectin-Conjugated Clarithromycin and Acetohydroxamic Acid-Loaded PLGA Nanoparticles: a Novel Approach for Effective Treatment of H. pylori . AAPS PharmSciTech 17, 1131–1140 (2016). https://doi.org/10.1208/s12249-015-0443-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-015-0443-5