
Abstract
A clear scientific and operational need exists for harmonized bioanalytical immunogenicity study reporting to facilitate communication of immunogenicity findings and expedient review by industry and health authorities. To address these key bioanalytical reporting gaps and provide a report structure for documenting immunogenicity results, this cross-industry group was formed to establish harmonized recommendations and a develop a submission template to facilitate agency filings. Provided here are recommendations for reporting clinical anti-drug antibody (ADA) assay results using ligand-binding assay technologies. This publication describes the essential bioanalytical report (BAR) elements such as the method, critical reagents and equipment, study samples, results, and data analysis, and provides a template for a suggested structure for the ADA BAR. This publication focuses on the content and presentation of the bioanalytical ADA sample analysis report. The interpretation of immunogenicity data, including the evaluation of the impact of ADA on safety, exposure, and efficacy, is out of scope of this publication.
Graphical Abstract

Similar content being viewed by others

Introduction
Throughout the industry, there has been a wide range of conventions employed for the presentation of immunogenicity results. The ultimate goal of any sample analysis report is to facilitate understanding of the work performed. In order to efficiently convey these results, it is of benefit to present this information in a more harmonized report format.
The reporting of immunogenicity results is further complicated by aspects unique to ADA assays, such as the selection of assay platform and instrument, the selection and utilization of critical reagents, the estimation of sensitivity with a semi-quantitative or qualitative assay, and the evaluation of drug and/or target tolerance. A discussion on immunogenicity assay result reporting at the 2017 AAPS NBC led to the establishment of working groups to address key bioanalytical reporting gaps for both binding ADA and neutralizing antibody (NAb) assay results. Working group members across multiple organizations including pharmaceutical, biotechnology, contract research organizations, and regulatory agencies discussed these gaps and inconsistencies in immunogenicity reporting, and are hence producing a series of publications focused on appropriate validation (1) and result reporting. The discussions related to the reporting of ADA results are presented here, including an example report template (Supplementary Material) to facilitate filings with health authorities. The companion to this series, which will focus on the unique aspects essential to immunogenicity BAR reporting of NAb assay results, will follow as a separate publication.
As each scientific organization will have unique operational requirements associated with results reporting for study data, this publication provides a general guide for standardizing the format of immunogenicity results presented in the ADA BAR. These recommendations are intended to harmonize the presentation of ADA data and facilitate report review by industry and regulatory authorities, and are in alignment with the documentation recommended by current guidance (e.g., Section VIII of 2019 FDA Guidance (2,3,4).
Regarding the BAR content, study blinding status as well as availability of individual subject and sample information determines the level of detail to be reported, as appropriate. A summary of ADA such as incidence, prevalence, persistence, or impact in the context of clinical results such as PK, PD, and safety is usually provided in documents such as the Clinical Study Report (CSR), or the Integrated Summary of Immunogenicity (ISI). It is recommended the ADA sample analysis reports be provided as appendices to the CSR, or if required, the ISI.
Finally, the authors have included a report template as Supplementary Material that may be used as a general guide for what may be included in a bioanalytical ADA sample analysis report. Each drug development program will have unique requirements, and therefore not every aspect for reporting addressed in this publication may apply. Although the examples cited here are typical to clinical ADA result reporting, many of the same principles may be applied to reporting nonclinical ADA results. It is highly recommended that each user includes those aspects of this guidance that best apply to their drug development program.
Bioanalytical Report Structure and Content
Statement of Compliance
While there is no formal regulatory guidance on the conduct of ADA testing, it is generally performed under the principles of GLP/GCP. It is recommended to include, at minimum, a statement regarding which guidelines were followed (e.g., GLP/GCP, sample analysis plan, relevant site SOPs) as well the extent of QC or Quality Assurance (QA) review. If QA review is conducted, a separate statement with specific details of the QA audit and associated signatures should be included.
Summary
The report summary provides a short review of the study information and ADA sample analysis. The summary includes information related to the drug name, study name and study number, study population and/or indication, as well as the method and its qualification or validation status, and study sample matrix. The number of samples received and analyzed, as well as the total number of samples that were positive in the screening and the confirmatory assays (if performed) for each clinical study, should be provided. The detection of any antibodies present prior to treatment (pre-existing antibodies) should be noted. The presence of pre-existing antibodies requires a careful assessment of post-treatment response (titer) to understand whether administration of the study drug led to a treatment-emergent (boosted) response. This interpretation is considered out of scope for the ADA BAR and should be included in the appropriate section of the CSR or ISI.
The format for reporting titer should clearly state the total final dilution applied to the sample, especially in the case that dilution beyond the assay minimum required dilution (MRD) is required. For example, if a dilution of 1:10 was applied during sample analysis (this being in addition to the assay MRD of 1:5), the final reported sample titer would be 1:50 (2019 FDA Guidance). If additional processing of samples is required, such as with acid dissociation, these dilutions should be accounted for in the MRD and final reported titer. Detailed examples of determination of MRD are provided in the Anti-Drug Antibody Validation Testing and Reporting Harmonization guidance (1).
The summary section may also provide details of sample characterization, such as isotype, domain specificity testing and any additional characterization(s) required due to specifics of the drug modality, such as bispecific molecules or antibody drug conjugate (ADC) drugs (1).
Bioanalytical Method
To properly evaluate the reported results, it is recommended that a brief summary of the study drug (biotherapeutic, gene therapy, ADC, monoclonal antibody therapy, etc.) and the method used for sample analysis be provided. The method summary can be presented in a tabular format and include information about assay platform (colorimetric vs. electrochemiluminescence) and assay format (sandwich, solution-based bridging, or direct/indirect assay). Any requirement for pre-treatment of samples, such as acid dissociation or immunoadsorption, added preservatives or enzyme inhibitors, and the associated data reduction required (raw response or normalized response) should be described. Additionally, method details and validation parameters should be summarized in a tabular format as noted in Section 5 of the example report template (Supplementary Material). This summary table should include a clear reference or link to the bioanalytical method and/or validation report for cross-referencing. The method summary table should also briefly list the study drug and sample matrix as well as a description of the positive control antibody recommended, including clonality, host and target species, and the final concentration for each positive control as determined in neat matrix.
System suitability controls such as negative, low, and high positive controls from each run are included to evaluate assay performance. During pre-study method development and validation, the low positive control should have been selected with an appropriate targeted failure rate to guarantee consistent sensitivity in-study (during the duration of sample analysis supporting the current study), (1, 5, 6). The MRD should be clearly defined as per industry-wide accepted guidance and be taken into consideration in the final reported dilution (titer or S/N) of the sample (1, 6, 7).
Critical validated method parameters, including screening and confirmatory cut point (and titration cut point, if applicable), assay sensitivity, drug tolerance, and target interference (in both screen and confirmatory assay tiers), matrix effect (or selectivity), and effect of hemolysis/lipemia on any of these parameters, should be summarized, if applicable (see Template Table 5-1). Multi-domain therapeutics that require assessment of more than one cut point or domain specificity assessment should be included. If any assay parameters (for example, cut point factor) changed in-study, these should be noted along with the original validated value.
Materials
The materials section should include a description of critical reagents and equipment and software used during sample analysis in tabular format as presented in Section 6 of the template.
Critical Reagents
Materials that are key to the performance of the method are considered critical reagents (8, 9). These include, but are not limited to, the study drug, positive controls, custom-labeled drug or detection reagent, and negative control matrix. All critical reagents should be listed in a table that includes available reagent description, reagent source, lot and catalog number, preparation date, concentration, expiration and/or retest date, and appropriate storage conditions. Documentation such as certificates of analysis for the drug substance or drug product or reagents unique to the assay, are not required, but may be included as an appendix to the report (10).
Positive Control Antibodies
A purified antibody with a defined concentration is preferable for the consistent evaluation of the surrogate positive control. In the case that the positive control antibody is not purified (such as with antibodies directed against toxin or anti-sera), any available information related to the positive control purity should be provided. A description of the positive control antibody clonality (monoclonal versus polyclonal), host and target species, and storage condition should be included.
Matrix and Controls
The negative control is a critical reagent as all sample results are evaluated against the assay cut point. Given the possible presence of interfering factors such as endogenous target, or the presence of a high false-positive individual within the negative control pool, the overall negative control results may be skewed by a non-representative sample. Therefore, the information from the individual lots that comprise the validated negative control pool should be summarized. This includes any assays needed for bridging the negative control pools used during sample analysis that are different from those used in method validation (e.g., when the original validated negative control pool is expended) (1). Therefore, it is recommended that a reference be provided to any documentation (e.g. notebook pages, report number) summarizing additional assays related to new negative control pools used during sample analysis.
The final qualified matrix pool used as the negative control should be included in Template Table 6.1. If the negative control pool is prepared from multiple individual matrix lots, the lot numbers of the individual matrix samples should be documented and retained in validation or study-related folders with the study records for cross reference, and therefore do not need to be individually listed in Table 6.1.
Equipment & Software
The equipment and software section should include the major equipment (e.g., plate reader, plate washer) and software only. Major equipment is critical to the performance of the assay, and is dependent on the assay being performed, and therefore does not include minor equipment such as freezers and incubators, used during sample analysis. If all equipment and software used are the same as those used in validation, this should be noted in the report for expedience and ease of reference. Ensure that specific equipment identification numbers are included to allow for study reconstruction. See Template, Section 6.2 (Supplementary Material).
Data Analysis
Describe the software used for the analysis of the results as well as a brief description of how the data were analyzed.
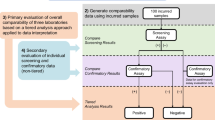
Data analysis should be performed and briefly described in accordance with the recommended tiered analysis including screening, confirmatory analysis, and characterization as described in Shankar (6) and Devanarayan (11) (Fig. 1). Any replicate outlier analysis, such as Grubb’s outlier analysis that was used in the assessment of positive or negative control samples should also be briefly described here. If an in-study cut point is required, provide a brief explanation in this section, including observed false-positive rate and data transformation. The details of in-study cut point calculation is discussed in the “Results” section.

Tiered analysis for reporting of immunogenicity results (assumes false- positive rate is reflective of validation cut point population)
Sample results must be reported in a consistent format that reflects how they were analyzed. For example, in cases where data are reported as raw responses such as ECL units (relative luminescence units or RLU), all statistical evaluations (e.g., Std. Dev.) must be performed with raw data RLU numbers. In cases where data are reported as a signal to noise (S/N) ratio, all assay evaluations must be performed using the S/N ratio.
A statement that describes calculation of results taking place prior to rounding of numbers, as well as designating the total number of significant figures to be reported should be included. In addition, a brief description of how the data were transferred to the sponsor and/or clinical database (e.g., according to specifications listed in the Data Transfer Agreement) may be included, if applicable. This clarifies why manual calculations from displayed values may differ in the final significant figure from the original value.
Study Sample Receipt and Storage
Sample receipt and storage of samples is summarized in Table 8-1. Supportive documentation does not need to be included within the report, but should be referenced and stored with study records for sample tracking purposes. This may include additional documentation such as sample manifests, temperature data logger information, and sample reconciliation, as available.
A brief description of the condition of samples upon receipt as well as their storage and any temperature excursions is critical. For sample tracking, it is recommended to include sample identification number and/or accession number, and time point or visit information (as provided in the protocol, laboratory manual, or data transfer agreement documentation). Samples that were received in poor condition, had a temperature excursion, or an unreconciled discrepancy should be noted. If backup sample aliquots are provided, ensure the correct vial identification number and/or accession number is reported, especially in the case that the original and backup vials were required during analysis. If investigational sample discrepancies have been resolved, a statement should be included to reflect that no further investigation is required.
Sample laboratory origination locations, dates of receipt, and study center of origin are not necessary, assuming there are systems in place to ensure chain of custody through the assigned sample identification number.
As antibodies have proven to typically be stable for several years when stored frozen (12, 13), long-term stability is not always performed for anti-drug antibodies in study samples. Information on any excursions from established process stability (freeze-thaw cycles, benchtop, or refrigerated storage) should be described.
Results
This section is an overall assessment of method performance and the sample testing data in context of the validation results. It is meant to document the assay performance and qualitative (positive/negative) and quasi-quantitative (titer or S/N as appropriate) results of sample testing.
This section should, at a minimum, cover the following aspects: in-study cut point (CP) assessment (if applicable), sample testing overview (i.e., run summary), control performance, sample results, repeat analysis, and impact on sample results from deviations that may have occurred during the conduct of sample testing.
In-study Cut Point Assessment
An analysis of screening false-positive rate (FPR) should be performed using the validated CPF to understand whether the specific study population differs from the validation population. Generally, ADA CPs (screening, confirmatory, and in some cases, titer) were established using samples from normal healthy individuals as these are readily available and of sufficient volume. It is recommended that the FPR assessment be performed for each study given that the study population may differ from the validation population or the same disease state in a previous study due to concomitant medications, severity of disease, demographics of enrolled population, etc. If a sufficient number of pre-dose samples are available, the FPR may be calculated by the formula: (B/(A+B)) × 100, where A = number of pre-dose/baseline samples screened negative, and B = number of pre-dose/baseline samples screened positive and confirmed negative. Each organization may have different requirements for an acceptable/unacceptable FPR; while a range of 2–11% has been suggested for the initial assessment (11), this range is sample-size dependent and should not be automatically relied upon, but instead assessed according to study needs (14). In the case that pre-existing ADA are present, a short summary addressing the prevalence of these antibodies and associated considerations for in-study cut point assessment should be included.
An unacceptable FPR may not be the only reason an in-study CP is established, but it is the most common. The reason for adopting an in-study cut point as well as a brief description of how it was determined (number of samples/days/analysts for each assessment), and the statistical method used to generate the in-study CP(s) inclusive of exclusion of known positives or outliers should be described. Additional examples of how to determine the need to establish an in-study cut point are detailed in Devanarayan (11). Any statistical reports generated as a result of CP reassessment should be included in the appendix.
The cut points used in study sample testing, as included in Template Table 5-1, must be clearly stated in the report.
Sample Testing Overview
This section is a summary of the number of passing runs/performed runs for each tier (pass rate), including, as applicable, domain characterization. A statement describing whether samples were analyzed within established stability parameters (i.e., freeze-thaw cycles, bench-top, or refrigerator stability) may be included with specifics of any exceptions detailed in the “Sample Results” section.
Template Table 13-1 includes the run number, tier type assessed, run date, pass/fail status, and associated comments (such as reason for failure, or if a repeat run was performed) as the recommended minimum information. It may be of benefit to evaluate the results of the screening tier separately from the confirmatory tier, as each result may provide useful information on assay performance.
Control Performance and Acceptance Criteria
For each tier, include a description of the number of negative control wells (or if applicable, sets of x wells) and the number of sets and levels of positive controls (LPC, HPC) tested. Provide a description of control acceptance criteria as listed in the bioanalytical plan and/or method validation report. The overall inter-assay precision is also described. If precision does not meet the a priori acceptance criteria, or if results are outside any established pre-study validation ranges, include a discussion of the outcome. Where outlier assessment was applied to controls —either the overall assessment, or as part of the plate acceptance (particularly for the negative control) — this information should be described or referenced. If a particular control batch (or batches) led to failure of multiple assay runs, the steps taken to resolve the failure should be outlined. As it is recommended to statistically set the low positive control level during validation to lead to a 1% assay failure rate (5), monitoring the LPC performance is suggested to ensure samples are correctly evaluated and assay performance is maintained during the study.
Two table options are provided in the template to summarize assay control performance. Tables 13-2 and 13-3 comprise screen/titer assay and confirmatory assay control performance, respectively. Alternately, Table 13-3 can be used alone for all assay runs to allow for the calculation of summary statistics of drug naïve controls, since these tend to be the same in all tiers. Both tables have the option to provide results as either signal or signal to noise (S/N) ratio as specified in the method. Rather than including a specific column for %CV, failed %CV acceptance may be formatted with italics or a footnote. Finally, Table 13-4 provides a summary option for titer assay control performance if a different set of positive controls, such as a series of dilutions, is used to monitor titer assay acceptability. All control performance tables include the run number and plate specific cut point to allow for traceability/assessment of pass/fail performance (8).
Sample Results
This section includes (1) whether and why any samples were not tested, (2) a summary of the total number of samples screened, confirmed positive, and subsequently titered, and (3) re-assay information. Explanatory text detailing how sample results are reported should be included, for example, “Samples that test negative in the screening or confirmatory tier are reported as confirmed negative” and “Samples assessed in the titer tier are reported as sample dilution x MRD.” Per guidance, titer results should account for all dilutions of the test sample (1, 6). Additionally, provide a description for reporting of samples that are confirmed positive in the screening and/or confirmatory assay and return a result below the screening or titer cut point in the titer analysis. If samples were also assessed in a domain specificity test, the overall summary should be provided (e.g., N of X samples showed specificity for Y domain of drug Z).
Template Table 13-5 provides the suggested minimum demographic columns (also dependent on data transfer agreement), all analyzed sample results (inclusive of all tiers), and applicable comments for each sample in a single row. The Sample Results section in the body of the report or Comments column in Table 13-5 may also include whether certain results should be interpreted with caution due to out of stability specifications or if the sample was lipemic/hemolyzed, or if the sample contains drug concentrations exceeding established ADA assay tolerance (when known and available). If any analyzed sample results are excluded, the exclusion should be justified and noted in the body of the report. The “Sample Results” section should also include a discussion on how any deviations from the method or study plan impacted individual sample results. Finally, laboratories and sponsors should be aware that efforts are ongoing to standardize reporting of ADA tiered data through controlled terminology (standardized data tabulation model, SDTM), under oversight by Clinical Data Interchange Standards Consortium (CDISC) (15).
Template Table 13-6 lists all samples that were reanalyzed, the reason for reanalysis, and the reported result (which is included in Table 13-5). Samples may be reanalyzed due to failed plate or sample acceptance criteria, however only samples from acceptable runs requiring repeat analysis should be listed. Inadvertent repeats should also be addressed with a clear explanation as to the reason for duplicate results and how the final reported result was determined. Data should be carefully evaluated for potential analytical or technical error.
Finally, as noted earlier, interpretation such as reporting of incidence, prevalence, or impact of ADA should be avoided in the BAR ADA report sample result discussions. However, it is recommended that an integrated table of available ADA results, inclusive of NAb data with drug concentration and pharmacodynamic data (as applicable) be included either in the CSR or the ISI.
Deviations
The section should detail significant deviations from either the bioanalytical plan or from the method, as well as an impact assessment of the deviations on the affected runs (as applicable), and overall study impact. Any investigations performed in association with the sample analysis must be detailed.
Archival
The archival or disposition of samples, data, and reports is described in accordance with current archival policy in a manner that facilitates information retrieval for future studies.
Discussion
Review of immunogenicity reports can be facilitated by providing a harmonized report structure and presentation of key assay parameters. Provided here are recommendations for essential aspects of reporting clinical ADA results using traditional technologies, with examples that may be applicable across a wide range of ADA assays. The recommendations and template are intended to provide guidance of the key parameters to include in the summary of ADA results, with the acknowledgement that each organization may have different requirements for results reporting. It is also acknowledged that when a CRO is performing analysis on behalf of a Sponsor, or when the study is blinded, all subject information may not be available during sample analysis. It is recommended that as much salient information be reported as possible, to allow for correct interpretation and the reconstruction or reproduction of original assay results, if needed.
For the purpose of illustration, the examples provided here were limited to those from traditional LBA assay platforms (ELISA and ECL), but alternative assay platforms are also being used for the analysis of ADA. Technology such as Gyros, liquid chromatography-mass spectrometry (LC-MS), and ELISPOT may pose unique reporting challenges. A brief explanation of the readouts and data format should be included to appropriately evaluate these results. For example, there is a distinction between colorimetric ELISA and ELISPOT readouts. Although both are reported in OD units, ELISA results report the total antibody present in a sample, whereas ELISPOT reports the total number of cells producing antibodies in the sample. Another example of differences between readouts is in the case of LC-MS compared to traditional LBA assays such as ELISA or ECL. LC-MS may report total mass or the mass shift as the final readout (as in the case of measurement of the toxin portion of an ADC), whereas in conventional LBA assays, the titer of ADA present is reported for positive samples.
The presence of pre-existing antibodies have been associated with PEGylated therapeutics (16), and have been implicated in modulation of vaccine response (17, 18). Appropriate measurement of pre-existing ADA requires evaluation of antibody responses within the same individuals prior to and after treatment, and is included in the CSR or ISI.
Additional characterization of the anti-drug antibody response such as isotyping or domain specificity testing may be required for specialty classes of biologics; these include gene therapy products, multi-domain biotherapeutics, and antibody drug conjugates. As with any assessment of ADA, providing sufficient detail to facilitate evaluation of the results is recommended. For example, immunogenicity assessment for gene therapy programs may require a separate assessment for the antibodies directed against the viral capsid (i.e., an adeno-associated virus vector), in tandem with an assessment of antibodies specific to the transgene product. The traditional screen, confirm, and titer tiers are still required here, and the details for each of these assays should be described in sufficient detail.
In cases where samples require pre-treatment such as acid dissociation, the details of these conditions should also be included to assist with result interpretation. In cases when an in-study cut point is required, the justification for in-study cut point as well as relevant sample analysis information needs to be provided. Inclusion of these relevant details in the summary report ensures proper evaluation of the final results.
Although interpretation of the ADA data is not within the scope of this report, provided in this guidance is a list of references for the proper evaluation of antibody responses in the context of drug PK, PD, and safety (as may be applicable) for the study CSR or ISI. For clinical studies, the ADA BAR sample analysis reports are intended to be appendices of the CSR or ISI. Final sample values reported should be presented clearly in the ADA Sample Analysis BAR, with sufficient information to allow for sample analysis reconstruction, and in the proper context to allow for accurate evaluation of results.
References
Myler H, Pedras-Vasconcelos J, Phillips K, et al. Anti-drug antibody validation testing and reporting harmonization. AAPS J. 2021;24(1):4. https://doi.org/10.1208/s12248-021-00649-y.
EMA. Guideline on immunogenicity assessment of therapeutic proteins. In: (CHMP) CfMPfHU, editor. London. 2017.
Mire-Sluis AR, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289(1-2):1–16. https://doi.org/10.1016/j.jim.2004.06.002.
Chamberlain P. Effective presentation of immunogenicity risk assessments and related data in regulatory dossiers. Bioanalysis. 2019;11(17):1581–92. https://doi.org/10.4155/bio-2018-0209.
Immunogenicity testing of therapeutic protein products —developing and validating assays for anti-drug antibody detection, guidance for industry. In: U.S. FDA CfDEaR, Center for Biologics Evaluation and Research, editor. MD, USA. 2019.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, Fiscella M, Gorovits B, Kirschner S, Moxness M, Parish T, Quarmby V, Smith H, Smith W, Zuckerman LA, Koren E. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81. https://doi.org/10.1016/j.jpba.2008.09.020.
Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, Quarmby V, Richards S, Schneider CK, Subramanyam M, Swanson S, Verthelyi D, Yim S, American Association of Pharmaceutical Scientists. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16(4):658–73. https://doi.org/10.1208/s12248-014-9599-2.
Azadeh M, et al. Quality controls in ligand binding assays: recommendations and best practices for preparation, qualification, maintenance of lot to lot consistency, and prevention of assay drift. AAPS J. 2019;21:89. https://doi.org/10.1208/s12248-019-0354-6.
King LE, Farley E, Imazato M, et al. Ligand binding assay critical reagents and their stability: recommendations and best practices from the global bioanalysis consortium harmonization team. AAPS J. 2014;16(3):E504–14.
(USP) USP. General chapter Immunogenicity assays - design and validation of immunogenicity assays to detect anti-drug antibodies. 2014.
Devanarayan V, et al. Recommendations for systematic statistical computation of immunogenicity cut points. AAPS J. 2017;19:1487–98.
Michaut L, Laurent N, Kentsch K, Spindeldreher S, Deckert-Salva F. Stability of anti-immunotherapeutic antibodies in frozen human serum samples. Bioanalysis. 2014;6(10):1395–407. https://doi.org/10.4155/bio.14.97.
Correia I. Stability of IgG isotypes in serum. mAbs. 2010;2(3):221–32. https://doi.org/10.4161/mabs.2.3.11788.
Tan CY, Steeno GS, You Z, Gaitonde P, Cai C-H, Kamerud J, et al. Criteria to reevaluate anti-drug antibody assay cut point suitability in the target population. AAPS J. 2020;22(2):1–3.
White J. Reporting immunogenicity raw data for nonclinical and clinical databases. AAPS Webinar 2021. https://www.pathlms.com/aaps/webinars/20723.
Xue L, et al. Recommendations for the assessment and management of pre-existing drug-reactive antibodies during biotherapeutic development. AAPS J. 2017;19(6):1576–86. https://doi.org/10.1208/s12248-017-0153-x.
Das S, Singh J, Shaman H, et al. Pre-existing antibody levels negatively correlate with antibody titers after a single dose of BBV152 vaccination. Nat Commun. 2022;13:3451. https://doi.org/10.1038/s41467-022-31170-1.
Dangi T, Sanchez S, Lew MH, Visvabharathy L, Richner J, Koralnik IJ, Penaloza-MacMaster P. Pre-existing immunity modulates responses to mRNA boosters. bioRxiv [Preprint]. 2022:2022.06.27.497248. https://doi.org/10.1101/2022.06.27.497248.
Acknowledgements
Acknowledgements are due to AAPS TPIFG team members. Joleen White, Ph.D., Bill and Melinda Gates Foundation. Boris Gorovits, Ph.D., Sana Biotechnology. Ronald Bowsher, Ph.D. B2S Biosciences. FDA : OBP Office of Biotechnology Products, OSIS Office of study Integrity and surveillance, OCP Office of Clinical Pharmacology.
Funding
Apart from the time investment, no other funding was involved in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Darshana Jani: she led and coordinated the project from its inception including co-writing the manuscript. Lead the scientific and technical discussion for all the topics.
Robin Marsden and Michele Gunsior have equal contribution in the preparation of the manuscript. Co-led the discussions for manuscript topics and creating the example template.
Laura Schild Hay and Bethany Ward assisted in organizing the meetings, maintaining the meeting minutes, and participated in scientific and technical discussion of the key topics.
All authors participated in key discussion about various components of manuscript, provided expert opinions, and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
All authors are affiliated with the companies as they are listed in the authors section.
Disclaimer
The views expressed in this article are those of the authors and do not reflect official policy of the respective companies or US FDA.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 63 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jani, D., Marsden, R., Gunsior, M. et al. Anti-drug Antibody Sample Testing and Reporting Harmonization. AAPS J 24, 113 (2022). https://doi.org/10.1208/s12248-022-00762-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-022-00762-6