
Abstract
Orally administered artemisinin-based combination therapy is the first-line treatment against uncomplicated P. falciparum malaria worldwide. However, the increasing prevalence of artemisinin resistance is threatening efforts to treat and eliminate malaria in Southeast Asia. This study aimed to characterize the exposure-response relationship of artesunate in patients with artemisinin sensitive and resistant malaria infections. Patients were recruited in Pailin, Cambodia (n = 39), and Wang Pha, Thailand (n = 40), and received either 2 mg/kg/day of artesunate mono-therapy for 7 consecutive days or 4 mg/kg/day of artesunate monotherapy for 3 consecutive days followed by mefloquine 15 and 10 mg/kg for 2 consecutive days. Plasma concentrations of artesunate and its active metabolite, dihydroartemisinin, and microscopy-based parasite densities were measured and evaluated using nonlinear mixed-effects modeling. All treatments were well tolerated with minor and transient adverse reactions. Patients in Cambodia had substantially slower parasite clearance compared to patients in Thailand. The pharmacokinetic properties of artesunate and dihydroartemisinin were well described by transit-compartment absorption followed by one-compartment disposition models. Parasite density was a significant covariate, and higher parasite densities were associated with increased absorption. Dihydroartemisinin-dependent parasite killing was described by a delayed sigmoidal Emax model, and a mixture function was implemented to differentiate between sensitive and resistant infections. This predicted that 84% and 16% of infections in Cambodia and Thailand, respectively, were artemisinin resistant. The final model was used to develop a simple diagnostic nomogram to identify patients with artemisinin-resistant infections. The nomogram showed > 80% specificity and sensitivity, and outperformed the current practice of day 3 positivity testing.
Similar content being viewed by others

INTRODUCTION
Malaria-related mortality has decreased significantly during the last decade, but there are still approximately 500,000 deaths each year (1). Artemisinin-based combination therapy (ACT) is the recommended first-line treatment for uncomplicated falciparum malaria (2). ACTs comprise a short-acting but potent artemisinin derivative combined with a long-acting antimalarial with a different mechanism of action. The artemisinin component is responsible for eliminating the majority of the parasite biomass during the first days of treatment, and its rapid and broad parasiticidal activity has been a major advantage compared to other antimalarial agents (3). However, this efficacy is threatened by the emerging artemisinin resistance in Southeast Asia (4,5,6,7). Artemisinin resistance has been manifested by a gradual increase in parasite clearance times over the last decade and has resulted in increasing rates of ACT treatment failure (8).
Artemisinin-resistant falciparum malaria has been confirmed in Cambodia, Laos, Myanmar, Thailand, and Vietnam. The recently discovered molecular marker (i.e., mutations in P. falciparum (Pf) kelch13) is associated with artemisinin resistance and correlated with increased parasite clearance times (7,9,10). Slower parasite clearance results in a higher parasite burden requiring to be eliminated by the ACT partner drug and an increased probability of selecting resistance. High failure rates have been observed recently after dihydroartemisinin-piperaquine treatment in Cambodia and Vietnam and after artesunate-mefloquine on the Thailand-Myanmar borders (11,12). Strategies to restore high cure rates include increasing dosage, duration, and frequency of artemisinin administration and use of triple combinations. These do not restore rapid parasite clearance in infections with artemisinin-resistant parasites (13).
The aim of this study was to characterize the population pharmacokinetic (PK) properties of artesunate (ARS) and its active metabolite, dihydroartemisinin (DHA), in patients with artemisinin sensitive and resistant falciparum infections, and then to develop a population exposure-response model to describe the time course of drug exposure and its relationship to the elimination of malaria parasites. The developed pharmacokinetic-pharmacodynamic (PK-PD) model was also used to develop a simple algorithm (nomogram) to identify patients with in vivo artemisinin-resistant infections.
METHODS
Study Design
The data used in this study were from two open-label randomized clinical trials; Pailin Referral Hospital in western Cambodia and the Shoklo Malaria Research Unit (SMRU) clinic in Wang Pha, Tak Province, north western Thailand. The trial which recruited patients between June 2007 and December 2009 aimed to assess the efficacy of artemisinin in two locations in the Greater Mekong subregion and is reported in full elsewhere (4). Ethical approval was obtained from the Ministry of Health in Cambodia, the Ethics Committee of the Faculty of Tropical Medicine of Mahidol University in Thailand, the Oxford Tropical Medicine Ethical Committee, the WHO Research Ethics Review Committee, and the Technical Review Group of the WHO Western Pacific Regional Office (ClinicalTrials.gov number, NCT00493363).
Briefly, 40 patients with uncomplicated falciparum malaria were recruited at each site. Non-pregnant adults and children (> 5 years) were enrolled in Pailin, and non-pregnant adults (> 16 years) were enrolled in Wang Pha. Patients were randomized (50:50) to receive once daily oral ARS monotherapy for 7 days (2 mg/kg/day) or once daily oral ARS monotherapy for 3 days (4 mg/kg/day) followed by mefloquine treatment (15 mg base/kg on day 4 and 10 mg base/kg on day 5). ARS was provided by Guilin Pharmaceutical (repacked and quality-controlled by Sanofi Synthelabo for distribution in Cambodia and by Atlantic for distribution in Thailand). Mefloquine was provided by Medochemie for Cambodia and Mepha Pharma for Thailand.
Venous plasma samples were obtained at 0 (pre-dose), 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, and 12 h post-dose on the 1st day of treatment. Opportunistic sparse sampling was performed after each dose throughout the study. Blood samples were collected in pre-chilled fluoride oxalate tubes in Pailin and in pre-chilled lithium heparin tubes in Wang Pha. Samples were centrifuged at 4°C and stored in liquid nitrogen until transported to the Department of Clinical Pharmacology, Mahidol-Oxford Tropical Medicine Research Unit, Thailand, for drug measurements. The laboratory is accredited according to ISO15189 and ISO15190 and participates in the Worldwide Antimalarial Resistance Network Pharmacology Proficiency Testing Programme (14,15). ARS and DHA plasma samples were quantified using high-throughput liquid chromatography-tandem mass spectrometry (16). The lower limit of quantification (LLOQ) was 1.2 and 2.0 ng/ml for ARS and DHA, respectively. Quality control samples at three levels (low, middle, and high) were analyzed within each batch of clinical samples to ensure precision and accuracy during routine clinical drug measurements. The relative bias (accuracy) was below 15%, and the coefficients of variation (precision) were lower than 5% for all quality control samples, which is well below the required precision of ± 15% according to US FDA regulatory guidelines (16).
Venous blood asexual and sexual parasite densities were assessed by microscopy. Parasite counts (thin or thick blood films) of asexual parasites and gametocytes were performed at 0 (pre-dose), 4, 8, and 12 h after the first dose and then every 6 h until two consecutive slides were negative for asexual parasites.
Population Pharmacokinetic and Pharmacodynamic Modeling
Estimation and simulation were performed using nonlinear mixed-effects modeling in the software NONMEM v.7.3 (ICON Development Solutions, Ellicott City, MD) (17). Post-processing, diagnostic plots, and automation were performed using Perl-Speaks-NONMEM (PsN), v. 4.5.3 (18,19), Xpose, v.4.5.3 (20,21), Pirana, v. 2.9.2 (22), and R, v. 3.2.4 (The R Foundation for Statistical Computing) (23,24).
Population Pharmacokinetics
Molar units of ARS and DHA concentration measurements were transformed into their natural logarithms and characterized simultaneously, assuming complete and irreversible in vivo conversion of ARS into DHA (25,26). The fraction of data below the LLOQ was high (~ 40%). Observations below LLOQ was therefore included in the analysis and modeled as categorical observations, conditioned on the underlying model predicted plasma concentrations using the Laplacian estimation method (i.e., likelihood-based M3-method) (27,28,29). Different disposition models were explored for the two compounds (one-, two-, and three-compartment models). The best performing disposition model was carried forward to evaluate different absorption models, i.e., first-order absorption with and without lag-time, zero-order absorption, sequential zero- and first-order absorption, and transit-compartment absorption model (30). Between-subject variability (BSV) was modeled exponentially as illustrated below (Eq. 1).
where θ i is the estimated individual parameter value, θ TV is the estimated population parameter value, and η i represents the BSV, assumed to be independent and symmetrically distributed around zero with a variance ω 2. BSV was estimated for all structural parameters initially but only maintained if they could be estimated with adequate precision (< 50% RSE). Correlations between individual parameters were also evaluated by estimating the off-diagonal elements in the covariance-variance matrix. Between-occasion variability (BOV) was explored between dose occasions on relevant absorption parameters. The unexplained residual variability (RUV) was estimated by separate additive error models for log-transformed ARS and DHA concentrations (i.e., essentially equal to exponential error models on an arithmetic scale).
Body weight (BW) was evaluated as an allometric function on all clearance and volume parameters, where clearance was scaled to a power of 0.75 and volume to the power of 1 (Eq. 2):
where BWi is the individual body weight, BWmed is the median body weight of the population, and γ is the allometric exponent. Potential malaria disease effects on absorption parameters were evaluated using observed parasitemia as a continuous time-dependent covariate. The covariate relationship was centered on the lowest observed parasite density (Log (PARAmin)) and evaluated manually using both linear and nonlinear implementations (i.e., linear, piecewise linear, power, exponential, and Emax functions). The Emax relationship was implemented according to Eq. 3 (MTT) and Eq. 4 (F).
where PARAMTT is the estimated linear effect of parasite density on MTT, PARAmaxF is the maximum effect of parasite density on F, PARA50F is the parasite density which produces 50% of the maximum covariate response, and Log (PARAi) is the individual parasite density. Relationships between model parameters and other potentially influential covariates (i.e., age, sex, baseline hemoglobin, baseline parasitemia, and temperature) were identified using correlation plots and subsequently evaluated by stepwise forward inclusion (p < 0.05) and backward exclusion (p < 0.01) covariate modeling using linear, piecewise linear, power, and exponential functions.
Population Pharmacodynamics
A PK-PD model was developed using a sequential approach, in which individual PK parameter estimates from the final PK model were used in the PK-PD model (31). Observed total parasite densities for each patient, i.e., asexual parasite count per microliter of blood multiplied by a theoretical blood volume (assuming 80 mL of blood per kilogram body weight), were transformed into their natural logarithms and modeled using the first-order conditional estimation method with interactions or with addition of the Laplacian option (likelihood-based M3 method) to evaluate the dynamic parasite effects of ARS/DHA. Parasite microscopy LLOQ was set to an observed parasite count of 16 per microliter. All the data below the LLOQ were explored with and without the application of the M3-method for censored observations (29). Parasite densities at day 4 and forward were censored in the mefloquine group since combination treatment started at this day. However, only 10.5% of patients had detectable asexual parasite counts at this time point.
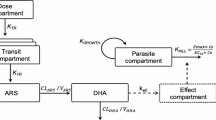
In the absence of any drug, the parasites were assumed to grow exponentially with a parasite multiplication rate (PMR) of approximately tenfold multiplication per asexual cycle of 48 h (32,33). The predicted plasma concentrations of DHA were used to link to the drug-dependent killing of parasites (K KILL) (Eq. 5) (34). Both direct and delayed concentration-response Emax models were evaluated (34). The delayed effect model links the central compartment of the PK model to a hypothetical intraerythrocytic effect compartment that exerts the PD effect (Fig. 1).
where Emax is the maximum parasite kill rate, Ce is the drug concentration in the effect compartment, and EC50 is the concentration which produces 50% of maximum killing effect.

Schematic representation of the final population pharmacokinetic-pharmacodynamic model of artesunate (ARS) and its active metabolite, dihydroartemisinin (DHA), in patients with uncomplicated malaria. Ce predicted DHA concentration in the effect compartment, CL elimination clearance, EC 50 the concentration which produces 50% of maximum parasite kill, Emax maximum parasite kill, K E0 effect compartment rate constant governing the delayed drug effect, K GROWTH parasite multiplication rate, fixed to tenfold multiplication per 48-h cycle, KTR first-order transit absorption rate constant [KTR = (n + 1)/mean transit time], V apparent volume of distribution
A mixture model was evaluated on Emax and/or EC50 to quantify the bimodal distribution of the observed parasite clearances. The probability of having a resistant infection was estimated for each site separately, and potentially influential covariates (i.e., site and study arm) were evaluated. Individual Emax values of the two mixture populations, one with low parasite-killing (resistant infections; EmaxR) and one with high/normal parasite-killing (sensitive infections; EmaxS) were parameterized as below (Eqs. 6 and 7) to prevent aliasing, i.e., to constrain all individual EmaxSi to be greater than the typical population Emax value for the resistant population.
where θ TVR and θ TVS are the estimated population parameter values for resistant and sensitive populations, respectively, with their associated variance (η).
Model Evaluation
Model selection was guided by plausible parameter estimates, precision of parameters, visual diagnostics, and minimum objective function value (OFV); proportional to minus twice the log-likelihood of the data, computed by NONMEM. A drop in OFV of 3.84, 6.63, and 10.83 or more was considered a significant improvement at P < 0.05, P < 0.01, and P < 0.001, respectively, between two hierarchical models after inclusion of one additional parameter (one degree of freedom). Visual predictive checks (VPCs) were performed (1000 simulations) to evaluate the predictive performance of the PK and PK-PD models. The 5th, 50th, and 95th percentiles of observed data for the PK model and for the PD model were overlaid with the model predicted 95% confidence intervals of the same percentiles. Diagnostic plots were used to evaluate the overall goodness of fit by plotting observed drug concentrations against the population and the individually predicted concentrations. The reliability of individual parameter estimates and goodness-of-fit plots were assessed by reported eta and epsilon shrinkages (35). Precision or parameter uncertainty in parameter estimates was obtained from a recently developed sampling importance resampling (SIR) procedure (36).
Simulation of Alternative Dosing Regimens for Patients with Resistant Infections
The final PK-PD model was also used to simulate different dosing scenarios to evaluate possible alternative dosing in patients with artemisinin-resistant infections. The following dosing scenarios were evaluated; 2 mg/kg of artesunate administered twice a day for 3 days (i.e., increased frequency of dosing), 8 mg/kg of artesunate administered once daily for 3 days (i.e., increased dosage), and 4 mg/kg of artesunate administered once daily for 5 days (i.e., increased treatment duration). All simulations assumed a total baseline parasitemia of 1011. The simulated residual parasite burden was evaluated 24 h post last dose for each of the evaluated dosing regimens.
Resistance Nomogram
The established PK-PD model was used to simulate data for the development of a static nomogram to identify patients with artemisinin-resistant falciparum infections. The development of this nomogram was based on the hypothesis that the ratio between baseline parasite density at enrollment and the parasites density at a later time point could be a more sensitive approach to identify patients with “slow” parasite clearance compared to the traditionally used microscopy-based day 3 positivity test.
A simulated dataset with 10,000 hypothetical patients with artemisinin-resistant (n = 5000) and artemisinin-sensitive (n = 5000) infections was created using the developed PK-PD model, and this was regarded as the reference “true” classification. A uniform baseline parasite density distribution of 1010 to 1012.5 parasites was applied for the simulations to mimic the observed clinical parasite density distribution. The individual parasite density ratio (Ratioi) was calculated for time point j, i.e., 24, 48, and 72 h post-dose as below (Eq. 8).
The calculated median ratio at each time point (i.e., 24, 48, or 72 h) was considered as the resistant/sensitive “cutoff” value. The simulated individual patient ratios were then compared to the “cutoff” values at each time point to categorize them as having a resistant or sensitive infection (e.g., the 48-h time point calculation is shown in Eq. 9).
However, in case the simulated parasite density was below the LLOQ at the evaluated time point, the ratio calculation was not performed and the patient was categorized as having a sensitive infection. The category outcome was compared to the simulated patient’s known classification, and a sensitivity analysis was performed according to the statistical metrics in Table I. A schematic overview of the above approach is presented in Fig. 2.

A schematic representation of the development of the resistance nomogram
Absolute parasite clearance time is directly proportional to initial parasite density, assuming that parasite clearance half-life is independent of initial parasite density (37). Thus, the performance of the nomogram (baseline-adapted nomogram) at different initial parasite densities was explored to avoid any potential bias. As such, 10,000 hypothetical patients with artemisinin-resistant (n = 5000) and artemisinin-sensitive (n = 5000) infections were simulated for each baseline parasitemia scenario (i.e., 10,000 hypothetical patients were simulated for each baseline parasite density of 108 up to 1013 with an integral of a half log), using the developed PK-PD model. The performance of the nomogram was evaluated for each individual baseline parasite density in the same manner as above. However, the optimal day of assessing the nomogram was evaluated for individual baseline parasite densities in order to develop the baseline-adaptive nomogram. For each individual baseline parasite density, a sensitivity analysis was performed to inform if the nomogram should use measurements on day 1, 2, or 3 for optimal performance.
The same simulated hypothetical patients (used during the static nomogram development) with artemisinin-resistant and artemisinin-sensitive infections (i.e., 1010 up to 1012.5 baseline parasite density) were used to evaluate the developed monograms (static and baseline adaptive nomogram) and also (1) a pure PK-PD model-based approach and (2) the commonly used day 3 positivity test as a measure of the proportion of resistant infections. For (1), individual parameters of the developed PK-PD model were fitted to the simulated data, and for (2), the simulated day 3 positivity test (i.e., above LLOQ at day 3) was obtained to get individual patient resistance classifications. The individual classification of sensitive and resistant patients described above was compared to the simulated patient’s known resistance status with a sensitivity analysis as described in Table I.
RESULTS
Population Pharmacokinetics
Seventy-nine patients in this study were analyzed (Table II), and the final PK model and parameter estimates are presented in Table III and Fig. 1. Simulation-based diagnostics for the parent compound, ARS, and its active metabolite, DHA, are presented in Fig. 3. The final PK model consisted of one-compartment disposition models for both ARS and DHA. A transit compartment absorption model, consisting of four transit compartments, was superior to all other absorption models. In the final model, the absorption rate constant (k a) and the transit-compartment rate constant (K TR) were fixed to be identical due to the inability to estimate them separately. BSV was estimated for all structural parameters, except for elimination clearance of ARS which was estimated with poor precision (> 50% RSE). Incorporating BOV had a significant impact on both MTT (∆OFV = − 127) and F (∆OFV = − 916). Body weight, implemented allometrically on all clearance and volume parameters, resulted in an improved model fit (∆OFV = − 6.5). Malaria infection had a significant impact on MTT (∆OFV = − 48.1), resulting in a linear 10% decrease in MTT with every log increase in parasite density. Observed parasite densities also had a significant impact on F (implemented as an Emax-type relationship; ∆OFV = − 72.4), resulting in increasing drug exposure with increasing parasite densities. Eta shrinkages (BSV) computed in the final PK model were relatively low (V ARS = 4.24%, V DHA = 0.01%, MTT = 37.8%, and F = 35.6%). Eta shrinkages (BOV) for MTT and F were around 30% for occasion 1–4 but higher than 50% for occasion 5–6, due to the relative sparseness of data at later dose occasions. Computed epsilon shrinkage was 20.0 and 21.0% for ARM and DHA, respectively.

Visual predictive check of final population pharmacokinetic model of artesunate (a) and its active metabolite dihydroartemisinin (b), and population pharmacodynamic model stratified by site (c; Pailin, Cambodia, and d; Wang Pha, Thailand), in patients with uncomplicated falciparum malaria. The open circles are observed data; solid black lines represent the 50th percentiles of observed data; dashed black lines represent the 5th and 95th percentiles of observed data for the pharmacokinetic model (a and b) and the pharmacodynamic model (c and d); shaded areas are the 95% confidence intervals of the simulated percentiles; vertical gray lines represent the lower limit of quantification (LLOQ) for ARS (1.2 ng/ml), DHA (2.0 ng/ml), and parasite density (107.93 [maximum parasite biomass for 16 parasite/μL]). The lower panels show the fraction of observed data below the LLOQ (open circles) overlaid with the 95% prediction interval of the fraction of simulated data below the LLOQ (shaded area)
Population Pharmacokinetics-Pharmacodynamic Relationships
The first-order conditional estimation method with interactions and the M3-method produced similar parameter estimates, with no bias in LOQ data when treated as missing or categorical data. Thus, the first-order conditional estimation method was used throughout modeling since the M3-method was somewhat less stable and produced substantially longer computational run-times. A delayed concentration-response model (i.e., hysteresis model) showed a significantly better model fit compared to the direct concentration-response model. The introduction of a mixture model on Emax resulted in a significantly better model fit and was able to describe the bimodality of the parasite clearance profiles and resulted in a significantly better model fit (ΔOFV = − 11.3). There was no significantly improved model fit when the mixture model was implemented on EC50 alone compared to a model without a mixture model (ΔOFV = − 2.53). Furthermore, there was no further improvement on model-fit when a mixture model was implemented on both Emax and EC50 compared to when implemented on Emax alone. Site was found to be correlated with the bimodal distribution, and it significantly improved the model when it was introduced as categorical covariate to the mixture probability (ΔOFV = − 14.0). No other covariates were found to be significant in this analysis. The final model estimated 84% of the patients in Pailin (Cambodia) to have artemisinin-resistant infections compared to only 16% of patients in Wang Pha (Thailand). The EC50 was estimated to be 9.81 ng/mL (34.5 nM), but the uncertainty in this parameter was relatively high (31.7% RSE). The half-life of effect compartment was 9.66 h. The final model demonstrated reasonable parameter precision (Table IV) and a good predictive performance (Fig. 3).
Simulation of Alternative Dosing Regimens for Patients with Resistant Infections
As expected, the predicted mean residual parasite density 24 h after the last dose was substantially lower in patients with sensitive infections (6.0 × 104) compared to those with resistant infections (5.1 × 107), assuming standard dosing (4 mg/kg of artesunate administered once daily for 3 days). Increased dosing frequency (2 mg/kg of artesunate administered twice daily for 3 days) resulted in negligibly improved residual resistant parasite densities (4.4 × 107). Similarly, a higher dosage (8 mg/kg of artesunate administered once daily for 3 days) had a minor impact on the residual resistant parasite densities (2.3 × 107). However, a substantial improvement was seen with increased treatment duration (4 mg/kg of artesunate administered once daily for 5 days) resulting in comparable residual parasite densities in patients with resistant infections (2.2 × 105) to that seen after standard treatment in patients with sensitive infections (6.0 × 104). Thus, the residual parasite density, to be eliminated by the partner drug, was considerably higher in patients with resistant infections compared to patients with sensitive infections and was not improved substantially by increased dosing frequency or increased dosage over 3 days. A standard dosage administered over 5 consecutive days was predicted to be the most effective method of reducing the increased burden of residual parasite densities in patients with resistant infections.
Resistance Nomogram
The evaluation of the developed static nomogram showed that the ratio between baseline parasite density and parasite density on day 2 post-dose (i.e., 48-h post-dose) performed better than when using parasite density measurements on day 1 or day 3 post-dose. The sensitivity was ~ 70% with a type II error rate (i.e., resistance incorrectly identified) of 29.2% (Table V).
The performance was improved further when the nomogram was optimized to take into account the observed parasite density at baseline (baseline-adapted nomogram).
For optimal performance, the nomogram should be performed on day 1 (24-h post-dose) for patients with baseline densities of 109 to 1010.5 (“cutoff” value of 1.46; sensitivity of 62.6 to 74.7%). For baseline densities of 1011 to 1012.0, optimal performance was found on day 2 (“cutoff” value of 2.93; sensitivity of 80.7 to 84.1%). The performance for patients with hyper-parasitemia (i.e., baseline parasite density ≥ 1012.0) was optimal when performed on day 3 (“cutoff” value of 4.34; sensitivity > 85%). The nomogram performance for low baseline density of < 109 was not satisfactory with sensitivity below 30.0%. Figure 4 illustrates the performance of the nomogram on day 1, 2, and 3 post-dose for each parasite baseline density.

Overall nomogram performance (i.e., sensitivity) versus baseline parasite density stratified on when the nomogram evaluation was performed (i.e., 24-h post-dose [red], 48-h post-dose [green], and 72-h post-dose [blue]) within this study context
The overall sensitivities of the developed predictive tools were 98, 80, 71, and 27% for applying the model-based analysis, baseline-adapted nomogram, static nomogram, and day 3 positivity test, respectively. Consistent with this, when the developed baseline-adapted nomogram was applied on the observed parasite density measurements from the clinical trial, sensitivity was predicted at 83% with accuracy and specificity of 81 and 87%, respectively.
DISCUSSION
The emergence of artemisinin resistance in Southeast Asia is of great concern, and it must be contained and then eliminated before spreading to other parts of the world. It is important to characterize the concentration-response relationship of artemisinin derivatives in resistant and sensitive malaria infections and to develop new tools to identify artemisinin resistant infections early and accurately in field settings.
Population Pharmacokinetics
A simple one-compartment model was employed to describe the concentration-time profiles of ARS and DHA, which was in agreement with previous studies (38). However, in some reports, peripheral compartment structures have been proposed for DHA. Differences are likely to reflect sampling methodologies and data censoring due to quantification issues (39,40). Malaria infection (measured by the parasite density) had a significant impact on both absorption rate and relative bioavailability of ARS, resulting in increased absorption during the acute malaria infection compared to the convalescent phase. Enzyme auto-induction cannot be excluded as an underlying cause to the time-dependent difference in exposure. However, previous detailed PK studies of oral and parenteral administration of ARS have demonstrated a clear malaria effect on the relative bioavailability thought to result from reduced first pass metabolism (40,41). The decreased absorption rate with increasing parasite density was unexpected and opposite to previous observations (39,42,43,44,45,46,47,48). The underlying mechanism of this effect cannot be elucidated from data collected in this study. Reported PK parameter estimates were in agreement with those previously reported and also with that obtained from a model-independent analysis of the same data (4).
Population Pharmacokinetics-Pharmacodynamics
Parasite quantification by microscopy is prone to both systemic and random errors, especially at low parasite densities due to observational bias (49,50). To minimize the effect of this potential observational bias, only the first parasite count of a series of low parasite counts was retained in the data (51). DHA, when implemented in an Emax model, showed a clear concentration-response relationship as described previously (33). A sub-population of patients having artemisinin-resistant infections was supported by the implementation of a mixture model on Emax. The resistance mechanism is not known currently, but it was clear from data presented here that Emax rather than EC50 (potency) was affected by this resistance mechanism. This was also supported by a recent clinical trial by Das et al. (13) demonstrating no clinical benefit from an increased dose (8 mg/kg/day), as could have been expected if the potency would have been affected.
A high proportion of patients in Cambodia (84%) and a lower proportion of patients in Thailand (16%) were estimated to have an artemisinin-resistant infection, which is in agreement with resistance prevalence reported at the time of the study period (4). Cambodia is a historical breeding ground for antimalarial drug resistance development. Since the time of sampling, artemisinin resistance and the related Pf kelch13 mutation have spread throughout the Greater Mekong sub-region and efforts are ongoing to contain/eliminate artemisinin-resistant malaria before resistant parasites spread further (9). In addition, molecular genotyping to assess antimalarial drug resistance selection reported that the majority of recent clinical isolates (2014–2015) from northeastern Thailand, Laos, and western Cambodia suggest a hard transnational selective sweep originating from Cambodia in 2008 (52). Thus, exposure-response resistance modeling conducted on these data should therefore be relevant for the Greater Mekong Sub-region.
There was a significant delay in drug effect, characterized by a theoretical effect compartment. The most likely explanation to this apparent and varying delay in drug effect might be the in vivo stage of development and synchronicity of the infection in falciparum malaria patients. Some patients may present with infections undergoing synchronous schizont rupture causing a rise in parasite numbers, while others may have declining parasitemias as a result of sequestration. Thus, the net-effect of drug-dependent parasite elimination will be different in patients presenting with malaria at different stages of the life cycle and therefore create an apparent delayed drug effect in certain patients. The in vivo EC50 value estimated here was 34.9 nM (9.92 ng/ml), which is 20–30-fold higher compared to in vitro EC50 values (range: 0.12–4.56 nM) from a variety of field isolates and laboratory strains (53,54,55). DHA is 93% protein-bound in patients with malaria infection, which could explain part of the differences seen between in vitro results and in vivo results obtained here (56). Furthermore, the EC50 value estimated in this study should be interpreted with caution due to the high uncertainty of the estimate. A substantial number of patients with recrudescent infections would be needed for a solid estimation of this parameter (57). Current methods of assessing artemisinin pharmacodynamics assume concentration-dependent parasite killing with or without hysteresis. These do not satisfactorily explain why increasing dosing frequency does not increase parasite killing substantially and therefore accelerate parasite clearance and augment cure rates (13). Saturation of splenic clearance with accumulation of dead parasites in the circulation has been proposed to explain these apparent contradictions (58,59) but there is no evidence for this (51). The constructs used in the current and previous studies may be oversimplifications. A second limitation is the pooling of artemisinin resistance in a single category whereas it is becoming increasingly apparent that there are significant phenotypic differences between infections with the different kelch mutations (9,51) and that other genetic factors may modify the phenotype.
Simulation of Alternative Dosing Regimens for Patients with Resistant Infections
The slower parasite clearance rates (decreased Emax) associated with artemisinin resistance mean that a higher parasite biomass has to be eliminated by the partner drug. This generates an increased risk of treatment failure and drives the development of partner drug resistance. Different dose regimens (i.e., increased dosing frequency, increased dosage, and increase duration of treatment) were explored at the population level to evaluate potentially improved treatment regimens in patients with artemisinin-resistant infections. Simulations performed here suggested that increased dosage and increased dosing frequency would not improve the parasite clearance substantially. These observations were in line with the outcome of a recent clinical trial conducted in a similar study site (i.e., Pailin, Cambodia, and Wang Pha, Thailand), demonstrating no clinical benefit of twice daily doses or an increased dosage of up to 8 mg/kg per day (13). The simulations indicate that increased dose duration of up to 5 days of consecutive treatment is required to ensure that the residual parasite burden is reduced effectively in patients with resistant malaria infections. Five days of treatment is expected to cover up to three parasite life cycles (> 96 h) compared to two life cycles covered by standard 3-day treatment. These predictions assume that PK-PD properties estimates based on the standard 3-day dosing regimen can be extrapolated to a 5-day dosing regimen. However, the pharmacokinetic samples collected here did not display time-dependent properties. Furthermore, patients are likely to be symptom-free and have relatively low parasite densities at day 3 so it is a reasonable assumption that there will be no major differences in the pharmacokinetics between day 3 and day 5. It is of course more difficult to extrapolate the parasite killing beyond that what can be measured by microscopy, but data presented here did not demonstrate any parasite density-dependent effects on the killing of parasites. These results are consistent with the excellent efficacy demonstrated in the recent TRAC trial in which a 3-day course of artesunate alone preceded the 3-day ACT regimen (9).
Resistance Nomogram
Microscopy-based day 3 parasite positivity has been used clinically to identify artemisinin-resistant infections. However, this analysis does not take into account the baseline parasite density which is directly proportional to the parasite clearance time, assuming similar drug-dependent elimination of parasites. The developed nomogram (baseline-adapted and unadjusted nomogram) performed better than the commonly used day 3 positivity test, and it proved to have high sensitivity in identifying patients with artemisinin-resistant infections. The kelch 13 genotype is a generally accepted molecular marker for artemisinin resistance, but it is difficult to implement a PCR-based test in field settings which often lack the necessary equipment. The developed nomogram suggested here is a sensitive and field adapted alternative to the day 3 positivity test, although it requires further validation in different epidemiological settings with different levels of immunity (60) and different kelch mutations, and thus different degrees of slow parasite clearance conferred. A simple decision tree for the implementation of the resistance nomogram based on this study’s results is proposed in Fig. 5.

Decision tree for the implementation of the nomogram in order to identify patients with artemisinin-resistant malaria infections in this study context
CONCLUSION
A PK-PD model describing the PK and PD properties of ARS treatment in patients with artemisinin-resistant and sensitive-falciparum malaria infections was developed. The developed model could distinguish adequately between patients with artemisinin resistant and sensitive infections and predicted a high proportion of patients with resistant infections in Cambodia. Simulations, using the final PK-PD model, suggested that increased treatment duration is needed in order to treat patients with resistant infections, to achieve equivalent residual parasite densities to those in patients with sensitive infections. A nomogram was developed to provide a basis for a field-adapted tool to identify patients with artemisinin-resistant infections. The proposed nomogram showed better performance compared to the commonly used day 3 positivity test and if validated in different epidemiological contexts could be a useful tool in field settings.
References
WHO. World Malaria report. Geneva: WHO; 2015.
WHO. Guidelines for the treatment of Malaria. Geneva: WHO; 2015.
Meshnick SR. Artemisinin: mechanisms of action, resistance and toxicity. Int J Parasitol. 2002;32(13):1655–60.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455–67.
Denis MB, Tsuyuoka R, Poravuth Y, Narann TS, Seila S, Lim C, et al. Surveillance of the efficacy of artesunate and mefloquine combination for the treatment of uncomplicated falciparum malaria in Cambodia. Tropical Med Int Health. 2006;11(9):1360–6.
Dondorp AM, Yeung S, White L, Nguon C, Day NPJ, Socheat D, et al. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8(4):272–80.
WHO. Global plan for artemisinin resistance containment (GPARC). Geneva: WHO; 2011.
Phyo AP, Nkhoma S, Stepniewska K, Ashley EA, Nair S, McGready R, et al. Emergence of artemisinin-resistant malaria on the western border of Thailand: a longitudinal study. Lancet. 2012;379(9830):1960–6.
Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371(5):411–23.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014;505(7481):50–5. https://doi.org/10.1038/nature12876.
Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, et al. Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis. 2016;16(3):357–65.
Phyo AP, Ashley EA, Anderson TJC, Bozdech Z, Carrara VI, Sriprawat K, et al. Declining efficacy of artemisinin combination therapy against P. falciparum malaria on the Thai–Myanmar border (2003–2013): the role of parasite genetic factors. Clin Infect Dis. 2016;63(6):784–91.
Das D, Tripura R, Phyo AP, Lwin KM, Tarning J, Lee SJ, et al. Effect of high-dose or split-dose artesunate on parasite clearance in artemisinin-resistant falciparum malaria. Clin Infect Dis. 2013;56(5):e48–58.
Lourens C, Lindegardh N, Barnes KI, Guerin PJ, Sibley CH, White NJ, et al. Benefits of a pharmacology antimalarial reference standard and proficiency testing program provided by the worldwide antimalarial resistance network (WWARN). Antimicrob Agents Chemother. 2014;58(7):3889–94.
Lindegårdh N, Dondorp AM, Singhasivanon P, White NJ, Day NPJ. Validation and application of a liquid chromatographic–mass spectrometric method for determination of artesunate in pharmaceutical samples. J Pharm Biomed Anal. 2007;45(1):149–53.
Hanpithakpong W, Kamanikom B, Dondorp AM, Singhasivanon P, White NJ, Day NPJ, et al. A liquid chromatographic-tandem mass spectrometric method for determination of artesunate and its metabolite dihydroartemisinin in human plasma. J Chromatogr B. 2008;876(1):61–8.
Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM user's guides. (1989-2009). Icon development solutions, Ellicott City, MD, USA; 2009.
Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)—a perl module for NONMEM related programming. Comput Methods Prog Biomed 2004;75(2):85–94.
Lindbom L, Pihlgren P, Jonsson EN. PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Prog Biomed. 2005;79(3):241–57.
Hooker AC, Karlsson MO, Wilkins JJ, Jonsson EN. Xpose4: tools for nonlinear mixed-effect model building and diagnostics. R package version 4.5.3. 2014.
Jonsson ENKM. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Prog Biomed. 1999;58(1):1–64.
Keizer RJKM, Hooker AC. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50.
R Core Team. R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2013.
Hadley Wickham WC. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2009.
Klayman D. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228(4703):1049–55.
Luo X-D, Shen C-C. The chemistry, pharmacology, and clinical applications of qinghaosu (artemisinin) and its derivatives. Med Res Rev. 1987;7(1):29–52.
Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35(4):401–21.
Jusko WJ. Use of pharmacokinetic data below lower limit of quantitation values. Pharm Res. 2012;29(9):2628–31.
Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS J. 2009;11(2):371–80.
Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711–26.
Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30(6):387–404.
White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113(8):1084–92.
Patel K, Simpson JA, Batty KT, Zaloumis S, Kirkpatrick CM. Modelling the time course of antimalarial parasite killing: a tour of animal and human models, translation and challenges. Br J Clin Pharmacol. 2015;79(1):97–107.
Patel K, Batty KT, Moore BR, Gibbons PL, Bulitta JB, Kirkpatrick CM. Mechanism-based model of parasite growth and dihydroartemisinin pharmacodynamics in murine malaria. Antimicrob Agents Chemother. 2013;57(1):508–16.
Savic RM, Karlsson MO. Importance of shrinkage in empirical Bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11(3):558–69.
Dosne A-G, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn. 2016;43(6):583–96.
Stepniewska K, Ashley E, Lee SJ, Anstey N, Barnes KI, Binh TQ, et al. In vivo parasitological measures of artemisinin susceptibility. J Infect Dis. 2010;201(4):570–9.
Morris CA, Onyamboko MA, Capparelli E, Koch MA, Atibu J, Lokomba V, et al. Population pharmacokinetics of artesunate and dihydroartemisinin in pregnant and non-pregnant women with malaria. Malar J. 2011;10(1):1–11.
Tan B, Naik H, Jang I-J, Yu K-S, Kirsch LE, Shin C-S, et al. Population pharmacokinetics of artesunate and dihydroartemisinin following single-and multiple-dosing of oral artesunate in healthy subjects. Malar J. 2009;8:304.
Tarning J, Rijken MJ, McGready R, Phyo AP, Hanpithakpong W, Day NP, et al. Population pharmacokinetics of dihydroartemisinin and piperaquine in pregnant and nonpregnant women with uncomplicated malaria. Antimicrob Agents Chemother. 2012;56(4):1997–2007.
Tarning J, Chotsiri P, Jullien V, Rijken MJ, Bergstrand M, Cammas M, et al. Population pharmacokinetic and pharmacodynamic modeling of amodiaquine and desethylamodiaquine in women with Plasmodium vivax malaria during and after pregnancy. Antimicrob Agents Chemother. 2012;56(11):5764–73.
Le Thi DT, Le NH, Nguyen CH, Phan Thi D, Na-Bangchang K. Pharmacokinetics of a five-day oral dihydroartemisinin monotherapy regimen in patients with uncomplicated falciparum malaria. Drug Metab Pharmacokinet. 2008;23(3):158–64.
Diem Thuy LT, Ngoc Hung L, Danh PT, Na-Bangchang K. Absence of time-dependent artesunate pharmacokinetics in healthy subjects during 5-day oral administration. Eur J Clin Pharmacol. 2008;64(10):993–8.
Gordi T, Xie R, Huong NV, Huong DX, Karlsson MO, Ashton M. A semiphysiological pharmacokinetic model for artemisinin in healthy subjects incorporating autoinduction of metabolism and saturable first-pass hepatic extraction. Br J Clin Pharmacol. 2005;59(2):189–98.
McGready R, Phyo AP, Rijken MJ, Tarning J, Lindegardh N, Hanpithakpon W, et al. Artesunate/dihydroartemisinin pharmacokinetics in acute falciparum malaria in pregnancy: absorption, bioavailability, disposition and disease effects. Br J Clin Pharmacol. 2012;73(3):467–77.
Khanh NX, de Vries PJ, Ha LD, van Boxtel CJ, Koopmans R, Kager PA. Declining concentrations of dihydroartemisinin in plasma during 5-day oral treatment with artesunate for falciparum malaria. Antimicrob Agents Chemother. 1999;43(3):690–2.
Binh TQ, Ilett KF, Batty KT, Davis TM, Hung NC, Powell SM, et al. Oral bioavailability of dihydroartemisinin in Vietnamese volunteers and in patients with falciparum malaria. Br J Clin Pharmacol. 2001;51(6):541–6.
Hong X, Liu CH, Huang XT, Huang TL, Ye SM, Ou WP, et al. Pharmacokinetics of dihydroartemisinin in Artekin tablets for single and repeated dosing in Chinese healthy volunteers. Biopharm Drug Dispos. 2008;29(4):237–44.
Ohrt C, O'Meara WP, Remich S, McEvoy P, Ogutu B, Mtalib R, et al. Pilot assessment of the sensitivity of the malaria thin film. Malar J. 2008;7:22.
Alves ER, Gomes LT, Assis-Oliveira FB, Silverio-Silva LR, Nery AF, Fontes CJ. Quantification of parasite density in 200 microscopic fields underestimates the parasitemia level in malaria patients. Trop Biomed. 2014;31(2):387–91.
White NJ. Malaria parasite clearance. Malar J. 2017;16(1):88.
Imwong M, Suwannasin K, Kunasol C, Sutawong K, Mayxay M, Rekol H, et al. The spread of artemisinin-resistant Plasmodium falciparum in the greater Mekong subregion: a molecular epidemiology observational study. Lancet Infect Dis. 2017;17(5):491–7.
Brockman A, Price RN, van Vugt M, Heppner DG, Walsh D, Sookto P, et al. Plasmodium falciparum antimalarial drug susceptibility on the north-western border of Thailand during five years of extensive use of artesunate-mefloquine. Trans R Soc Trop Med Hyg. 2000;94(5):537–44.
Chaijaroenkul W, Na Bangchang K, Mungthin M, Ward SA. In vitro antimalarial drug susceptibility in Thai border areas from 1998–2003. Malar J. 2005;4(1):1–7.
Jambou R, Legrand E, Niang M, Khim N, Lim P, Volney B, et al. Resistance of plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet 2005;366(9501):1960–3.
Batty KT, Ilett KF, Davis TME. Protein binding and α : β anomer ratio of dihydroartemisinin in vivo. Br J Clin Pharmacol. 2004;57(4):529–33.
Hien TT, White NJ, Thuy-Nhien NT, Hoa NT, Thuan PD, Tarning J, et al. Estimation of the in-vivo minimum inhibitory concentration of cipargamin in uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2016;61(2). https://doi.org/10.1128/AAC.01940-16.
Kay K, Hodel EM, Hastings IM. Altering antimalarial drug regimens may dramatically enhance and restore drug effectiveness. Antimicrob Agents Chemother 2015;59(10):6419–27. https://doi.org/10.1128/AAC.00482-15.
Hastings IM, Kay K, Hodel EM. How robust are malaria parasite clearance rates as indicators of drug effectiveness and resistance? Antimicrob Agents Chemother. 2015;59(10):6428–36.
Ataide R, Ashley EA, Powell R, Chan JA, Malloy MJ, O'Flaherty K, et al. Host immunity to Plasmodium falciparum and the assessment of emerging artemisinin resistance in a multinational cohort. Proc Natl Acad Sci U S A. 2017;114(13):3515–20.
Acknowledgements
The Wellcome Trust-Mahidol University-Oxford Tropical Medicine Research Programme is supported by the Wellcome Trust of Great Britain. The trial was supported by grants from the Wellcome Trust of Great Britain, the Li Ka Shing Foundation, the Bill and Melinda Gates Foundation, and the US Agency for International Development. Jesmin Permala was supported by a scholarship from Ministry of Higher Education (MOHE), Malaysia and University Terknologi MARA, (UiTM) Malaysia. Pascal Ringwald is a staff member of the World Health Organization, and he alone is responsible for the views expressed in this publication and they do not necessarily represent the decisions, policy, or views of the World Health Organization.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lohy Das, J., Dondorp, A.M., Nosten, F. et al. Population Pharmacokinetic and Pharmacodynamic Modeling of Artemisinin Resistance in Southeast Asia. AAPS J 19, 1842–1854 (2017). https://doi.org/10.1208/s12248-017-0141-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-017-0141-1