
Abstract
The nuclear enzyme poly(ADP-ribose) polymerase (PARP) is a key component of molecular mechanisms leading to cell death or survival after an ischemic insult. Oxidative stress damages DNA, and breaks in the DNA strands activate PARP enzyme, leading to poly(ADP-ribosyl)ation of nuclear proteins. In this study, we investigated PARP activation using immunodetection of PAR polymers in the brain of neonatal rat pups subjected to unilateral focal ischemia with reperfusion. PARP activation was detected in the ischemic core between 2 and 18 h, and in the penumbra between 24 and 48 h in the middle cerebral artery (MCA) territory but also in territories of the anterior and posterior cerebral artery, and in white matter tracts. The intranuclear accumulation of PAR in cells preceded a positive terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick-end labeling, suggesting that PARP activation may actually contribute to delayed cell death. Pretreatment with 3-aminobenzamide (3-AB, 10 mg/kg) strongly reduced PARP activation and cell death. These data suggest that PARP activation represents, in the immature brain, the early sign of ischemic cell death. This raises the possibility of the use of PARP inhibitors not only immediately postischemia but perhaps also later to reduce ischemic lesion in the MCA territory and its connected structures.
Similar content being viewed by others

Main
Cerebral infarction during the perinatal period remains a major cause of mortality and morbidity in newborn infants (1, 2). Survivors of perinatal asphyxia have moderate to severe brain injury (3) for which there currently is no promising therapy. Animal research on various models mimicking hypoxic-ischemic encephalopathy is therefore required to describe the molecular pathways involved in cell death and represents the first step to better delineate the possible field of application and best timing of administration for potential neuroprotective drugs (4).
Recent studies reported that the nuclear enzyme PARP-1 (EC 2.4.2.30) was a key component of molecular mechanisms leading to cell death or survival after an ischemic insult (5–7). PARP is activated in response to DNA damage, and adds multiple ADP-ribose groups to various nuclear proteins (such as histones, topoisomerase, DNA-polymerase, transcriptional factors, and PARP itself) involved in DNA repair (8, 9). However, PARP overactivation by major DNA damage can deplete tissue stores of ATP and NAD+ and may lead to cell death by energy failure (10–12). PARP inactivation by caspase-3-mediated cleavage during the early stage of apoptosis facilitates DNA alteration and nuclear disassembly and ensures the completion of the apoptotic process, which is energy dependent (6, 13, 14). PARP is then at the parting of the ways of major biochemical pathways regulating DNA repair or cell death. Recently, PARP has been demonstrated to be an exciting molecular target for neuroprotective drugs, some of which have already shown promising results in animal models (15–17) (for review, see Ref. 7). Because of the complex regulation of PARP activity during ischemia, PARP may seem to be protective or deleterious, depending on the animal model (6, 18). Conversely, modulation of PARP activity could avoid or delete one type of cell death but precipitate another (19).
In an attempt to better understand the pathophysiological role of PARP activation, the aims of the present study were to determine the following: 1) the presence of increased quantities of functional PARP as evidenced by intranuclear accumulation of PAR polymers; 2) its distribution in the ipsilateral hemisphere in relation with subsequent cell death; and 3) the effect of PARP inhibition on cell death occurrence, in a model of neonatal stroke.
MATERIALS AND METHODS
Animal model of cerebral ischemia.
All animal experimentation was carried out with the highest standards of care, approved by the French Ethical Science Committee (statement no. 04223). Ischemia was performed in 7-d-old Wistar rats (weight, 17–21 g) of both sexes (R. Janvier, Le Genest-St-Isle, France), as previously described (20). Rat pups were anesthetized with intraperitoneal injection of chloral hydrate (350 mg/kg). After 15 min, rats were positioned on their back and a median incision was made in the neck to expose left common carotid artery. Rats were then placed on the right side and an oblique skin incision was made between the ear and the eye. After excision of the temporal muscle, the cranial bone was removed from the frontal suture to a level below the zygomatic arch. Then, the left middle cerebral artery, exposed just after its apparition over the rhinal fissure, was coagulated at the inferior cerebral vein level. After this procedure, a clip was placed to occlude the left common carotid artery and was removed after 50 min. Carotid blood flow restoration was verified with the aid of a binocular loupe. Both neck and cranial skin incisions were then closed. During surgery, body temperature was maintained at 37–38°C. After waking, rat pups were transferred to their mother for long-time survival. Naive and sham age-matched rats served as controls.
In vivo administration of PARP inhibitor.
Three-aminobenzamide (3-AB, Sigma Chemical, St. Louis, MO, U.S.A.), one of the most frequently used agents to inhibit PARP, at a concentration of 10 mg/kg was dissolved in 0.9% NaCl and administrated intraperitoneally immediately after the clip was placed on the left common carotid artery. This dose has been previously demonstrated to significantly reduce the cortical infarct volume (17). Five animals were injected similarly with equivalent volume of the vehicle in the same schedule. Rat pups were then killed at 6, 24, and 48 h or 20 d after reperfusion (five at each time point).
Tissue preparation.
Control rats and ischemic rats at 2, 6, 12, 18, 24, 48, and 72 h after reperfusion (four at each time point) were anesthetized (chloral hydrate, 300 mg/kg) and perfused via the ascending aorta with warm heparinized saline followed by phosphate-buffer (PB, 0.12 M, pH 7.4) containing 4% paraformaldehyde. Brains were then removed, kept for 2 h in the same fixative solution, and placed in 0.12 M PB containing 10% sucrose for 3 d. Brains were rapidly frozen in isopentane (−40°C), and stored at −70°C until used. Coronal cryostat sections (20 μm thick) were collected on gelatin-coated slides. Some sections were stained with cresyl violet to delineate the cortical infarct.
Measurement of infarct and cavity volume.
On each section stained with cresyl violet (at 1-mm intervals), area of infarction (at 48 h) or cavity (at 3 wk) was measured using an image analyzer (IMSTAR, Paris, France). The volume was calculated by integrating the areas. Data are presented as mean ± SEM (n = 6).
Silver staining procedure.
The method of Gallyas et al.(21) was used to visualize both degenerating terminals and cell bodies of neurons, and lysosomes that bind silver (i.e. become “argyrophilic”). Briefly, the staining procedure included alkaline pretreatment, silver impregnation, and development at pH between 5.5 and 6.3, then washing in acetic acid, dehydration, and mounting in Permount.
In situ labeling of fragmented DNA.
Coronal cryostat sections were processed for TUNEL assay as previously reported (22). Briefly, sections were incubated with terminal deoxynucleotidyl transferase (0.2 U/μL; Invitrogen, Carlsbad, CA, U.S.A.) and biotin-16-dUTP (20 μM; Roche, Meylan, France), then visualized with streptavidin-biotin-peroxidase complex, hydrogen peroxide, and diaminobenzidine. Cells exhibiting DNA fragmentation (TUNEL-positive) were counted in the cortical infarct at a 40× magnification in at least three separate experiments by counting cells in three distinct section areas delineated by an ocular grid. Number of cells was calculated per 0.2 mm2. Data are presented as mean ± SEM. Statistical comparisons between two groups (untreated and 3-AB-treated ischemic rats) were made by two-tailed t test.
PAR polymer immunohistochemistry.
Rabbit antibodies against PAR polymer (minimum subunit length of 8) were purchased from BIOMOL Research Laboratories (Tebu, France). Sections were incubated with the primary antibody (1:200 dilution) in PBS 0.1 M containing 0.5% gelatin and 0.3% triton-X100 overnight at room temperature. The secondary anti-rabbit biotinylated antibodies (1:400 dilution, DAKO, Trappes, France) were visualized by the avidin-biotin peroxidase (Elite ABC kit, Vectastain, AbCys, Paris, France) and diaminobenzidine. Nonspecific peroxidase activity was abrogated by incubating the sections in 1% hydrogen peroxide in PBS 0.1 M at the appropriate stage. As negative controls, alternative sections were incubated in the absence of primary antibody. Several sections were double-immunostained for glial fibrillary acidic protein (GFAP, Roche) and revealed by the avidin-biotin alkaline phosphatase (ABC kit, Vectastain) followed by immunodetection of PAR polymer, as described above. Sections used in the figures are representative of the four animals analyzed in each group at each time point. PAR-positive nuclei were counted as described above.
Gel electrophoresis of nuclear proteins.
Nuclear extracts of cortical infarct tissue obtained from ischemic and 3-AB (3-aminobenzamide) treated rats (n = 3 each) at 48 h reperfusion were prepared as described previously (23). Briefly, tissues (50 mg) were solubilized in 500 μL of buffer containing 10 mM HEPES (pH 7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM DTT, 0.5 mM phenylmethyl sulfonyl fluoride (PMSF), and the protease inhibitor cocktail (Roche). Homogenates were kept on ice for 10 min and NP-40 (2.5% final) was added, then centrifuged at 8000 ×g for 2 min. The cleared cytosolic fraction was removed and the pelleted nuclei were resuspended in 100 μL of buffer containing 20 mM HEPES (pH 7.9), 0.4 M NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 10 mM DTT, 0.5 mM PMSF, and protease inhibitors. The nuclear fraction was shaken vigorously for 20 min at 4°C. The nuclear suspension was cleared by centrifugation at 13,000 ×g for 5 min at 4°C. Sample proteins (50 μg) were resolved on a 7.5% SDS-PAGE gel and electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked with 5% nonfat dry milk in TBS containing 0.1% Tween 20 and were probed with anti-PAR antibodies (1:3000 dilution) overnight at 4°C. After incubation with goat anti-rabbit-horseradish peroxidase (1:75,000, Amersham Pharmacia Biotech, Saclay, France), proteins with PAR polymers were revealed with enhanced chemiluminescence (ECL+, Amersham Pharmacia Biotech). Quantitative analysis was performed by the “quantity one” program (GelDoc 2000, Bio-Rad S A, Marnes-la-Coquette, France).
RESULTS
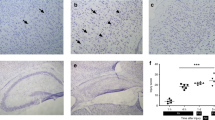
Permanent left MCA occlusion in association with transient occlusion of the left common carotid artery produced a reproducible well-delineated cortical lesion with an infarct volume of 64 ± 8 mm3 at 48 h of reperfusion. Within the cortical infarct, a core and a so-called penumbra zone (Fig. 1a) have been distinguished, and the former evolved into a cavity 7 d after recovery (Fig. 1b).

Histopathological changes after ischemia on postnatal d 7. Representative cresyl violet-stained brain sections from animals killed at 48 h and 7 d of recovery. (a) The cortical infarct at 48 h. Note that the core is pale compared with the penumbra (P). (b) At 7 d of recovery, the core disappears and leads to a cavity (indicated by *). C, core;P, penumbra. Bar represents 200 μm.
Distribution of PAR polymers in cells and nerve fiber.
In the cerebral cortex of control and sham animals, no poly(ADP-ribosyl)ation was detected (Fig. 2a). In all ischemic rat pups, PAR-positive cells, identified by a strong immunoreactivity in the nuclei, were observed as early as after 2 h of reperfusion in the cortical infarct. Their number increased in the ischemic core and peaked at 6 h (Table 1 and Fig. 2b); PAR-positive nuclei were maintained in the core but decreased with time reperfusion (Table 1). From 24 to 48 h, PAR-immunostained nuclei were mainly observed within the penumbra (arrowheads in Fig. 2c). PAR staining was predominantly nuclear and involved 83% of cells with explicit nuclear condensation and segmentation (white arrow in Fig. 2d). At 72 h, labeling of PAR-positive nuclei was restricted to a few cells (three to five) in the margin of the penumbra. From this time point, the immunoreactivity of PAR completely disappeared in the ipsilateral cortex. In double-staining experiments, accumulation of PAR polymers was detected in neurons, during the first 18 h of reperfusion, and in several glial cells present in the penumbra at 48 h of recovery (not shown).

Accumulation of PAR polymers in the ischemic cortex. (a) Contralateral cortex. (b) PAR immunostained nuclei in the ipsilateral cortex (core of the infarct) at 6 h of reperfusion (arrowheads at example). (c) PAR immunostained nuclei in the penumbra (P) at 24 h of reperfusion. Arrowheads show PAR-positive nuclei and arrows demonstrate fragmented PAR-positive nuclei. Note that no PAR-positive nuclei are detected in the core (C) of the infarct. (d) High magnification of PAR-labeled nuclei. Note the presence of both large (white arrow), with characteristic features of apoptosis (nuclear condensation and segmented chromatin), and small PAR-positive nuclei (arrows). Some could represent fragmented nuclei. Bars represents 24 (a–c) and 8 (d) μm.
A transient staining of PAR polymers in several cells was detected in the Sommer sector (CA1-CA3 field) of the hippocampus between 18 and 24 h of recovery (Fig. 3a), in the central amygdaloid nucleus at 24 h, and in the ventrolateral thalamus nucleus at 48 h of reperfusion (Fig. 3c), but not in the contralateral hemisphere. In many of these cells, the cytoplasm was immunostained. Staining disappeared at later time point of recovery (72 h). In numerous ischemic rat pups the cerebral periventricular white matter contained enhanced PAR immunoreactivity compared with contralateral side. This appeared as a punctuate staining along fibers in the corpus callosum (sometimes crossing the midline but never extending laterally), external capsule (not shown) and fimbria at 24 h of reperfusion (arrows in Fig. 3, b and d).

Accumulation of PAR polymers in the ipsilateral hemisphere. PAR-positive nuclei could be detected in the CA1 field of the hippocampus (a) between 18 and 24 h, and the ventrolateral thalamus nucleus (c) at 48 h of recovery. Note that in the thalamus, PAR staining was detected either in the nuclei (arrows) or the cytoplasm (arrowheads indicated labeled cells with a clear nucleus). PAR staining in the corpus callosum (b) and the fimbria (d). Note that the labeling is punctuate and followed the fibers (arrows) Arrowheads indicate twi PAR-positive nuclei (b). Bar represents 24 μm.
Distribution of delayed cell death.
With the selective Gallyas silver staining, several silver-impregnated cell bodies in the ipsilateral cortex, hippocampus, thalamus, amygdala, and degenerating processes in corpus callosum were observed (not shown). In sham-operated controls, as in the contralateral cortex of ischemic brains, occasional TUNEL-positive nuclei (two to three per coronal section) were seen in the middle layers (II and III) of the cortex. This may represent a very low rate of programmed cell death occurring in the P7 brain. In contrast, TUNEL-positive nuclei significantly increased with reperfusion time between 6 and 48 h in the ischemic core (Table 1), the time point for which the infarct was stabilized. At this later time, the number of TUNEL-positive nuclei was found to be higher in the core than in the penumbra (Table 1). DNA fragmentation was also detected in the CA1-CA3 fields of the ipsilateral hippocampus at 24 (Fig. 4a) and 48 h, in the central amygdaloid nucleus at 24–48 h, and in the ventrolateral thalamus nucleus at 48 h of reperfusion (not shown), and corpus callosum (Fig. 4b).

Representative photomicrographs (of four independent sets of samples) depict TUNEL staining in the ischemic hemisphere of animals treated with saline (a and b) and 10 mg/kg of 3-AB (c and d). CA2 field of the hippocampus (a–c); corpus callosum (b–d). Bar represents 24 μm.
PARP inhibition reduces cell death.
Three-AB administration led to a significant decrease in the damaged volume, as previously described (17). A significant reduction (49% and 41%) in the number of TUNEL-positive nuclei was observed in the ischemic core and penumbra, respectively, in animals treated with the PARP inhibitor (Table 1) at 48 h of reperfusion. This reduction coincided with a 52 ± 4% decrease in PAR signal as quantified after gel electrophoresis of nuclear proteins, However, immunohistochemistry demonstrated that 3-AB induced a delayed time-course of PAR-positive cells, but the maximum of their number was reduced compared with ischemic untreated rats (Table 1). Furthermore, the nuclear labeling was lighter compared with those found in untreated pups (not shown). PAR polymers and TUNEL labeling were not detected in the CA1-CA3 fields of the hippocampus (Fig. 4c), or in the thalamus, amygdala, and white matter tracts (Fig. 4d) at 48 h of reperfusion after 3-AB treatment.
When brains were examined 3 wk after the ischemic onset, ipsilateral hemisphere exhibited a large cavity (36. 8 ± 10.4 mm3, n = 5) in the full-thickness of the frontoparietal cortex (star in Fig. 5a), an enlarged lateral ventricle (LV) and a sclerosed hippocampus (white arrows in Fig. 5e). In 3-AB treated rats, the cortical cavity was very significantly reduced (4.7 ± 0.9 mm3, n = 5) and the LV and hippocampus appeared normal (Fig. 5, b–d).

Histopathological changes of the left cerebral hemisphere after ischemia and 3 wk of reperfusion. (a) A large cavity (star) in an ischemic rat. (b, c) Typical examples of reduced cavity (star) in two animals treated with 10 mg/kg of the PARP inhibitor, 3-AB. (d) The CA2 area of the hippocampus in a 3-AB-treated animal. Note that the pyramidal CA2 layer is normal (black arrow,also in c) compared with the hypercellular and sclerosed CA2 field from an ischemic rat (white arrows in e). Bar represents 192 μm.
DISCUSSION
Activation of the DNA repair-associated enzyme PARP by genomic injury may have divergent results: either DNA repair or consequences related to cellular energy depletion and death. In the current study, we report that neonatal ischemia induces an early and transient intranuclear accumulation of PAR, which leads to subsequent cell death in the ipsilateral hemisphere of rat pups, suggesting an important role of PARP in the pathogenesis of brain damage.
Ischemia in immature brain first triggers an acute cellular response accompanied by production of PAR polymers, suggesting increased amounts of functional enzyme and activation of DNA repair, as previously reported in brains from cardiac arrest patients (24). Poly(ADP-ribosyl)ation of nuclear proteins has been reported to be an immediate covalent and transient post-translational modification of nuclear proteins induced by DNA damage (25). In our model, production of PAR polymers was first observed within the ischemic core (2–12 h), and later within the penumbra (between 24 and 72 h). Early involvement of poly(ADP-ribosyl)ation was reported after focal (10, 11) and global (18) ischemia in adult rats and after cardiac arrest in humans (26). However, detection of PAR polymers as early as 5 min (10) and 1 (18) and 2 h (11) after injury, gradually decreased to not be observed after several (6–16) hours of recovery. By comparison with neurons of adults, immature neurons may synthesize a higher quantity of PARP enzyme, better able to repair DNA damage than terminally differentiated neurons. An age-dependent decline of cellular poly(ADP-ribosyl)ation had previously been found in lymphocytes from rats and humans (27). This burst of PAR synthesis may only be detectable during a short time period and occurred, before the onset of DNA cleavage into high molecular weight (300 and 50 kbp) fragments, the first irreversible event leading to the inevitable cellular demise at 6 h of reperfusion (22). Similarly, PAR polymers were observed in nuclei before a positive TUNEL signal, and the number of PAR-positive nuclei decreases concurrently with an increase of TUNEL-positive nuclei (Table 1), whatever the structure studied. Again, these data suggested that PARP might be activated to repair early and small DNA damage not easily detected, but not excessive DNA strand breaks leading to ischemic death (28). PARP activation was shown to be expressed within condensed and segmented nuclei (Fig. 2d). Increased poly(ADP-ribosyl)ation has been reported in extracts of apoptotic cells (29) and in karyorrhectic neurons of the pontine nucleus in patients with pontosubicular neuron necrosis, a special form of perinatal hypoxic brain damage (30). Apoptosis is associated with effector caspase activation, and PARP cleavage by caspase-3 has been shown to preserve energy stores necessary for apoptotic events (31). Caspase-3 activation (DEVDase activity and immunodetection) and PARP cleavage, now considered as biochemical criterions to characterize apoptotic cell death were found in the cortical infarct of P7 rat pups (31a). Taken together, these data suggested that PARP is regularly activated during neuronal apoptosis. Numerous in vivo and in vitro studies have demonstrated that PARP overactivation causes neuronal injury, whereas PARP inhibitors decrease ischemic damage (for reviews, see Refs. 6 and 7). In contrast to the above, another functional role for PARP activation has been recently reported in a rat model of brief sublethal global ischemia (18). Under these conditions, PARP activation, without subsequent NAD+ depletion (possibly occurring more frequently and at a younger age than in severe ischemia) was beneficial, whereas systemic administration of 3-AB significantly increased the number of dying neurons containing DNA strand breaks in the hippocampal CA1 region. This article underlines that it is not possible to prejudge whether PARP activation is beneficial or not in a given model.
The distribution of regional cell death, demonstrated by DNA fragmentation, indicated that neonatal cerebral ischemia produced neuronal loss first in the cortex and secondary in remote structures containing extensive synaptic connections with it (hippocampus, striatum and thalamus (32, 33) as well as damage to white matter tracts (34). Such lesions involved portions of the MCA territory but also territories of the anterior (ACA) and posterior (PCA) cerebral artery, but did not extended to the contralateral cortex.
Inhibition of PARP activation with 3-AB, one of the first-generation PARP inhibitors, displaying low toxicity in vivo and highly selective for PARP compared with mono (ADP-ribose) transferase (35), significantly decreases PARP activation and cell death in the cortex but also inhibits delayed cell death in remote structures that undergo secondary degeneration induced by cortical infarct. The presence of poly(ADP-ribosyl)ation in neurites in the white matter suggests that PARP likely may play a direct role in modulating damage to these structures, particularly when ischemic injury was more severe. HI-mediated damage to the white matter is important in term human infants and it is particularly prominent in premature infants, resulting in periventricular cerebral leukomalacia. Injury to the white matter in periventricular cerebral leukomalacia is a major contributor to the subsequent development of cerebral palsy (36).
The current study demonstrates the strong involvement of poly(ADP-ribosyl)ation in the development of cerebral infarction, which represents the early sign of cell death implicating neurons, glial cells, and probably oligodendrocytes. Nevertheless, the timing of PARP activation depends on the infarct zone studied, and can be delayed until 72 h in the penumbra. This raises the possibility of the use of PARP inhibitors, not only immediately postischemia but perhaps later to reduce ischemic lesion in the MCA territory and its connected structures. This hypothesis should be tested with proper pharmacologic protocols in animal models.
Abbreviations
- PAR:
-
poly (ADP-ribose)
- PARP:
-
poly (ADP-ribose) polymerase
- TUNEL:
-
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick-end labeling
- MCA:
-
middle cerebral artery
References
Sarnat H, Sarnat M 1976 Neonatal encephalopathy following fetal distress. Arch Dis Child 33: 696–705
Volpe J 1987 Neurology of the newborn. In: Volpe J (ed) Neurology of the Newborn. WB Saunders, Philadelphia, pp 236–280.
Berger R, Garnier Y 1999 Pathophysiology of perinatal brain damage. Brain Res Rev 30: 107–134
STAIR 1999 Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 30: 2752–2758
Scovassi A, Poirier G 1999 Poly(ADP-ribosylation) and apoptosis. Mol Cell Biochem 199: 125–137
Ha H, Snyder S 2000 Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol Dis 7: 225–239
Bürkle A 2001 Physiology and pathophysiology of poly(ADP-ribosyl)ation. Bioessays 23: 795–806
Pieper A, Verma A, Zhang J, Snyder S 1999 Poly(ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 20: 171–181
Javier Oliver F, Menissier-de Murcia J, de Murcia G 1999 Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. Am J Hum Genet 64: 1282–1288
Endres M, Wang Z, Namura S, Waeber C, Moskowitz M 1997 Ischemic brain injury is mediated by the activation of poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab 17: 1143–1151
Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, Ueda K 1998 Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. J Cereb Blood Flow Metab 18: 991–997
Ha H, Snyder S 1999 Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 23: 13978–13982
Lazebnik Y, Kaufmann S, Denoyers S, Poirier G, Earnhaw W 1994 Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371: 346–347
Eliasson M, Sampei K, Mandir A, Hurn PD, Traystman R, Bao J, Pieper A, Wang Z, Dawson T, Snyder S, Dawson V 1997 Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095
Takahashi K, Greenberg J, Jackson P, Maclin K, Zhang J 1997 Neuroprotective effects of inhibiting poly(ADP-ribose) synthase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab 17: 1137–1142
Lo E, Bosque-Hamilton P, Meng W 1998 Inhibition of poly(ADP-ribose) polymerase. Reduction of ischemic injury and attenuation of NMDA-induced neurotransmitter dysregulation. Stroke 29: 830–836
Ducrocq S, Benjelloun N, Plotkine M, Ben-Ari Y, Charriaut-Marlangue C 2000 Poly (ADP-ribose) synthase inhibition reduces ischemic injury and inflammation in neonatal rat brain. J Neurochem 74: 2504–2511
Nagayama T, Simon R, Chen D, Henshall D, Pei W, Stetler R, Chen J 2000 Activation of poly(ADP-ribose) polymerase in the rat hippocampus may contribute to cellular recovery following sublethal transient global ischemia. J Neurochem 74: 1636–1645
Lee Y, Shacter E 1999 Oxidative stress inhibits apoptosis in human lymphoma cells. J Biol Chem 274: 19792–19798
Renolleau S, Aggoun-Zouaoui D, Ben-Ari Y, Charriaut-Marlangue C 1998 A model of transient unilateral focal ischemia with reperfusion in the P7 neonatal rat: morphological changes indicative of apoptosis. Stroke 29: 1454–1461
Gallyas F, Güldner F, Zoltay G, Wolff J 1990 Golgi-like demonstration of “dark” neurons with an argyrophilic III method for experimental neuropathology. Acta Neuropathol 79: 620–628
Charriaut-Marlangue C, Richard E, Ben-Ari Y 1999 DNA damage and DNA damage-inducible protein Gadd45 following ischemia in the P7 neonatal rat. Dev Brain Res 116: 133–140
Glasgow J, Wood T, Perez-Polo J 2000 Identification and characterization of Nuclear factor kB binding sites in the murine bcl-x promoter. J Neurochem 75: 1377–1389
Love S, Barber R, Wilcock G 1998 DNA fragmentation and repair in ischemic brain injury in man. Neuropathol Appl Neurobiol 24: 134–135
Shall S, de Murcia G 2000 Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model?. Mutat Res 460: 1–15
Love S, Barber R, Wilcock G 1999 Neuronal accumulation of poly(ADP-ribose) after brain ischemia. Neuropathol Appl Neurobiol 25: 98–103
Grube K, Bürkle A 1992 Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc Natl Acad Sci U S A 89: 11759–11763
Hou S, Tu Y, Buchan A, Huang Z, Preston E, Rasquinha I, Robertson G, MacManus J 1997 Increase in DNA lesions and the DNA damage indicator Gadd45 following transient ischemia. Biochem Cell Biol 75: 383–392
Negri C, Donzelli M, Bernardi R, Rossi L, Burkle A, Scovassi A 1997 Multiparametric staining to identify apoptotic human cells. Exp Cell Res 234: 174–177
Meng S, Itoh M, Obonai T, Takashima S 2000 Roles of glutamate transporter and receptors, poly (ADP-ribose) polymerase, and transforming growth factor-beta1 in pontosubicular neuron necrosis. J Child Neurol 15: 362–369
Nicholson D, Ali A, Thornberry N, Vaillancourt J, Ding C, Gallant M, Gareau Y, Griffin P, Labelle M, Lazebnik Y 1995 Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376: 37–43
Benjelloun N, Joly L-M, Palmier B, Plotkine M, Charriaut-Marlangue C 2003 Apoptotic mitochrondrial pathway in neuron and astrocyte after neonatal hypoxia-ischemia in the rat brain. Neuropathol Appl Neurobiol, in press
Rice J, Vannucci R, Brierley J 1981 The influence of immaturity on hypoxia-ischemia brain damage in the rat. Ann Neurol 9: 131–141
Tamura A, Tahira Y, Nagashima H, Kirino T, Gotoh O, Hojo S, Sano K 1991 Thalamic atrophy following cerebral infarction in the territory of the middle cerebral artery. Stroke 22: 615–618
Ness JK, Romanko MJ, Rothstein RP, Wood TL, Levison SW 2001 Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev Neurosci 23: 203–208
Szabo C, Dawson V 1998 Role of poly(ADP-ribose) synthase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci 19: 287–298
Volpe J (ed) 1995 Neurology of the Newborn. WB Saunders, Philadelphia
Acknowledgements
The authors thank J. Doublet (Service Audiovisuel, CHU St-Anne, Paris) for photomicrography.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Joly, LM., Benjelloun, N., Plotkine, M. et al. Distribution of Poly(ADP-ribosyl)ation and Cell Death After Cerebral Ischemia in the Neonatal Rat. Pediatr Res 53, 776–782 (2003). https://doi.org/10.1203/01.PDR.0000059751.00465.F6
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000059751.00465.F6
