
Abstract
The PI3K/AKT/mTOR and RAF/MEK/ERK pathways are commonly activated by mutations and chromosomal translocation in vital targets. The PI3K/AKT/mTOR signaling pathway is dysregulated in nearly all kinds of neoplasms, with the component in this pathway alternations. RAF/MEK/ERK signaling cascades are used to conduct signaling from the cell surface to the nucleus to mediate gene expression, cell cycle processes and apoptosis. RAS, B-Raf, PI3K, and PTEN are frequent upstream alternative sites. These mutations resulted in activated cell growth and downregulated cell apoptosis. The two pathways interact with each other to participate in tumorigenesis. PTEN alterations suppress RAF/MEK/ERK pathway activity via AKT phosphorylation and RAS inhibition. Several inhibitors targeting major components of these two pathways have been supported by the FDA. Dozens of agents in these two pathways have attracted great attention and have been assessed in clinical trials. The combination of small molecular inhibitors with traditional regimens has also been explored. Furthermore, dual inhibitors provide new insight into antitumor activity. This review will further comprehensively describe the genetic alterations in normal patients and tumor patients and discuss the role of targeted inhibitors in malignant neoplasm therapy. We hope this review will promote a comprehensive understanding of the role of the PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathways in facilitating tumors and will help direct drug selection for tumor therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the discovery of oncogenes and antioncogenes in the 1980s, the novel targeted treatment of imatinib was first approved in 2001 by the FDA [1,2,3]. The requirement of new anticancer medicines to the molecular groundwork is significantly higher than that of traditional chemotherapeutic drugs, of which their molecular mechanism of action was not identified empirically [3]. Kinases and phosphatases have key roles in controlling cellular functions [4]. There are currently seventy-one kinase inhibitors approved by the Food and Drug Administration (FDA) and 16 attached inhibitors approved by other countries and regions’ regulatory agencies [5]. The PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathways are composed of kinase cascades that are managed by phosphorylation and dephosphorylation by particular kinases, phosphatases, and proteins regulating the exchange [6] (Fig. 1). The PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathways have been extensively studied over the past 25 years [7]. Enormous breakthroughs in component detection and the mechanisms of how the components relay on the signals and mutations result in aberrant signaling and uncontrolled proliferation diseases. Broader perspectives and feedback loops have been identified, leading to more choice in blocking the pathways.

Schematic of the PI3K/AKT/mTOR and Raf/MEK/ERK signaling pathways. Growth factors, hormones, cytokines, GPCRs, and mitogens activate receptor tyrosine kinases (RTKs) recruiting PI3K to attach to the plasma membrane, where PI3K catalyzes PI (4,5) P2 to PI (3,4,5) P3. PTEN suppressed the process, and PTEN mutations could induce abnormal activation. PI (3,4,5) P3 promotes AKT activation via the activity of PDK1 and mTORC2. AKT activation induced cell cycle progression, cell growth, cell apoptosis, survival, glucose metabolism, protein synthesis, signal triggering and transduction, and phosphorylation of the downstream substrate TSC2. AKT activation suppressed the activity of TSC2 to promote the production of Rheb complex, resulting in mTORC1 activation. mTORC1 activation facilitates the initiation of eukaryotic protein translation. 4E-binding protein 1 (4E-BP1) activation enhanced the release of eukaryotic translation initiation factor 4E (eIF4E). RTK activation further accelerates guanine exchange factor to load RAS with GTP. RAS–GTP dimers recruit RAFs or RAF/MEK heterodimers to membranes, where tetramers consisting of RAF and MEK promote RAF activation. MEK activation is initiated by docking on RAF dimers, which further facilitate ERK phosphorylation. RTKs, receptor tyrosine kinase; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PTEN, phosphatase and tensin homolog; PDK1,3-pphosphoinositide-dependent kinase 1; mTOR, mechanistic target of rapamycin; TSC1, tuberous sclerosis 1; TSC2, tuberous sclerosis 2; 4E-BP, eIF4E-binding protein; GRB10, growth factor receptor-bound protein 10; IGF-1R, insulin-like growth factor 1 receptor; mLST8, mammalian lethal with SEC thirteen 8; RICTOR, rapamycin-insensitive companion of mTOR; S6K, ribosomal S6 kinase; FLCN, folliculin; ULK1, UNC-51-like kinase 1; RAPTOR, regulatory-associated protein of mTOR; RICTOR, rapamycin-insensitive companion of mTOR.
PI3K/AKT/mTOR signaling is a crucial intracellular pathway in regulating fundamental cellular functions, including but not limited to regulating cell growth, motility, survival, metabolism, and angiogenesis. Hyperactivation of the PI3K/AKT/mTOR pathway occurs in nearly all malignant neoplasms [8]. A previous study showed that single nucleotide polymorphisms (SNPs) in the PI3K/AKT/mTOR pathway are connected to the distant metastasis of carcinomas [8]. This pathway also has a vital role in promoting cell apoptosis through inhibition of related genes such as p53, caspase 3, Fas receptor (CD95) and TNF receptor (TNFR1) [9, 10].
AKT could serve as the targeted effector to the cell surface for activation. Two sites (T308 and S473) of AKT could be phosphorylated. In addition to phosphotidylinositide-dependent kinases (PDKs), AKT can be phosphorylated by mTOR. This signaling activation contributed to the growth of cells by suppressing autophagy via the activation of mechanistic target of rapamycin (mTOR) [11, 12]. MTOR can inhibit the initiation of autophagy via distinct usual pathways [13, 14]. In addition, PI3K/AKT/mTOR lead to epithelial-mesenchymal transition (EMT) in chemotherapy resistance and metastasis in malignant tumor cells [15,16,17].
The RAF/MEK/ERK pathway is a critical signaling pathway in transmitting signals from membranes to the nucleus [18]. The RAF family serine/threonine (ser/thr) protein kinases (Raf-1(C-Raf), B-Raf, A-Raf) activate MEK by phosphorylation, and MEK phosphorylation promotes ERK 1 and 2 phosphorylation at their residues. RAS GTPases and growth factor receptors, such as epidermal growth factor receptor (EGFR), control the activation of the RAF/MEK/ERK signaling pathway. Similar to PI3K/AKT/mTOR, activation of RAF/MEK/ERK signaling pathways was observed in a large fraction of solid cancers [6]. These observations contribute to the development of inhibitors targeting kinases containing the new RAF and MEK kinases approved by the FDA. Extracellular signal-regulated kinases (ERK1/2) are a subfamily of mitogen-activated protein kinases (MAPKs) that facilitate the culmination of signal transduction and regulate transcription. ERK1/2 are phosphorylated by the upstream MAPK/ERK kinases MEK1/2, which are tyrosine/threonine protein kinases that are necessary for proliferation and regular growth in human cells.
Both pathways share common inputs and can also be activated via RAS. In addition, when one pathway is suppressed, the other pathway may offer compensatory effectiveness [19]. MTOR, a downstream molecule phosphorylated by AKT, is inhibited, and PI3K can stimulate MAPK through RAS. These results showed that these two pathways serve as a complex network and provide a method for dual therapeutic compounds that can simultaneously block both pathways [20]. In addition to mTOR, several nodes of the PI3K/AKT/mTOR and RAF/MEK/ERK pathways are well known to interact with each other, and mounting evidence indicates that dual blockade of both pathways might contribute to anticancer effects [21]. Mutations observed in the genes of the pathway or in upstream receptors that activate these pathways [22]. The particular combination of phosphoinositide 3-kinase or phosphatidylinositol-3 kinase (PI3K) and MEK inhibitors is generally being evaluated in several clinical studies in various kinds of cancers.
PI3K/AKT/mTOR and RAF/MEK/ERK contain various kinases regulated by phosphorylation and dephosphorylation via relevant kinases. These two signaling pathways are considered vital oncogenic signaling pathways in tumorigenesis. In the current review, we describe the critical role of the PI3K/AKT/mTOR and RAF/MEK/ERK signaling pathways in carcinoma initiation and tumor development as potential strategies for tumor therapy, summarizing the recent doubts about targeting the pathway in monotherapy and combination therapy.
Overview of the PI3K/AKT/mTOR pathway and its role
The signaling pathway composed of PI3K, protein kinase B (AKT), and mTOR is a part of a complicated signaling cascade comprising distinct upstream regulators and downstream effectors, which play critical roles in the formation processes of human cancers [23]. Prior evidence has identified that hyperactivation of PI3K/AKT/mTOR signaling promotes tumorigenesis. PI3K was first identified as a lipid kinase in the 1980s in the Cantley group’s study. Furthermore, the first clone and report of TOR was completed in 1991 by Hall, and its mammalian homolog mTOR was developed three years later [24].
Vital mouse models serving as key genetic evidence to identify the imperative roles of the PI3K/AKT/mTOR signaling pathway in promoting tumorigenesis have been constructed in recent years, contributing to the comprehension of the signaling by the recognition of whole modules via various methods and supplying valuable information for patients to confirm activation of the pathways in human carcinomas by deep sequencing, proteomics, reversed-phase protein arrays (RPPA) and bioinformatics approaches [25]. Different therapeutic agents targeting different components in the PI3K/AKT/mTOR signaling pathway have been designed and applied as anticancer agents [26].
PI3Ks promoted the transfer from PIP2 to PIP3 via phosphorylation of phosphatidylinositol. PIP3 is the basis of multiple downstream targets of the PI3K/AKT/mTOR pathway [27]. PI3Ks contain three classes: PI3Ks class I, PI3Ks class II and PI3Ks class III, which are separated by their structure, reaction mechanism, and characteristic [28,29,30,31]. Both Class IA and Class IB can be generally activated by G protein-coupled receptors (GPCRs), either via Gβγ protein or indirectly via RAS [32]. In addition, Class IA could also be regulated by receptor tyrosine kinases (RTKs) and upstream oncogenes. Class IA PI3Ks contain a regulatory subunit (p85α, p85β, p85γ) and a catalytic unit (p110α, p110β, p110δ, p110γ), which belong to heterodimers [29, 33] (Table 1). The regulatory subunit can be activated by the catalytic subunit [32]. After stimulation or subsequent activation, class IA PI3Ks can be recruited to the cytomembrane via the p85 subunit for motif phosphorylation. The activation of the p110 catalytic subunit in turn activated downstream signals [34]. P85 combined with the receptors, and p110 catalyzed the formation of PIP3 by adding an additional phosphate on PIP2. PTEN promoted the transfer of PI (3,4,5) P3 back to PI (4,5) P2 to reduce PIP3 by intrinsic lipid phosphatase. The N-terminal region of AKT docked to PI (3,4,5) P3 contributes to the translocation to the cytomembrane, resulting in AKT activation with two vital amino acid residues phosphorylated [35]. AKT induced the phosphorylation of PRAS40 and tuberous sclerosis complex (TSC2) to alleviate the inhibitory effect on mTORC1 to induce mTOR activation. In addition to mTORC1, mTORC2 is another complex of mTOR. MTOR is a kind of ser/thr protein kinase in PI3Ks kinases [36]. The Ragulator/Rag GTPase complex acts as a regulator in controlling mTORC1 activity. S6K1 and 4E-BP1 are downstream effectors of mTOR, which mediates protein synthesis [37]. AKT is a key node that transduces signals from mTORC2 to mTORC1, while mTORC1 could be regulated independently of mTORC2. MTORC1 activation by amino acids is mediated by Ras-related GTP binding (RAG) GTPases. Amino acids activate mTORC1 via Rag GTPases, which are recruited to lysosomes by the Ragulator complex (MAPK and TOR activator). Downregulated cellular energy promotes AMPK activation to trigger Raptor phosphorylation to inhibit mTORC1 action [38]. MTORC1 activation was promoted by ERK-dependent Raptor phosphorylation by RAS/MAPK activation. More alternations occurring in this pathway according to transcription, protein production, and other factors were discussed in next paragraph.
Dysregulation of the PI3K/AKT/mTOR pathway in neoplasms mediated by genetic alterations
Gene mutagenesis-induced overactivation of the PI3K/AKT/mTOR pathway in neoplasms
Referencing previous studies, the frequency of the PIK3CA mutated gene varies from 10 to 15% in human cancers [39]. PIK3CA mutations are found in nearly all neoplasm types, such as mammary tumors, colorectal carcinoma, esophageal cancer, gallbladder carcinoma, non-small cell lung cancer (NSCLC), ovarian cancer, and gastric carcinoma [40]. PIK3CA mutations in breast carcinoma are the most prevalent. H1047R, E545K, E542K, N345K, and H1047 L were the top five mutations that accounted for three-quarters of all PIK3CA mutations [41]. PIK3CA gene amplification is frequent in gastric carcinoma (36.4%), thyroid adenocarcinoma (30%), prostatic cancer (28%), ovarian cancer (13.3–29.8%), and cervical carcinoma (9.0–80%). Various positions were observed to be mutated in the PIK3CB gene, including lung carcinoma, thyroid cancer, and lymphoma [42]. These studies implied that the function of the PI3K isoform was extremely different. Clinically, upregulated PIK3CA expression was significantly related to neoplasm invasiveness, poor patient survival and lymph node metastasis [43].
AKT is a ser/thr kinase in downstream effectors of the PI3K/AKT/mTOR signaling pathway and includes three subtypes: AKT1, AKT2 and AKT3 [44]. AKT1 is observed in the majority of tissues, AKT2 is mainly found in organisms with high sensitivity to insulin, and AKT3 is expressed in the brain and testicles [45, 46]. PI3K facilitates Akt1 in cytoplasm transport to interact with PIP3 on the cytomembrane, leading to Akt1 phosphorylation and activation [47]. In addition, negatively regulated PI3K activation can inhibit Akt1 via PTEN phosphorylation. In conclusion, positive regulation of PI3K signaling and negative regulation of PTEN signaling can induce Akt1 activation in human cancers. Akt1 facilitates cell proliferation, survival, and metabolism via its downstream effectors BAD, FOXO1, and TSC1/2 [9]. Genomic alterations of Akt1 could directly promote the activation of Akt1 without phosphoinositide.
Mutations in Akt2 and Akt3 disrupt the role of pleckstrin homology (PH) and kinase domain (KD), resulting in AKT oncogenic activation [48]. Akt1 amplification is frequently reported to promote cisplatin resistance in epithelial cancers, such as gastric carcinoma, breast neoplasm, gallbladder tumor, NSCLC, and SCLC [49]. Akt2 gene copy number gain was found in ovarian cancer, pancreatic carcinoma, liver cancer, colorectal carcinoma, gastric carcinoma and breast neoplasm [50]. Akt3 gene alterations are rare in carcinomas. The Akt1-E17K mutation existing in the PH domain has been observed frequently in breast carcinoma, ovarian carcinoma, and malignant meningioma with increased oncogenicity, particularly by facilitating AKT binding with PIP3, which promotes the action of AKT [51]. The The Akt1-E17K mutation in mouse models contributed to mammary hyperplasia and resulted in lung epithelium disorder, demonstrating that Akt-activating mutation plays an oncogenic role in promoting tumorigenesis [52]. The other two subtypes of AKT-E17K mutations were less frequent than The Akt1-E17K mutations. The Akt2-E17K mutation is commonly observed in hyperinsulin mucoglycemia with dysregulated insulin production [53]. The Akt3-E17K somatic mutation was observed in human malignant melanoma [52]. Mutations in kinases manage the development of tumors and developmental disorders by promoting kinase activation [54].
The mTOR activation mutations increase its kinase activity, contributing to overactive downstream pro-proliferation signaling pathways [55]. The mutation rate in metastatic cancer was 3% (329/10,336) in the MSK IMPACT Clinical Sequencing Cohort, which is similar to that (2.9%, 292/10,194) of the China Pancancer Cohort (OrigiMed2020). The rate of nonsynonymous mTOR mutations was approximately 10% in malignant melanoma patients, and nonsynonymous mTOR mutations were connected to a poor prognosis [56]. The mTOR mutation was found in a wide variety of malignant tumors, including lung, renal cell, endometrium, colorectal and squamous carcinoma [57]. HEAT repeat, FAT domain, and KD mutations promote the oncogenic role of mTOR in tumorigenesis [58]. Rictor and mTOR are prevalently observed in cancer, while mutations in mSin1, mLST8 and Raptor are not common in human cancers [59]. Rictor amplification is a selection criterion for potential mTOR inhibition treatment.
Deletion of genes promotes hyperactivity of the PI3K/AKT/mTOR pathway
PTEN, the most common tumor suppressor gene, is the major PI(3,4,5)P3 kinase that antagonizes PI3K phosphorylation, and its loss could contribute to uncontrolled PI3K signal transduction [60, 61]. PTEN deletion in different neoplasms has been found in either genetic or epigenetic mechanisms [62,63,64]. Moreover, the deletion of PTEN leads to its loss efficiency and is closely correlated with worse prognosis, drug resistance and advanced tumor stages. PTEN deletion is frequently found in solid tumors and hematologic malignancies, including prostate cancer, diffuse large B-cell lymphoma, glioma, endometrial carcinoma, hepatopancreatic ductal malignancy, and invasive bladder cancer [65,66,67,68,69,70,71]. PTEN neddylation is promoted by Nedd8 interaction at high glucose levels, which does not lead to an increase in PTEN accumulation in the nucleus [72]. PTEN in the nucleus could promote RAD51 expression, which results in DNA double-strand breaks.
PTEN mutations plays an important role in PI3K pathway activation in tumor occurrence and contributes to a majority of activates in tumor prognosis [73]. PTEN mutations presumably lead to the hyperactivation of all PI3K-mediated pathways. However, PTEN mutations were not observed in some neoplasms. In head and neck cancers, 15% of PTEN mutations was found [74]. In breast carcinomas, only 3% of PTEN mutations was identified [73]. It is clear that some other signaling pathway existed besides PTEN mutations, activation and deletions to constitutively activated PI3K activity. The interaction between RAS and PI3K was supported by the vivo data. However, RAS and PTEN mutations is mutually exclusive in neoplasm of endometrium and malignant melanomas. PTEN mutations were commonly observed in spongioblastoma but rarely seen in carcinoma of pancreas, lung and colorectum [75,76,77,78,79,80]. The mutations of RAS are opposite in these neoplasms. Compared with Pten+/− mice, RAS mutations was commonly observed in Pten+/+ mice in according to chemically induced skin neoplasms. Lacking RAS mutations results in second Pten allele loss. The results demonstrated RAS and PTEN may have synergistic effect in carcinogenesis.
PTEN promoter methylation induced PTEN transcription reduction that correlated with the relapse and recurrence of gastric cancer via PI3K/AKT/mTOR signaling pathway hyperactivation [81,82,83]. Increased PTEN promoter methylation could decrease PTEN transcription, resulting in worse survival [82]. P53 can be stabilized by PTEN and promote its transcription. PTEN promoter deficiency demonstrated a defect in stabilizing and binding p53, contributing to PTEN reduction and alleviating its suppressive function in sporadic cancers and Cowden syndrome patients [84]. Pin1, polycomb group protein EZH2, AKT activation, and RAC1P29S negatively mediate the transcription and function of PTEN [85,86,87,88]. Previous studies have indicated that mutations in the PTEN promoter are dispensable for common cancers. However, the potential role of the PTEN promoter in cancers needs more in-depth investigation.
The effect of deletion of other genes besides PTEN in the hyperactivity of PI3K/AKT/mTOR pathway
Besides PTEN deletion alone, deletion of both Lkb1 and Pten genes and mTOR excessive activation result in the development of ovarian carcinoma [89]. PTEN inactivation by S-nitrosylation could also induced PI3K/AKT Activation. PARK2 depletion mediated AMPK activity and promoted oxygen reaction leading to PTEN S-nitrosylation [90]. Inositol polyphosphate 4-phosphatase type II (INPP4B) acts as a negative regulator in PI3K activity, which promotes PIP2 to generate PIP3. INPP4B mutation led to the silence of PI3K signaling [91]. In mice kidney neoplasms, TSC2 exon 3 deletion contributed to the hyperactivity of mTOR, which resulted in Hes1 overexpression [92].
Hyperactivation of PI3K/AKT/mTOR signaling driven by gene fusion
Gene fusion is made by linking parts of two different genes during DNA from one chromosome moves to another chromosome, leading to fusion protein production. Fusion protein production has been found in colorectal cancer, myofibroma, B-lymphoblastic leukemia, glioblastoma, NSCLC, and thyroid carcinoma [93]. Gene fusions are commonly found in the PI3K/AKT/mTOR axis in cancer development. In the landscape of recurrent kinase fusions in solid tumors, the proportion of gene fusions was broadly different among various tumors, indicating diversity in the etiology of these neoplasms. PI3KCA and AKT fusions have been widely researched, and their functions are involved in PI3K/AKT/mTOR hyperactivation [94]. TBL1XR1–PIK3CA fusions were shown in invasive breast cancer and prostate cancer, which were driven by hormones [95]. Juxtaposing the promoter of the TBL1XR1 gene to the 5’ end of the intact PIK3CA coding sequence contributes to TBL1XR1–PIK3CA fusions, which causes an increase in PIK3CA mRNA [96]. LAMTOR1-Akt1 was observed in epithelioid cancer patients with tumorigenic driver function. In addition, BCAM-Akt2 and RPS6KC1–Akt3 were reported and validated in ovarian carcinoma and mammary cancer, respectively [97]. Hyperactivation and alterations of AKT proteins and their upstream and downstream effectors were generally researched in neoplasms of adults and pediatric malignancies, and only rare AKT fusions have been described [98,99,100,101,102,103,104,105,106].
In humans, the mTOR-TP53BP1 fusion gene is easily observed in colorectal carcinoma, mammary neoplasm, ovarian carcinoma, and lung carcinoma and regulates PI3K/AKT/mTOR signaling pathway activation. TFE3-mTOR, FKBP-mTOR, CHD1-mTOR, mTOR-CASZ1 and mTOR-TP53BP1 were found in cancers. However, their roles in tumorigenesis need further exploration [107, 108].
Transcriptional modifications drove PI3K/AKT/mTOR signaling pathway hyperactivation in cancer
In addition to genetic alterations leading to stable oncogenicity to promote oncogenesis and chemotherapy resistance, the production of proteins and expression of mRNA in PI3K/AKT/mTOR signaling pathways also participate in neoplasm growth [109]. Various effectors are involved in regulating the PI3K/AKT/mTOR pathway, such as promoter modification, microRNAs (miRNAs), and transcriptional control. MicroRNAs (miRNAs), small noncoding RNAs, regulate a third of the functional genomics at the posttranslational level. Upregulated Rictor expression was present in glioblastoma, advanced hepatocellular carcinoma (HC), SCLC, prostate carcinoma, cervical cancer, glioblastoma, mammary neoplasm, colorectal carcinoma, endometriosis, melanoma and esophageal squamous cell carcinoma (ESCC) [110, 111].
Distinct factors facilitate the signals in the pathways to regulate gene expression Promoter methylation could decrease the transcription of a targeted gene. PTEN promoter methylation is one of the most frequent sites in the PI3K/AKT/mTOR signaling pathway. TSC2 and TSC1 promoter methylation was significantly increased in breast carcinoma, oral squamous cell carcinoma, and tuberous sclerosis complex [112, 113]. The data show for the first time that methylation of the TSC2 promoter might cause a complete loss of tuberin in TSC2 cells and that the pathogenesis of angiomyolipomas might also originate from epigenetic defects in smooth muscle cells. TSC2 promoter methylation promoted the abnormal proliferation of smooth muscle-like cells, leading to TSC [114]. Both PI3K/AKT/mTOR and RAF/MEK/ERK can be activated by TBX1 suppression in thyroid adenocarcinoma, which is induced by its promoter methylation [115].
In human NSCLC cells, a decrease in miR-192-5p induces tumor development. Downregulation of miR-143, miR-145, and miR-101 in Burkitt’s lymphoma promotes tumor cell growth [116]. MiR-19a and miR-96 reduction suppress tumor cell proliferation through the PI3K/AKT pathway in HC cells [117]. MiR-425/489 is a target gene for the long noncoding RNA MHENC [118]. MiR-425/489 is increased in melanocytes. Knocking down MHENCR significantly inhibits melanocyte growth and causes cell cycle arrest and cell apoptosis.
Yang et al. reported that miR-1297 bound to the 3’ end of Meg3 and that miR-1297 could also bind to PTEN in testicular germ cell neoplasms [119]. The effect of miR-1297 on the 3’ end of PTEN mRNA expression could be suppressed by PTEN expression, leading to deactivation of AKT and reduction of cell proliferation. PTEN mRNA polyadenylation is frequently observed to regulate miRNA-mediated PTEN [120]. Polyadenylation of PTEN mRNAs is a dynamic process and results in different isoforms with distinct 3′UTRs [121]. Considering the lack of specificity of transcription factors in controlling PI3K/AKT/mTOR signaling expression, we did not discuss transcription factors and transcription modification in this review.
Inhibitors of the PI3K/AKT/mTOR pathway
This pathway is one of the most common dysregulated pathways in tumors and a key signal in regulating tumor cell proliferation and apoptosis [122, 123]. The molecules in this pathway have drawn comprehensive attention in recent years. Many agents targeting components in this signaling pathway have been researched and assessed in animals and humans.
PI3K inhibitors
PI3K inhibitors were the primary agents developed and investigated in the PI3K/AKT/mTOR signaling pathways (Fig. 2) [124]. Various PI3K inhibitors were in development and can be separated into three categories based on their pharmacokinetic effect and interaction with ATP: pan-PI3K inhibitors, isoform-specific PI3K inhibitors and dual PI3K/mTOR inhibitors. Clinical trials have presented advanced antitumor treatment effects (Table 2) [125]. Compared with isoform-specific inhibition, class IA pan-PI3K inhibitors have been more comprehensively researched [126]. The off- and on-target effects of blocking all isoforms caused by pan-Class I inhibitors limited their use; however, their antitumor role was not influenced [127].

Inhibitors of the PI3K/AKT/mTOR pathway. Various classes of agents target different effectors of the PI3K/AKT/mTOR pathway
Pan-PI3K inhibitors
Buparlisib (BKM120) is an ATP-competitive pan class l PI3K inhibitor that can also affect mTOR and Vps34 at higher doses [128]. Buparlisib has already been assessed in a phase 3 trial [129]. Buparlisib exhibited superior antitumor effects in human cells in vitro [130, 131]. In vivo, buparlisib presents excellent oral bioavailability and superior antitumor activity in mouse models. In a dose-escalation study of buparlisib, the most frequent buparlisib-related adverse effects (AEs) included rash, hyperglycemia, diarrhea, anorexia, mood alteration, decreased appetite, nausea and abnormal hepatic function [132]. In a phase I trial, the AEs increased in patients treated with buparlisib (40 mg daily) combined with standard mFOLFOX6 compared with either buparlisib or mFOLFOX6 [133]. In patients treated bevacizumab with BKM120, no encouraging efficacy was observed in glioblastoma tumors when bevacizumab was combined with BKM120 [134].
Based on the safety dose in solid tumors treated with chemotherapy combined with BKM120, the combination of BKM120 at a safe dose and radiotherapy was applied to advanced non-small cell lung carcinoma [135]. This therapeutic regimen was well tolerated, and hypoxia could be improved by PI3K inhibition, which indicated that BKM120 may act as a radiosensitizer. The maximum tolerated dose of BKM120 was 100 mg/day in solid tumors; however, the maximum tolerated dose of BKM120 was 80 mg/day in leukemias. Leukemia patients treated with the maximum tolerated dose demonstrated modest efficacy. BKM120 monotherapy demonstrated promising efficacy and manageable AEs in advanced ESCC patients in a phase II study [136]. However, buparlisib monotherapy was connected to an unfavorable safety profile and unsatisfactory anticancer activity in advanced or recurrent endometrial carcinoma [137]. The study was withdrawn before recruitment was finished because of AEs. In the BELLE-3 trial, the combination of buparlisib with fulvestrant was not recommended in postmenopausal, hormone receptor (HR)-positive, HER2-negative, advanced breast cancer for the safety profile [138]. The use of buparlisib monotherapy in recurrent glioblastoma was limited by efficacy. Patients treated with buparlisib in combination with paclitaxel had longer survival times in relapsed or metastatic head and neck squamous cell carcinoma patients [139]. Then, buparlisib plus paclitaxel was recommended as a second-line therapy.
Copanlisib (BAY 80 − 6946) is a panclass I PI3K inhibitor that can inhibit all four PI3K class-I isoform activations [140]. The maximum tolerated dose of copanlisib monotherapy in patients with advanced solid tumors and non-Hodgkin lymphoma (NHL) was determined in the NCT00962611 clinical trial [141]. Copanlisib has promising antitumor activity in these patients, particularly in NHL patients. The most common AEs related to copanlisib included hyperglycemia, nausea, and hypertension. Grade ≥ 3 AEs related to copanlisib were hyperglycemia, hypertension, and rash. Another study developed on advanced or refractory solid tumors, and copanlisib was well tolerated in Japanese patients [142]. The most common toxicities in Japanese patients were hyperglycemia and hypertension. The maximum tolerated dose of copanlisib in Chinese patients with relapsed or refractory NHLs was at a lower dose and presented promising effects [143]. The COUP-1 trial assessed the effectiveness and AEs of copanlisib in combination with rituximab in marginal zone lymphoma patients, which demonstrated the potential for the clinical and translational use of copanlisib [144]. In a phase II study of copanlisib in various lymphomas, the results showed that intravenous copanlisib could offer promising efficacy and manageable toxicity [145]. Further studies on copanlisib in peripheral T-cell and mantle cell lymphomas were carried out. In PIK3CA mutation patients, the over response rate was 16% with tolerable AEs, including hyperglycemia (76%), fatigue (48%), diarrhea (44%), hypertension (40%), and nausea (40%). The long-term efficacy and toxicity of copanlisib in patients with relapsed or refractory indolent lymphoma exhibited progression-free survival, and overall survival was 12.5 months and 42.6 months, respectively. Approximately twenty-six patients received copanlisib four over one year [146]. In phase III of Copanlisib plus rituximab in indolent NHL, Copanlisib plus rituximab could improve progression-free survival compared with placebo plus rituximab [147]. Overall, serious AEs were largely unchanged and tolerable, with no new cases or grade 5 events in the undergoning and completed clinical trials. Treatment-emergent AEs caused by copanlisib did not contribute to increased incidence or worsening prognosis. The results in the study demonstrated that intravenous copanlisib led to a sustained, intensive treatment response without increasing treatment-emergent AEs, similar to other orally administered PI3K inhibitors [148].
In addition to buparlisib (BKM120) and copanlisib (BAY 80 − 6946), LY294002, pictilisib (GDC 0941), pilaralisib (SAR 245,408 and XL 147), SF1126, ZSTK474, rigosertib (ON-01910) and CH5132799 are pan class l PI3K inhibitors with ongoing clinical studies [149]. Copanlisib (BAY 80 − 6946) is the only pan-PI3K inhibitor approved for tumor therapy and is a potential treatment for malignat solid tumors and hematologic malignancies [141]. In 2017, the FDA administered copanlisib to patients with recurrent follicular lymphoma who were treated with two or more previous systemic therapies based on the outcomes of the CHRONOS-1 trial [146].
Isoform-selective PI3K inhibitors
Compared with pan-PI3K inhibitors, isoform-selective PI3K inhibitors target one of the isoforms in PI3K, which decreases AEs and enhances targets [150]. Patients treated with isoform-selective PI3K may be selected and identified with sensitivity and resistance markers [151]. Preclinical trials of isoform-specific PI3K inhibitors in cancer are limited. GS-1101 (idelalisib/CAL-101), IPI145 (duvelisib, INK-1197) and alpelisib (BYL719) have been approved by the FDA for tumor therapy. In 2014, idelalisib was approved for treating chronic lymphocytic leukemia (CLL), relapsed follicular B-cell NHL, and relapsed small lymphocytic lymphoma (SLL) [152]. In 2018, duvelisib was approved in relapsed or refractory CLL or SLL after more than two previous therapies [153]. In 2019, alpelisib (a PI3KCA inhibitor) was approved for treating HR-positive, EGFR-negative, PI3K mutation, advanced or metastatic mammary neoplasms [154]. Based on the outcomes of the CBYL719 × 2101 trial, alpelisib showed promising results and tolerable toxicity in PIK3CA-mutant tumor patients, demonstrating that isoform-selective PI3K inhibitors combined with other antitumor regimens may be efficient in treating PIK3CA-mutant tumors [155]. The newest PI3K inhibitor approved by the FDA was umbralisib in 2021, which is efficient in treating lymphoma according to the UTX-TGR-205 trial [156].
Dual PI3K/mTOR inhibitors
Considering that both PI3K and mTOR are members of the same PIKK super family of kinases accompany with similar structural isoforms and responses, inhibitors inhibiting both PI3K and mTOR targets were found through research on mTOR inhibitors [157]. The administration of BEZ235 to improve glucocorticoid resistance in pediatric T-ALL and the use of PKI-587 to decrease cancer cell proliferation in T-ALL demonstrated that dual PI3K/mTOR inhibitors significantly improved the treatment effect compared with inhibiting mTOR or PI3K alone [158,159,160].
In a phase Ib dose-finding study of BEZ235, diarrhea (58%), mucositis (58%), and nausea (42%) were the most frequent toxicities of all grades. Mucositis was the most common grade 3 AE [161]. No grade 4 BEZ235-related toxicity was observed, and no drug-related hospitalizations or deaths were found. BEZ235 monotherapy at 300 mg BID orally was recommended in a phase II study, which is well tolerated, and no objective responses were found [162]. Further studies of BEZ235 have been developed in monotherapy or combination with other agents in various solid tumors and hematologic malignancies [163]. Although the outcomes of these studies showed that BEZ235 is generally tolerated, the clinical response is restricted [164]. There are no new clinical trials of BEZ235 developed on solid tumors. Some acute lymphoblastic leukemia patients may have some alterations in the PI3K/AKT/mTOR signaling pathway, and BEZ235 had promising effects in this small subset of patients [165]. AML did not benefit from BEZ235 treatment. In metastatic renal cell carcinoma (RCC), apitolisib (GDC-0980) was less effective than everolimus [166]. Increased toxicity was observed in the trials.
Voxtalisib (SAR245409, XL765) is a potent inhibitor targeting class-I PI3Ks, mTORC1 and mTORC2 [167]. In vitro, voxtalisib inhibited the phosphorylation of PI3K and controlled mTOR effector incorporation in malignant tumor cells [168]. In a phase Ib study of voxtalisib in patients with advanced malignant tumors, pimasertib (90 mg) plus voxtalisib (70 mg) demonstrated poor long-term tolerability and no faverable survival in advanced solid tumor patients [169]. Diarrhea (75%), fatigue (57%), and nausea (50%) were frequently observed in the pimasertib plus voxtalisib group. In relapsed or refractory NHL or CLL, voxtalisib was tolerable when it was given at 50 mg orally twice a day [170]. Voxtalisib (40 mg b.i.d.) plus temozolomide with or without radiotherapy presented a favorable safety profile in patients with high-grade gliomas. Among these patients, 4% had a partial response, and 68% had stable disease.
Compared with voxtalisib and BEZ235, PQR309 (bimiralisib) showed a better ability to transfer the brain blood barrier (BBB) [171]. PQR309 monotherapy or in combination with other small molecular inhibitors demonstrated promising antitumor activity in lymphomas [172]. In a phase I dose-escalation study, dose-limiting toxicities were not exhibited in advanced solid lymphoma patients. Seventy precention patients were found to have grade 3 treatment emergent AEs, and approximately 30% of patients had grade 4 treatment emergent AEs. 28% of patients discontinued treatment. PQR530 and PQR620 also had a better ability to transfer the BBB [173].
GDC-0084 is another oral and brain-penetrant dual PI3K/mTOR inhibitor. The usual PI3K/mTOR-related AEs were observed after GDC-0084 treatment. The first phase I study of GDC-0084 in patients with progressive or recurrent glioma identified that GDC-0084 could cross the blood‒brain barrier [174]. The results indicated that GDC-0084 is a potential compound in brain metastatic mammary neoplasms with a dysregulativity of PI3K/mTOR signals conferred by PIK3CA mutations.
Many dual inhibitors have been developed and applied in xenograft mouse models and malignant tumor cell lines with promising effects on tumors. PKI-587 improved the radiosensitivity and oxaliplatin sensitivity of HC via the PI3K/AKT/mTOR pathways to reduce DNA damage repair [175]. The application of PKI-587 led to broad spectrum cancer cell stasis and cell apoptosis. PIK3CA mutations caused by activation of the WNT/β-catenin signaling pathway may decrease colorectal cancer cell sensitivity to the dual PI3K/mTOR inhibitor PKI-587 [176]. PKI-587 plus cofetuzumab pelidotin in metastatic triple-negative breast cancer (TNBC) showed moderate toxicity and promising clinical activity [177]. PKI-587 combined with carboplatin and paclitaxel in clear cell ovarian cancer exhibited similar trends [178]. The administration of PKI-587 is well tolerated by weekly intravenous infusion rather than daily oral use in recurrent endometrial cancer [179]. The clinical response in the PKI-587/stathmin-low arm was slightly satisfactory, while that of the gedatolisib/stathmin-high arm did not meet the clinical benefit response criteria.
In addition to the mentioned small molecule inhibitors, dual PI3K/mTOR inhibitors such as GSK2126458, SF1126, LY294002, PF-04691502, LY302341, and PWT33597 have shown favorable antitumor efficacy in various malignant neoplasms [180]. In a cellular assay, GSK2126458, an oral inhibitor, inhibited the growth and aggression of pancreatic cancer cells [181]. SF1126, an Arg-Gly-Asp (RGD)-conjugated LY294002 prodrug, can inhibit both the PI3K-Akt-mTOR and BRD4 cascades. In colorectal cell lines and primary human colon cancer cells from human tumors, SF1126 inhibited cell growth and apoptosis and blocked the cell cycle [182]. Administration of SF1126 led to tumor angiogenesis in tumor tissues. Combining different chemotherapeutic agents with PF-04691502 promoted breast cancer cell apoptosis [183]. PF-04691502 could also promote radiosensitivity of gastroenteropancreatic neuroendocrine tumors. BGT226, an imidazoquinoline derivative, is an ATP-competitive dual PI3K/mTORC1/C2 inhibitor [184]. LY302341 presented promising antitumor activity in esophageal adenocarcinoma [185]. Clinical research on PWT33597 in advanced malignancies has been developed, but no results have been posted.
AKT inhibitors
AKT is a kind of effector in the PI3K/AKT/mTOR pathway of tumors and is a potential target in treating cancer [186]. The AKT kinase family includes three isoforms, Akt1, Akt2, and Akt3 (Table 2).
Phosphorylation and dephosphorylation of AKT regulate the activation of Akt-dependent behavior [187]. ATP-competitive inhibitors promote dephosphorylation of AKT activity by inhibiting ATP activity [188]. Allosteric inhibitors interact with the AKT substrate by inducing conformational transitions of their enzymatic structure [189]. Irreversible inhibitors are uncommon AKT inhibitors. Both ATP-competitive inhibitors and allosteric inhibitors present potential effects in cancer cells [190]. ATP-competitive inhibitors contributed to AKT activation via inhibition of the pH and exposure of the ATP-binding pocket. Allosteric inhibitors block AKT on the plasmalemma and inhibit AKT activity [191].
In the first human study of capivasertib, capivasertib monotherapy showed clinical significance in Akt1 E17K-mutant metastatic breast cancer patients with estrogen receptor (ER) positivity [192]. Gastrointestinal events (diarrhea, vomiting, nausea) were the most frequent AEs related to capivasertib treatment in the D3610C00001 trial [193]. Approximately sixty patients experienced grade ≥ 3 AEs, including hyperglycemia, diarrhea, and maculopapular rash. The combination of capivasertib and enzalutamide is tolerable in metastatic castration-resistant prostate cancer and has a favorable prognosis. In the PAKT trial, capivasertib plus first-line paclitaxel therapy for metastatic TNBC led to extensively better progression-free survival and overall survival, which identified the role of capivasertib in TNBC treatment [194]. According to the FAKTION trial, the median progression-free survival of metastatic, ER-positive breast cancer patients treated with capivasertib plus fulvestrant was 10.3 months compared with 4.8 months in patients treated with fulvestrant plus placebo [195].
Ipatasertib (GDC-0068), which targets AKT signaling, showed acceptable AEs and a favorable prognosis in AKT-activated tumors [196]. No difference was found between the survival of ipatasertib/mFOLFOX6 and placebo/mFOLFOX6 (NCT01896531). In the IPATential150 trial, ipatasertib plus abiraterone prolonged the survival of metastatic prostate cancer patients with PTEN loss compared with standard regimens plus placebo (approximately 19 months vs. approximately 17 months) [197]. 70% of patients administered ipatasertib plus abiraterone had grade ≥ 3 AEs. Toxicity related to placebo plus abiraterone resulted in discontinuation in 5% (28/546) of prostate cancer patients. However, 21% (116/551) of patients in this trial were treated with ipatasertib plus abiraterone because of AEs. Ipatasertib plus paclitaxel prolonged survival compared with paclitaxel monotherapy (6.2 months vs. 4.9 months). In cohort B of the IPATunity130 randomized phase 3 trial, adding ipatasertib to paclitaxel did not have a promising effect in PI3K pathway-mutant HR-positive unresectable locally advanced/metastatic malignant mammary tumor patients. Referencing AEs, the administration of ipatasertib decreased the proportion of diarrhea, neutrophil count decrease, neutropenia, peripheral neuropathy, and peripheral sensory neuropathy [198].
SPY 2 is a phase II study elevating the response of MK-2206 in combination with standard taxane- and anthracycline-based neoadjuvant therapy [199]. In the I-SPY 2 trial, MK-2206 contributed to higher complete response rates in HR-negative and HER2-positive breast cancer [200]. The median progression-free survival and overall survival for recurrent endometrial cancer patients treated with MK-2206 were 2.0 months and 8.4 months, respectively. Rash, fatigue, nausea, hyperglycemia, diarrhea, fever, and vomiting were the most common AEs related to MK-2206. Compared with standard chemotherapy regimens, the activity of MK-2206 in recurrent endometrial cancer was limited [201]. Administration of MK-2206 demonstrated amazing effectiveness in uterine serous cancer.
mTOR inhibitors
The mechanistic target of rapamycin (mTOR) is a ser/thr kinase that belongs to the PIKK family [202]. MTOR contains two multiprotein complexes called mTORC1 and mTORC2 [203]. MTORC1 participates in multiple growth factor signals to promote cell growth, whereas mTORC2 is primarily responsible for cell proliferation and survival. Rapamycin is a widely known mTOR inhibitor that has been developed in various solid tumors and hematologic malignancies [204]. Rapamycin was primarily separated from the soil on Rapanui Island and could be used as an anti-fungal agent [205]. Rapamycin integrates antitumor cell growth and acts as an immune suppressant. Rapamycin directly combines with FKBP12 targets, which immediate rapamycin to regulate mTORC1 activity [206]. Two allosteric mTORC1 inhibitors, temsirolimus (CCI-779) and everolimus (RAD001), have been approved for the treatment of cancer [207]. Both temsirolimus and everolimus are derivatives from rapamycin [208]. The activity mechanisms of temsirolimus and everolimus were similar to that of rapamycin, which binds to FKBP12 to encourage mTOR activity [209]. Temsirolimus and everolimus disturbed Raptor binding to mTOR, contributing to mTORC1 isoform decomposition and mTORC1 inactivation [210].
A phase I trial of temsirolimus demonstrated a higher overall response rate (ORR) of advanced RCC [211]. A phase II trial exhibited promising effects in patients with metastatic renal cell carcinoma [212]. A phase III trial of temsirolimus plus other chemotherapies promoted the survival of metastatic RCC patients [213]. Overall, temsirolimus is a potent agent in treating RCC [214]. Temsirolimus plus sorafenib in advanced HC demonstrated acceptable toxicity. No grade 5 events were observed. The most frequent drug-related grade ≥ 3 AEs were hypophosphatemia, thrombocytopenia, and rash in this phase II trial. The median time to progression of advanced HC patients who received temsirolimus plus sorafenib was 3.7 months, with 14% of patients reaching a time to progression of at least 6 months [213]. The median overall survival was 8.8 months. Everolimus is another well-researched derivative from rapamycin, including advanced RCC, HR-positive/HER2-negative mammary neoplasms, pancreatic neuroendocrine tumors, and ependy malignant cell astrocytoma [215]. Everolimus monotherapy plus capecitabine and oxaliplatin is presently being evaluated in phase I and phase II clinical trials of patients with gastric carcinoma [216]. The first-generation mTOR inhibitors temsirolimus and everolimus can inhibit mTORC1, resulting in activation of the MAPK signaling pathway via PI3K. Second-generation mTOR inhibitors inhibited both TORC1 and mTORC2 [217]. AZD8055 is a second-generation mTOR inhibitor that inhibits protein synthesis in HCs, inhibits malignant mammary tumor cell proliferation and reduces tamoxifen resistance [218].
AZD2014 is another second-generation mTOR inhibitor that attenuates myeloid-derived suppressor cell recruitment and blocks cell proliferation in ovarian cancer [219]. AZD2014 increased docetaxel sensitivity and overcame docetaxel resistance in prostate carcinoma cells [220]. AZD2014 in tumors has been developed in phase I and II clinical trials. In a phase I/II randomized clinical trial, AZD2014 plus anastrozole showed therapeutic benefit and had manageable toxicity in women with HR-positive recurrent or metastatic carcinoma of the endometrium [221]. In the AcSé-ESMART trial, the absence of clinical response and deficient target management in the AZD2014-treated adult subgroup led to the termination of the study [222]. However, a phase II study claimed that dual mTORC1 and mTORC2 inhibitors were not superior to mTORC1 inhibitors in relapsed or refractory diffuse large B-cell lymphoma [223]. Second-generation mTOR inhibitors did not confer advantages over second-generation mTOR inhibitors. Approximately 30% treated with mTORC1 inhibitors had a clinical response. In diffuse large B-cell lymphoma, 6% of patients achieved partial response, with no complete response [224]. 20% of patients reached stable disease after six cycles of treatment. The effect of AZD2014 in metastatic clear cell RCC is unclear. Currently, several dual mTORC1/2 inhibitors are undergoing clinical tests for adult and pediatric tumor therapy, such as OSI-027, CC-223, and MLN0128 [225].
Overview of the RAF/MEK/ERK signaling pathway and its deregulation in cancer
Function of the RAF/MEK/ERK signaling pathway
RAF/MEK/ERK transfer signals from receptors on the cytomembrane to regulate transcription, which promotes protein synthesis and other functions (Fig. 1) [226]. This pathway has been comprehensively researched because of its role in regulating cell apoptosis, which makes inhibitors targeting the components in this pathway have potential antitumor effects. The RAF/MEK/ERK pathway, also known as the MAPK pathway, transmits signals from receptors on the cell surface to the nucleus to promote transcription [227]. This pathway could be activated in various cancers by overexpression of B7-H3, upregulated expression of decidual protein induced by progesterone, overexpression of HOXA3, and aberrant expression of the COX2/PGE axis [228]. This signaling pathway also has enormous effects on promoting tumor cell apoptosis by DR5 expression, ALDH6A1 decrease, and KIF15 expression [226]. The pathway participates in tumor cell growth, cell cycle block, apoptosis, cell adhesion and differentiation.
Construction of the RAF/MEK/ERK signaling pathway
The role of RAS in regulating the RAF/MEK/ERK signaling pathway
The complexity of this pathway was gradually observed with the academic paper published, including many factors in the transcription factor, apoptosis-promoting, and caspase executioner families [229]. Transcription of RAF genes could improve the phosphorylation of downstream proteins via MEK and ERK to control cancer cell apoptosis. The pathway positively or negatively regulates cancer cell apoptosis via different signals [230]. Alternations at upstream receptors, RAS, B-Raf and other genes contribute to abnormal RAF activation, which results in unnatural signaling pathway activity [231]. RAS, the key regulator and upstream protein of the RAF/MEK/ERK pathway, consists of four small GTPases [232]. RAS activation induced by epidermal growth factor promoted GTPase binding with upstream receptor tyrosine kinase signaling. The RAS gene family includes N-Ras, H-Ras, and K-Ras, which are the most commonly activated in human neoplasms. RAS alterations have been reported to be associated with poor overall survival. S-phase could be induced by V-Raf, indicating that RAF could be regulated by RAS or in parallel with RAS [233]. It was also confirmed by studies of Drosophila and C. elegans that RAF is understream of RTKs and RAS [234].
The cascade signaling in the RAF/MEK/ERK signaling pathway
RAF included three isoforms: A, B and C. The supporting evidence for C-Raf serving as a cellular oncogene was not sufficient. After stimulation of receptors on the cell surface for RAF activation, C-Raf was phosphorylated at the S43, S259 and S621 sites, which maintained kinase activity. A-Raf is rarely found to be mutated in neoplasms and is one of the weakest effectors in promoting MEK1. B-Raf is the strongest isoform that promotes MEK activity [235,236,237,238,239,240,241,242]. ERK is a downstream gene of a stable module stimulated by RAF ser/thr kinases. RAF activates ERK1/2 by stimulating MAPK/ERK kinase (MEK)1/2 dual-specificity protein kinases [243].
Collectively, the signaling pathway is a critical pathway in tumors and could act as a promising approach in therapy. Inhibitors in the pathway have been developed and are undergoing clinical trials [244].
Mutations in the RAF/MEK/ERK Signaling Cascade
Gene alterations in the signaling cascade can be divided into two categories: driver of neoplasms (RAF mutations) or indicator of worse survival (MEK and ERK mutations) [245]. In this section, mutations in the RAF/MEK/ERK signaling pathway are described.
RAF mutations
As mentioned above, V-Raf serves as a ser/thr protein kinase, of which C-Raf is rarely mutated in tumors [246]. The role of RAF was identified until the observation of BRAF(V600E). Alterations in BRAF are frequently observed, while the frequency of C-Raf, A-Raf, and KRAS mutations is much lower than that of BRAF [247]. BRAF mutations frequently occur in melanoma, thyroid adenocarcinoma, colon carcinoma, gallbladder carcinoma, ovary cancer, and lung carcinoma. RAF mutations in tumors are frequent in their specific regions of protein and can be separated into several subgroups according to the way they trigger the pathway [248]. Multiple groups of RAF alterations have been observed. The abnormal activation of RAF mimics the phosphorylation of the activation, which belongs to the first group [248]. The second group promoted RAF to relieve the autoinhibitory effect. The third group had no effect on its activity. They promoted the RAF/MEK/ERK signaling pathway via their wild-type counterparts. The first group of RAF mutations occurred in V600 mutations, including V600D/E/R/K, and V600E was the most common site based on the Catalog of Somatic Mutations in Cancer (COSMIC) database. The activity of B-Raf does not rely on RAS [249]. The second group of RAF mutations are commonly observed in the activation loop and Gly-rich loop, which break the autoinhibitory status and consist of an activation loop and Gly-rich loop. Class II RAF mutations could work as medium kinase activity and contribute to dimer formation. The mutations also triggered dimer ERK activation. Third group RAF mutations existed in the Gly-rich loop, the DFG motif, the catalytic loop, or the C-spine. RAF mutations lead to the inactivation of some kinases, and transactivating normal RAF can activate ERK by promoting dimerization affinity.
B-Raf is considered the primary event but is not enough for tumor formation. BRAF mutations may lead to abnormal signaling activation and protein overexpression, which contribute to cell cycle blockade [250]. In hematologic malignancies, including AMLs and acute lymphocytic leukemias (ALLs), the constitutive abnormal activation of the signaling pathway was observed without any distinct alterations [251]. Unrecognized mutations exist in the signals of the pathway, and some kinase deficiency or excess may promote the activation of the pathway. Furthermore, ERK overexpression is significantly associated with worse survival in AML and ALL patients [252]. RAF and MEK inhibitors are promising agents in some pathological subgroups of AML and ALL patients. Mutations in the signaling pathway need further exploration [253].
MEK and ERK mutations
MEK and ERK mutations are not frequently observed in tumors. Furthermore, MEK and ERK mutations did not co-occur with RAF mutations, which may demonstrate that the role of MEK and ERK mutations in neoplasms was not similar to that of RAF [245]. MEK mutations were correlated with resistance to RAF and MEK small molecule inhibitors [254, 255]. MEK deficiency facilitates resistance to RAF inhibitors by promoting ERK signaling flux. MEK mutations induced the activation of MEK signaling by disturbing its activity mediated by regulatory helix A. The activation of MEK signaling could also be turned on by MEK mutations by promoting MEK homodimerization [254, 256]. In consideration of the different mechanisms of the two types of MEK mutations and MEK signaling activation, MEK mutations presented distinct sensitivities to MEK inhibitors in animal models and clinical trials, which were similar to RAF mutations.
RAF/MEK/ERK pathway inhibitors
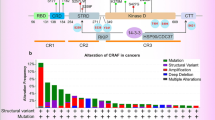
The RAF/MEK/ERK signaling pathway is critical in tumor cell growth, cell apoptosis, cell differentiation and cellular metabolism [257]. GTP-bound Ras can recruit RAF to the plasma membrane and facilitate only substrates, Mek1 and Mek2 phosphorylation [258]. Mek1 and Mek2 promote Erk1 and Erk2 activation, enabling Erk1 and Erk2 to phosphorylate more than 70 substrates containing nuclear transcription factors [259]. Based on the mechanisms and functions of the RAF/MEK/ERK signaling pathway, small molecular inhibitors targeting the signaling pathway have demonstrated excellent treatment effects in cancer patients (Fig. 3) [260]. Inhibitors of the RAF/MEK/ERK signaling pathway are potential agents in tumor therapy, and many compounds have been developed in clinical trials and preclinical studies [257]. Tour kinds RAF and MEK inhibitors have been approved. No ERK inhibitors have been approved.

Comprehensive understanding and agent direction for targeting the RAF/MEK/ERK signaling pathway in cancer treatment
RAF and MEK inhibitors
A series of RAF inhibitors are undergonig clinicals, and some are in preclinical research (Table 3). Generally, RAF inhibitors demonstrated a higher ORR in clinical cancer patients than MEK inhibitors, which may be associated with the extensive effect of RAF inhibitors that inhibit ERK activation [261]. In contrast, MEK inhibitors inhibited MEK in cancer or normal cells rather than other targets. Many inhibitors were initially considered as single target drugs, but with the pharmacodynamics of drug development, their effects on multiple targets were found [226]. This observation has no influence on their effect on tumor inhibition.
GSK2118436 (dabrafenib, Tafinlar), PLX-4032 (zelboraf, RG7204, and vemurafenib), encorafenib (Braftovi and LGX818), and sorafenib (BAY 43-9006) have been approved. It has been reported that sorafenib is approved for treating RCC and HC, while it is not a pure RAF inhibitor [262, 263]. Sorafenib has been generally considered a first-line treatment method for patients with advanced HC. Early identification of patients who would benefit from sorafenib is essential because many HC patients do not benefit from sorafenib treatment and experience intolerant toxicity [264, 265]. In phase 3 studies in patients treated with sorafenib, more serious AEs and fatal AEs were observed in patients receiving sorafenib plus nontargeted chemotherapy than in those treated with placebo or nontargeted chemotherapy [265, 266]. MEK inhibitors and other inhibitors have been used in treating advanced HC, but they have shown better effects than sorafenib [267]. It was suggested that sorafenib is a multiple target inhibitor that could suppress RAF and other targets [268].
PLX-4720 and PLX-4032 (vemurafenib and RG7204) are two inhibitors produced by Plexxikon/Roche [269]. PLX-4720 is a specific mutant B-Raf inhibitor that has not been researched in clinical trials [269, 270]. PLX-4032 was identified as a potential and selected B-Raf inhibitor of BRAF mutation signaling in 2010 and was approved by the FDA in 2017 [271]. PLX4032 showed a certain effect in melanoma patients with mutant B-RAF in phase 1–3 trials. Overall survival and progression-free survival were also prolonged in untreated melanoma patients [272]. The disease-free survival of melanoma patients was 7 months, which could be improved by PLX-4032 treatment [273]. Approximately 30% of patients treated with PLX-4032 maintained a stable disease status. The results showed that both intrinsic and acquired resistance could influence the clinical efficacy of PLX4032. Overall, the outcomes demonstrated that to prolong the survival of mutant B-RAF melanomas, more efficient small molecule inhibitors are essential. It is vital to improve PLX4032 activity to increase the clinical response and recognize the mechanisms of BRAF mutation in tumors. PLX-4720 is designed as a highly selective BRAF mutation inhibitor that can distinguish between mutant and wild-type proteins [269, 270]. Preclinically, PLX-4720 is efficient in suppressing the progression of colorectal cancer cells and melanoma cells with the V600E mutation. This in tumor cells was considered to be related to more aggressive activity and poor survival. In cellular assays, the IC50 for PLX-4720 in BRAF mutations is significantly lower than that in wild-type patients [274]. In the cell lines, the BRAF status could be detected. The IC50 of PXL-4720 was approximately 100 lower than that of sorafenib in malignant meningioma and colorectal neoplasms with the BRAF V600E mutation. In RAS mutations without BRAF mutations in cell lines, the IC50 of PLX-4720 was similar to that of sorafenib in colorectal cancers and NSCLC. PLX-4720 is a potential agent in patients with B-Raf mutations [274].
Trametinib (GSK1120212, Mekinist, and JTP 74,057) and cobimetinib (GDC-0973, Cotellic, and XL518) are approved for tumors with the BRAF V600E mutation and could be used as monotherapy or plus other therapies [275]. These inhibitors had a high specificity and could strongly suppress the MEK signaling pathway in tumor and normal cells, which do not block ATP.
Trametinib was approved in 2017 for treating unresectable BRAF V600E or V600K mutation melanoma patients [276]. The effect of trametinib is undergoing clinical trials of other neoplasms [277]. The median overall survival of locally recurrent pancreatic cancer patients treated with stereotactic body radiotherapy (SBRT) plus pembrolizumab and trametinib was 24.9 months compared with 22.4 months in patients treated with SBRT plus gemcitabine [278]. There were no therapy-related deaths reported. Compared with SBRT plus gemcitabine regimens, less neutropenia or thrombocytopenia was found in SBRT plus pembrolizumab and trametinib. SBRT plus pembrolizumab and trametinib may be a potential approach to patients with relapsed pancreatic carcinoma. Selumetinib (AZD6244, Arry-142,886) and MEK162/ARRY-483,162 (binimetinib and Mektovi) are two other agents approved by the FDA in 2016 and 2020, respectively [279, 280]. In addition to these small molecule inhibitors, dozens of MEK inhibitors are in the clinic, and some are in preclinical trials, such as refametinib (BAY86-9766, RDEA119), pimasertib (AS703026, MSC1936369B), mirdametinib (PD-0325901), CI-1040 (PD184352) and RO4987655 (Table 3).
ERK inhibitors
Some agents targeting the terminal kinase ERK have been developed, but most have been in preclinical trials, and few have been developed in clinical trials. Their effect in RAF or MEK-mutated tumors is unclear [281, 282]. Similar to MEK inhibitors, ERK inhibitors have no selectivity in tumor cells and normal cells, which may result in severe toxicity and unfavorable survival. ERK inhibitors combined with RAF inhibitors, as synergetic agents, may have amazing antitumor activity [281, 282]. In MAPK-related tumors with BRAF mutations, BRAF and MEK inhibitors improved survival. Furthermore, resistance to MAPK inhibitors results in abnormal regulation of ERK, and the phosphorylation of ERK increases [283]. The application of an ERK1/2 kinase inhibitor could relieve MAPK inhibitor resistance in tumor cells with BRAF mutations.
Overall, the application of RAF/MEK/ERK inhibitors contributes to targeted therapy for tumors, and the effect of RAF/MEK/ERK inhibitors facilitates the understanding of RAF mutations in tumor therapy. Comprehensive investigation of the relationship of RAF/MEK/ERK mutations and wild-type counterparts provides new insight into novel allosteric targets to help the treatment of neoplasms.
Cross-talk between the PI3K/AKT/mTOR and RAF/MEK/ERK pathways
Apoptosis induced by the RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways is regulated by key factors that are regulated by the phosphorylation of ERK or AKT [284]. HER2 overexpression results in abnormal activation of the RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways in breast cancer [257]. Furthermore, class III RTK uptake in upstream clouds also leads to the activation of the two pathways in AML. The suppressor p53 plays a vital role in both the PI3K/AKT/mTOR and RAF/MEK/ERK pathways [251]. The activity of p53 could be mediated by the PI3K/AKT/mTOR and RAF/MEK/ERK pathways. Approximately 90% BRAF mutations, 45% PTEN phosphatase-related gene depletion and 45% AKT amplification are found in melanomas. All these mutations contributed to the activation of AKT, which indicated poor survival. In addition to these mutations, signaling from RAS and the cell surface also leads to PI3K phosphorylation, contributing to AKT activation. Cosuppression of the RAF/MEK/ERK and PI3K/AKT/mTOR signaling pathways by RAF plus AKT inhibitors or mTOR inhibitors led to synergistic inhibition [285]. Dactolisib is an approved dual PI3K/mTOR inhibitor, and selumetinib is an approved MEK inhibitor [286]. Dactolisib combined with selumetinib resulted in the synergistic action of lung carcinoma with KRAS and PIK3CA mutations. The treatment response of a RAF kinase, sorafenib, could be enhanced by the administration of mTOR inhibitors in a hepatoma carcinoma cell tumor xenograft model [287, 288]. Dactolisib improved the treatment effect of Raf inhibitors in differentiated and medullary thyroid cancers.
Cell growth and protein production were suppressed by inhibiting MEK and mTOR simultaneously in human NSCLC cells [289]. Inhibiting both MEK and mTOR suppressed ribosomal biogenesis and was related to obstruction of translation [290, 291]. The results were identified in a mouse xenograft model. These results indicated that inhibiting both pathways could not only promote cell apoptosis but also converge to mediate the production of proteins. ERK mediates the initial translation activity via Mnk1/2 and p90Rsk phosphorylation [285]. Phosphorylated 4EBP1 is inhibited in BRAF-mutant cells [292]. AKT and mTOR were suppressed, and the activity of 4EBP1 was inhibited. It is clear that the translation of some mRNAs was promoted in cells with BRAF mutations. The production of certain proteins may result in synergistic responses. More potent mechanisms need further exploration.
Improving the effect of targeting the RAF/MEK/ERK and PI3K/AKT/mTOR pathways by simultaneous treatment
With the development of RAF/MEK/ERK and PI3K/AKT/mTOR pathways, combinations of RAF and PI3K/AKT/mTOR or MEK and PI3K/AKT/mTOR inhibitors are undergoing clinical trials. The combination application of the agents in advanced solid neoplasms was well tolerated and was a single agent. The clinical response and long-term survival were observed. The signals from the RAF/MEK/ERK signaling pathway negated PI3K/AKT/mTOR activation. Both signaling pathways are regulated by RAS and RTK on the surface and stimulated by second messengers. ERK phosphorylation was promoted by mTORC1 dephosphorylation of its residues in malignant mammary patients after therapy. The RAF/MEK/ERK signaling pathway was activated in patients with TNBC treated with buparlisib. The MEK inhibitor combined with buparlisib demonstrated a superior antitumor effect, indicating that inhibiting the PI3K/AKT/mTOR and MAPK/MEK/ERK pathways could have synergistic action [293, 294]. In contrast, PI3K inhibitors also play vital roles in RAF/MEK/ERK signaling pathway dysregulation. The alterations in PTEN may upregulate AKT activity, which attenuates apoptosis-induced BRAF inhibition. However, PI3K inhibitors plus BRAF inhibitors demonstrated promising effects compared with single inhibitor treatment [295, 296]. An AKT inhibitor (GSK2141795) and MEK inhibitor (GSK1120212) combination have been used in cancer patients. The study was completed in 2017, but no results were posted. The primary clinical response to PI3K/mTOR inhibitors (gedatolisib) plus MEK inhibitor (PD-0325901) was presented in ovarian carcinoma and endometrial carcinoma. According to the potential effect of the agents combination, MEK1/2 inhibitor (MEK162) plus the PI3K/mTOR dual inhibitor (BEZ-235) was also evaluated in selected advanced solid cancer patients, which included EGFR mutant NSCLC patients with response to EGFR inhibitors, TNBC, pancreatic carcinoma, colon carcinoma and melanoma with KRAS, NRAS, and/or BRAF mutations patients. The combination of a dual PI3K/mTOR inhibitor (BEZ-235) and a mTOR inhibitor (RAD-001) was evaluated in a phase 1 clinical trial. A MEK inhibitor (MSC1936369B) combined with a PI3K/mTOR inhibitor (SAR245409) demonstrated severe toxicity and poor clinical response [169].
Conclusion
Both the PI3K/AKT/mTOR and RAF/MEK/ERK pathways are frequently involved in cancer therapy. Both PI3K/AKT/mTOR and RAF/MEK/ERK can be regulated by p53, which contributes to cell proliferation, drug resistance, cell cycle progression and tumor metastasis [71]. Many oncogenes are trapped by retroviruses, including RAS, PI3K, AKT, Src, Abl, RAF, Fos, and Jun [229]. These genes in the two pathways have been found to be frequently and aberrantly mediated. Alternations in upstream receptors serving to PI3K/AKT/mTOR and RAF/MEK/ERK activation have also been widely discussed in the development of tumors [257]. Mutations upstream result in multiple abnormal downstream activation. These factors lead to the complicated treatment of tumors and targeted drug failure because single-target small molecular inhibitors may not suppress additional downstream factor activity. In addition, more subsequent mutations may be acquired, which contribute to resistance and tumor cell anti-apoptosis. The results indicated that mutations and additional activation should be taken into consideration during the development of inhibitors targeting these genes, which may relieve drug resistance. Although the mechanisms of the PI3K/AKT/mTOR and RAF/MEK/ERK pathways are different, the two pathways share many downstream targets that may promote cell proliferation and facilitate drug resistance in an alternative way. In addition to mutations, abnormal activation is another reason for tumor development and drug resistance. This reason also leads to limited effectiveness in patients treated with inhibitors.
In the RAF/MEK/ERK pathway, MEK inhibitors initially demonstrated the most specificity [230]. Some neoplasm patients benefit from MEK inhibitors, but some do not. MEK activation may not be the only target responsible for the disease. This limited the efficiency of MEK inhibitors. Therefore, the combination of MEK inhibitors with chemotherapy or radiotherapy may demonstrate promising effects. MEK inhibitor monotherapy promoted cell apoptosis and induced AEs. Therefore, the tolerance of the combination therapy deserves more attention [245]. A similar situation was observed in selective RAF inhibitors, while some Raf inhibitors have multiple targets. In addition to suppressing BRAF, sorafenib also inhibited VEGFR and PDGFR to block the cell cycle, promote cell apoptosis and relieve drug resistance. This promiscuous nature of sorafenib makes it effective in certain cancers. In addition to combination with chemotherapy and radiotherapy, dual inhibitors are another hot spot, such as RAF and PI3K inhibitors. These targets are upstream of pathways, and mutations are frequent, which may result in exciting effectiveness. The trouble is that RAS activation leads to Raf-1 activation, which may have a limited response to the inhibitors [230]. Therefore, inhibitors in combination with traditional drug/physical treatment are still widely researched. Likewise, dual PI3K and mTOR inhibitors may be a potential antitumor approach compared with either PI3K or mTOR inhibitors. Dozens of dual PI3K and mTOR inhibitors are undergoing preclinical assays and clinical trials. The effectiveness and toxicity are expected.
Thus, the concepts of the two pathways were summarized. First, with the wide understanding of the PI3K/AKT/mTOR and RAF/MEK/ERK pathways, hundreds of inhibitors have been developed. Some of them demonstrated promising effects and moderate toxicity in particular cancers, while some were abolished for certain reasons. Some potential mechanisms that are unknown with further exploration are essential. Second, crosstalk between PI3K/AKT/mTOR and RAF/MEK/ERK is complex and not fully discussed. The targets in the two pathways dependent or independently regulated cell proliferation, apoptosis, and other characteristics. Therefore, multiple target inhibitors are desirable. Third, some inhibitors were abolished, and some clinical trials were withdrawn because of their toxicity and AEs. How to solve this is another crucial question.
The PI3K/AKT/mTOR and RAF/MEK/ERK pathways are intriguing aspects of human cancer therapy and are two complex cascades containing many targets. Further studies on reducing toxicity and improving effectiveness should be conducted. Accumulated studies on these two pathways will provide new hope for cancer patients.
References
Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec. (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1(7):493–502. https://doi.org/10.1038/nrd839.
Nørøxe DS, Poulsen HS, Lassen U. Hallmarks of glioblastoma: a systematic review. ESMO Open. 2016;1(6):e000144. https://doi.org/10.1136/esmoopen-2016-000144.
Passirani C, Vessières A, La Regina G, Link W, Silvestri R. Modulating undruggable targets to overcome cancer therapy resistance. Drug Resist Updat. 2022;60:100788. https://doi.org/10.1016/j.drup.2021.100788.
Bononi A, Agnoletto C, De Marchi E, Marchi S, Patergnani S, Bonora M, et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011;2011:329098. https://doi.org/10.4061/2011/329098.
Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36(7):422–39. https://doi.org/10.1016/j.tips.2015.04.005.
Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging. 2011;3(3):192–222. https://doi.org/10.18632/aging.100296.
Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–41. https://doi.org/10.1016/j.ejmech.2016.01.012.
Le Rhun E, Bertrand N, Dumont A, Tresch E, Le Deley MC, Mailliez A, et al. Identification of single nucleotide polymorphisms of the PI3K-AKT-mTOR pathway as a risk factor of central nervous system metastasis in metastatic breast cancer. Eur J Cancer. 2017;87:189–98. https://doi.org/10.1016/j.ejca.2017.10.006.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381–405. https://doi.org/10.1016/j.cell.2017.04.001.
Daniel PM, Filiz G, Brown DV, Christie M, Waring PM, Zhang Y, et al. PI3K activation in neural stem cells drives tumorigenesis which can be ameliorated by targeting the cAMP response element binding protein. Neuro Oncol. 2018;20(10):1344–55. https://doi.org/10.1093/neuonc/noy068.
Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–95. https://doi.org/10.1016/j.febslet.2010.01.017.
Dossou AS, Basu A. The emerging roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel). 2019;11(10). https://doi.org/10.3390/cancers11101422.
Lin YT, Wang HC, Hsu YC, Cho CL, Yang MY, Chien CY. Capsaicin induces autophagy and apoptosis in human nasopharyngeal carcinoma cells by downregulating the PI3K/AKT/mTOR pathway. Int J Mol Sci. 2017;18(7). https://doi.org/10.3390/ijms18071343.
Sohn EJ, Park HT. Natural agents mediated autophagic signal networks in cancer. Cancer Cell Int. 2017;17:110. https://doi.org/10.1186/s12935-017-0486-7.
Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li JH, et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int J Cancer. 2008;122(2):433–43. https://doi.org/10.1002/ijc.23049.
Yu LL, Dai N, Yu HG, Sun LM, Si JM. Akt associates with nuclear factor kappaB and plays an important role in chemoresistance of gastric cancer cells. Oncol Rep. 2010;24(1):113–9. https://doi.org/10.3892/or_00000835.
Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 2015;9(4):317–24. https://doi.org/10.1080/19336918.2015.1016686.
Dey S, Singh AK, Singh AK, Rawat K, Banerjee J, Agnihotri V, et al. Critical pathways of oral squamous cell carcinoma: molecular biomarker and therapeutic intervention. Med Oncol. 2022;39(3):30. https://doi.org/10.1007/s12032-021-01633-4.
Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–8. https://doi.org/10.1016/j.tibs.2011.03.006.
Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–25. https://doi.org/10.1158/1078-0432.CCR-11-2381.
Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71(6):1395–409. https://doi.org/10.1007/s00280-013-2121-1.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3(9):954–87. https://doi.org/10.18632/oncotarget.652.
Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25(9):545–55. https://doi.org/10.1016/j.tcb.2015.06.002.
Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189(4):1177–201. https://doi.org/10.1534/genetics.111.133363.
Hassan B, Akcakanat A, Holder AM, Meric-Bernstam F. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am. 2013;22(4):641–64. https://doi.org/10.1016/j.soc.2013.06.008.
Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in Cancer. Front Oncol. 2014;4:64. https://doi.org/10.3389/fonc.2014.00064.
Tariq K, Luikart BW. Striking a balance: PIP2 and PIP3 signaling in neuronal health and disease. Explor Neuroprotective Ther. 2021;1:86–100. https://doi.org/10.37349/ent.2021.00008.
Verret B, Cortes J, Bachelot T, Andre F, Arnedos M. Efficacy of PI3K inhibitors in advanced breast cancer. Ann Oncol. 2019;30 Suppl 10:x12–12x20. https://doi.org/10.1093/annonc/mdz381.
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–56. https://doi.org/10.1038/nrd4204.
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–41. https://doi.org/10.1038/nrm2882.
Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9(6):667–76. https://doi.org/10.1023/B:APPT.0000045801.15585.dd.
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627–44. https://doi.org/10.1038/nrd2926.
Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for Cancer Treatment. Annu Rev Med. 2016;67:11–28. https://doi.org/10.1146/annurev-med-062913-051343.
Margaria JP, Ratto E, Gozzelino L, Li H, Hirsch E. Class II PI3Ks at the intersection between Signal transduction and membrane trafficking. Biomolecules. 2019;9(3). https://doi.org/10.3390/biom9030104.
Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. https://doi.org/10.1146/annurev.pathol.4.110807.092311.
Hermida MA, Dinesh Kumar J, Leslie NR. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul. 2017;65:5–15. https://doi.org/10.1016/j.jbior.2017.06.003.
Ben-Sahra I, Manning BD. mTORC1 signaling and the metabolic control of cell growth. Curr Opin Cell Biol. 2017;45:72–82. https://doi.org/10.1016/j.ceb.2017.02.012.
Takahara T, Amemiya Y, Sugiyama R, Maki M, Shibata H. Amino acid-dependent control of mTORC1 signaling: a variety of regulatory modes. J Biomed Sci. 2020;27(1):87. https://doi.org/10.1186/s12929-020-00679-2.
Liu SY, Chen W, Chughtai EA, Qiao Z, Jiang JT, Li SM, et al. PIK3CA gene mutations in Northwest Chinese esophageal squamous cell carcinoma. World J Gastroenterol. 2017;23(14):2585–91. https://doi.org/10.3748/wjg.v23.i14.2585.
Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol. 2010;347:21–41. https://doi.org/10.1007/82_2010_68.
Martínez-Sáez O, Chic N, Pascual T, Adamo B, Vidal M, González-Farré B, et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020;22(1):45. https://doi.org/10.1186/s13058-020-01284-9.
Shi J, Yao D, Liu W, Wang N, Lv H, Zhang G, et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer. 2012;12:50. https://doi.org/10.1186/1471-2407-12-50.
Liu JF, Zhou XK, Chen JH, Yi G, Chen HG, Ba MC, et al. Up-regulation of PIK3CA promotes metastasis in gastric carcinoma. World J Gastroenterol. 2010;16(39):4986–91. https://doi.org/10.3748/wjg.v16.i39.4986.
Huang R, Dai Q, Yang R, Duan Y, Zhao Q, Haybaeck J, et al. A review: PI3K/AKT/mTOR signaling pathway and its regulated eukaryotic translation initiation factors may be a potential therapeutic target in esophageal squamous cell carcinoma. Front Oncol. 2022;12:817916. https://doi.org/10.3389/fonc.2022.817916.
Miricescu D, Totan A, Stanescu-Spinu II, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast Cancer: from Molecular Landscape to clinical aspects. Int J Mol Sci. 2020;22(1). https://doi.org/10.3390/ijms22010173.
Yudushkin I. Getting the akt together: guiding intracellular akt activity by PI3K. Biomolecules. 2019;9(2). https://doi.org/10.3390/biom9020067.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20(2):74–88. https://doi.org/10.1038/s41568-019-0216-7.
Parikh C, Janakiraman V, Wu WI, Foo CK, Kljavin NM, Chaudhuri S, et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci U S A. 2012;109(47):19368–73. https://doi.org/10.1073/pnas.1204384109.
Abotaleb M, Samuel SM, Varghese E, Varghese S, Kubatka P, Liskova A, et al. Flavonoids in Cancer and apoptosis. Cancers (Basel). 2018;11(1). https://doi.org/10.3390/cancers11010028.
Rychahou PG, Kang J, Gulhati P, Doan HQ, Chen LA, Xiao SY, et al. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc Natl Acad Sci U S A. 2008;105(51):20315–20. https://doi.org/10.1073/pnas.0810715105.
López-Cortés A, Leone PE, Freire-Paspuel B, Arcos-Villacís N, Guevara-Ramírez P, Rosales F, et al. Mutational analysis of oncogenic AKT1 Gene Associated with breast Cancer risk in the high Altitude ecuadorian Mestizo Population. Biomed Res Int. 2018;2018:7463832. https://doi.org/10.1155/2018/7463832.
Malanga D, Belmonte S, Colelli F, Scarfò M, De Marco C, Oliveira DM, et al. AKT1E17K is oncogenic in mouse lung and cooperates with Chemical Carcinogens in inducing Lung Cancer. PLoS ONE. 2016;11(2):e0147334. https://doi.org/10.1371/journal.pone.0147334.
Mancini ML, Lien EC, Toker A. Oncogenic AKT1(E17K) mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis. Oncotarget. 2016;7(14):17301–13. https://doi.org/10.18632/oncotarget.8191.
McDonell LM, Kernohan KD, Boycott KM, Sawyer SL. Receptor tyrosine kinase mutations in developmental syndromes and cancer: two sides of the same coin. Hum Mol Genet. 2015;24(R1):R60–6. https://doi.org/10.1093/hmg/ddv254.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. https://doi.org/10.1016/j.cell.2012.03.017.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13. https://doi.org/10.1038/nm.4333.
Peng Y, Wang Y, Zhou C, Mei W, Zeng C. PI3K/Akt/mTOR pathway and its role in Cancer therapeutics: are we making Headway. Front Oncol. 2022;12:819128. https://doi.org/10.3389/fonc.2022.819128.
Tian T, Li X, Zhang J. mTOR Signaling in Cancer and mTOR inhibitors in solid Tumor Targeting Therapy. Int J Mol Sci. 2019;20(3). https://doi.org/10.3390/ijms20030755.
Kim LC, Rhee CH, Chen J. RICTOR amplification promotes NSCLC cell proliferation through formation and activation of mTORC2 at the expense of mTORC1. Mol Cancer Res. 2020;18(11):1675–84. https://doi.org/10.1158/1541-7786.MCR-20-0262.
Sekulić A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60(13):3504–13.
Álvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: it’s all about diversity. Semin Cancer Biol. 2019;59:66–79. https://doi.org/10.1016/j.semcancer.2019.02.001.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–7. https://doi.org/10.1126/science.275.5308.1943.
Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, et al. P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci U S A. 1997;94(17):9052–7. https://doi.org/10.1073/pnas.94.17.9052.
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13(5):283–96. https://doi.org/10.1038/nrm3330.
Tachibana M, Shibakita M, Ohno S, Kinugasa S, Yoshimura H, Ueda S, et al. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer. 2002;94(7):1955–60. https://doi.org/10.1002/cncr.0678.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27. https://doi.org/10.1016/j.ccr.2004.06.022.
Sawai H, Yasuda A, Ochi N, Ma J, Matsuo Y, Wakasugi T, et al. Loss of PTEN expression is associated with colorectal cancer liver metastasis and poor patient survival. BMC Gastroenterol. 2008;8:56. https://doi.org/10.1186/1471-230X-8-56.
Bucheit AD, Chen G, Siroy A, Tetzlaff M, Broaddus R, Milton D, et al. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin Cancer Res. 2014;20(21):5527–36. https://doi.org/10.1158/1078-0432.CCR-14-1027.
Mithal P, Allott E, Gerber L, Reid J, Welbourn W, Tikishvili E, et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int J Urol. 2014;21(12):1209–14. https://doi.org/10.1111/iju.12571.
da Costa AA, D’Almeida Costa F, Ribeiro AR, Guimarães AP, Chinen LT, Lopes CA, et al. Low PTEN expression is associated with worse overall survival in head and neck squamous cell carcinoma patients treated with chemotherapy and cetuximab. Int J Clin Oncol. 2015;20(2):282–9. https://doi.org/10.1007/s10147-014-0707-1.
Wang X, Cao X, Sun R, Tang C, Tzankov A, Zhang J, et al. Clinical significance of PTEN deletion, mutation, and loss of PTEN expression in De Novo diffuse large B-Cell lymphoma. Neoplasia. 2018;20(6):574–93. https://doi.org/10.1016/j.neo.2018.03.002.
Xie P, Peng Z, Chen Y, Li H, Du M, Tan Y, et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021;31(3):291–311. https://doi.org/10.1038/s41422-020-00443-z.
Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6(3):184–92. https://doi.org/10.1038/nrc1819.
Wadhwa B, Makhdoomi U, Vishwakarma R, Malik F. Protein kinase B: emerging mechanisms of isoform-specific regulation of cellular signaling in cancer. Anticancer Drugs. 2017;28(6):569–80. https://doi.org/10.1097/CAD.0000000000000496.
Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of ras. Nature. 1994;370(6490):527–32. https://doi.org/10.1038/370527a0.
Tsao H, Zhang X, Fowlkes K, Haluska FG. Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 2000;60(7):1800–4.
Ikeda T, Yoshinaga K, Suzuki A, Sakurada A, Ohmori H, Horii A. Anticorresponding mutations of the KRAS and PTEN genes in human endometrial cancer. Oncol Rep. 2000;7(3):567–70. https://doi.org/10.3892/or.7.3.567.
Liu W, James CD, Frederick L, Alderete BE, Jenkins RB. PTEN/MMAC1 mutations and EGFR amplification in glioblastomas. Cancer Res. 1997;57(23):5254–7.
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. https://doi.org/10.1038/nrc969.
Mao JH, To MD, Perez-Losada J, Wu D, Del Rosario R, Balmain A. Mutually exclusive mutations of the Pten and ras pathways in skin tumor progression. Genes Dev. 2004;18(15):1800–5. https://doi.org/10.1101/gad.1213804.
Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009;69(7):2766–74. https://doi.org/10.1158/0008-5472.CAN-08-3070.
Mueller S, Phillips J, Onar-Thomas A, Romero E, Zheng S, Wiencke JK, et al. PTEN promoter methylation and activation of the PI3K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro Oncol. 2012;14(9):1146–52. https://doi.org/10.1093/neuonc/nos140.
Feng J, Dang Y, Zhang W, Zhao X, Zhang C, Hou Z, et al. PTEN arginine methylation by PRMT6 suppresses PI3K-AKT signaling and modulates pre-mRNA splicing. Proc Natl Acad Sci U S A. 2019;116(14):6868–77. https://doi.org/10.1073/pnas.1811028116.
Tang Y, Eng C. PTEN autoregulates its expression by stabilization of p53 in a phosphatase-independent manner. Cancer Res. 2006;66(2):736–42. https://doi.org/10.1158/0008-5472.CAN-05-1557.
Kim MR, Choi HK, Cho KB, Kim HS, Kang KW. Involvement of Pin1 induction in epithelial-mesenchymal transition of tamoxifen-resistant breast cancer cells. Cancer Sci. 2009;100(10):1834–41. https://doi.org/10.1111/j.1349-7006.2009.01260.x.
Tsukamoto T, Hama S, Kogure K, Tsuchiya H. Selenate induces epithelial-mesenchymal transition in a colorectal carcinoma cell line by AKT activation. Exp Cell Res. 2013;319(13):1913–21. https://doi.org/10.1016/j.yexcr.2013.05.031.
Gan L, Xu M, Hua R, Tan C, Zhang J, Gong Y, et al. The polycomb group protein EZH2 induces epithelial-mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J Hematol Oncol. 2018;11(1):9. https://doi.org/10.1186/s13045-017-0547-3.
Lionarons DA, Hancock DC, Rana S, East P, Moore C, Murillo MM, et al. RAC1P29S induces a mesenchymal phenotypic switch via serum response factor to promote Melanoma Development and Therapy Resistance. Cancer Cell. 2019;36(1):68–83.e9. https://doi.org/10.1016/j.ccell.2019.05.015.
Tanwar PS, Mohapatra G, Chiang S, Engler DA, Zhang L, Kaneko-Tarui T, et al. Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis. 2014;35(3):546–53. https://doi.org/10.1093/carcin/bgt357.
Gupta A, Anjomani-Virmouni S, Koundouros N, Dimitriadi M, Choo-Wing R, Valle A, et al. PARK2 depletion connects Energy and oxidative stress to PI3K/Akt activation via PTEN S-Nitrosylation. Mol Cell. 2017;65(6):999–1013.e7. https://doi.org/10.1016/j.molcel.2017.02.019.
Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988–1004. https://doi.org/10.1038/nrd1902.
Pollizzi K, Malinowska-Kolodziej I, Doughty C, Betz C, Ma J, Goto J, et al. A hypomorphic allele of Tsc2 highlights the role of TSC1/TSC2 in signaling to AKT and models mild human TSC2 alleles. Hum Mol Genet. 2009;18(13):2378–87. https://doi.org/10.1093/hmg/ddp176.
Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. https://doi.org/10.1038/ncomms5846.
Parker BC, Zhang W. Fusion genes in solid tumors: an emerging target for cancer diagnosis and treatment. Chin J Cancer. 2013;32(11):594–603. https://doi.org/10.5732/cjc.013.10178.
Kraya AA, Maxwell KN, Eiva MA, Wubbenhorst B, Pluta J, Feldman M, et al. PTEN loss and BRCA1 promoter Hypermethylation negatively predict for immunogenicity in BRCA-Deficient ovarian Cancer. JCO Precis Oncol. 2022;6:e2100159. https://doi.org/10.1200/PO.21.00159.
Yun JW, Yang L, Park HY, Lee CW, Cha H, Shin HT, et al. Dysregulation of cancer genes by recurrent intergenic fusions. Genome Biol. 2020;21(1):166. https://doi.org/10.1186/s13059-020-02076-2.
Slotkin EK, Diolaiti D, Shukla NN, Dela Cruz FS, Clark JJ, Gundem G, et al. Patient-Driven Discovery, Therapeutic Targeting, and post-clinical validation of a Novel AKT1 Fusion-Driven Cancer. Cancer Discov. 2019;9(5):605–16. https://doi.org/10.1158/2159-8290.CD-18-0953.
Toretsky JA, Thakar M, Eskenazi AE, Frantz CN. Phosphoinositide 3-hydroxide kinase blockade enhances apoptosis in the Ewing’s sarcoma family of tumors. Cancer Res. 1999;59(22):5745–50.
Cen L, Arnoczky KJ, Hsieh FC, Lin HJ, Qualman SJ, Yu S, et al. Phosphorylation profiles of protein kinases in alveolar and embryonal rhabdomyosarcoma. Mod Pathol. 2007;20(9):936–46. https://doi.org/10.1038/modpathol.3800834.
Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67(2):735–45. https://doi.org/10.1158/0008-5472.CAN-06-2201.
Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–9. https://doi.org/10.1038/nature11154.
Perry JA, Kiezun A, Tonzi P, Van Allen EM, Carter SL, Baca SC, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A. 2014;111(51):E5564–73. https://doi.org/10.1073/pnas.1419260111.
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46(5):444–50. https://doi.org/10.1038/ng.2938.
Kannan K, Coarfa C, Chao PW, Luo L, Wang Y, Brinegar AE, et al. Recurrent BCAM-AKT2 fusion gene leads to a constitutively activated AKT2 fusion kinase in high-grade serous ovarian carcinoma. Proc Natl Acad Sci U S A. 2015;112(11):E1272–7. https://doi.org/10.1073/pnas.1501735112.
Mosquera JM, Varma S, Pauli C, MacDonald TY, Yashinskie JJ, Varga Z, et al. MAGI3-AKT3 fusion in breast cancer amended. Nature. 2015;520(7547):E11–2. https://doi.org/10.1038/nature14265.
Matissek KJ, Onozato ML, Sun S, Zheng Z, Schultz A, Lee J, et al. Expressed gene fusions as frequent drivers of poor outcomes in hormone receptor-positive breast Cancer. Cancer Discov. 2018;8(3):336–53. https://doi.org/10.1158/2159-8290.CD-17-0535.
Alaei-Mahabadi B, Bhadury J, Karlsson JW, Nilsson JA, Larsson E. Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers. Proc Natl Acad Sci U S A. 2016;113(48):13768–73. https://doi.org/10.1073/pnas.1606220113.
Yu YP, Liu P, Nelson J, Hamilton RL, Bhargava R, Michalopoulos G, et al. Identification of recurrent fusion genes across multiple cancer types. Sci Rep. 2019;9(1):1074. https://doi.org/10.1038/s41598-019-38550-6.
Lesma E, Sirchia SM, Ancona S, Carelli S, Bosari S, Ghelma F, et al. The methylation of the TSC2 promoter underlies the abnormal growth of TSC2 angiomyolipoma-derived smooth muscle cells. Am J Pathol. 2009;174(6):2150–9. https://doi.org/10.2353/ajpath.2009.080799.
Kamran S, Sinniah A, Abdulghani M, Alshawsh MA. Therapeutic potential of certain terpenoids as Anticancer Agents: a scoping review. Cancers (Basel). 2022;14(5). https://doi.org/10.3390/cancers14051100.
Cantile M, Di Bonito M, Tracey De Bellis M, Botti G. Functional Interaction among lncRNA HOTAIR and MicroRNAs in Cancer and Other Human Diseases. Cancers (Basel). 2021;13(3). https://doi.org/10.3390/cancers13030570.
Jiang WG, Sampson J, Martin TA, Lee-Jones L, Watkins G, Douglas-Jones A, et al. Tuberin and hamartin are aberrantly expressed and linked to clinical outcome in human breast cancer: the role of promoter methylation of TSC genes. Eur J Cancer. 2005;41(11):1628–36. https://doi.org/10.1016/j.ejca.2005.03.023.
Xu Z, Wang M, Wang L, Wang Y, Zhao X, Rao Q, et al. Aberrant expression of TSC2 gene in the newly diagnosed acute leukemia. Leuk Res. 2009;33(7):891–7. https://doi.org/10.1016/j.leukres.2009.01.041.
Chakraborty S, Mohiyuddin SM, Gopinath KS, Kumar A. Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer. 2008;8:163. https://doi.org/10.1186/1471-2407-8-163.
Wang N, Li Y, Wei J, Pu J, Liu R, Yang Q, et al. TBX1 functions as a tumor suppressor in thyroid Cancer through inhibiting the activities of the PI3K/AKT and MAPK/ERK pathways. Thyroid. 2019;29(3):378–94. https://doi.org/10.1089/thy.2018.0312.
Akao Y, Nakagawa Y, Kitade Y, Kinoshita T, Naoe T. Downregulation of microRNAs-143 and – 145 in B-cell malignancies. Cancer Sci. 2007;98(12):1914–20. https://doi.org/10.1111/j.1349-7006.2007.00618.x.
Baik SH, Lee J, Lee YS, Jang JY, Kim CW. ANT2 shRNA downregulates miR-19a and miR-96 through the PI3K/Akt pathway and suppresses tumor growth in hepatocellular carcinoma cells. Exp Mol Med. 2016;48:e222. https://doi.org/10.1038/emm.2015.126.
Chen X, Dong H, Liu S, Yu L, Yan D, Yao X, et al. Long noncoding RNA MHENCR promotes melanoma progression via regulating miR-425/489-mediated PI3K-Akt pathway. Am J Transl Res. 2017;9(1):90–102.
Yang NQ, Luo XJ, Zhang J, Wang GM, Guo JM. Crosstalk between Meg3 and miR-1297 regulates growth of testicular germ cell tumor through PTEN/PI3K/AKT pathway. Am J Transl Res. 2016;8(2):1091–9.
Thivierge C, Tseng HW, Mayya VK, Lussier C, Gravel SP, Duchaine TF. Alternative polyadenylation confers Pten mRNAs stability and resistance to microRNAs. Nucleic Acids Res. 2018;46(19):10340–52. https://doi.org/10.1093/nar/gky666.
Jafari Najaf Abadi MH, Shafabakhsh R, Asemi Z, Mirzaei HR, Sahebnasagh R, Mirzaei H, et al. CFIm25 and alternative polyadenylation: conflicting roles in cancer. Cancer Lett. 2019;459:112–21. https://doi.org/10.1016/j.canlet.2019.114430.
Liu R, Chen Y, Liu G, Li C, Song Y, Cao Z, et al. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020;11(9):797. https://doi.org/10.1038/s41419-020-02998-6.
Rascio F, Spadaccino F, Rocchetti MT, Castellano G, Stallone G, Netti GS, et al. The pathogenic role of PI3K/AKT pathway in Cancer Onset and Drug Resistance: an updated review. Cancers (Basel). 2021;13(16). https://doi.org/10.3390/cancers13163949.
Mishra R, Patel H, Alanazi S, Kilroy MK, Garrett JT. PI3K inhibitors in Cancer: clinical implications and adverse Effects. Int J Mol Sci. 2021;22(7). https://doi.org/10.3390/ijms22073464.
Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6(1):88. https://doi.org/10.1186/1756-8722-6-88.
Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36(10):1170–6. https://doi.org/10.1038/aps.2015.71.
Meng D, He W, Zhang Y, Liang Z, Zheng J, Zhang X, et al. Development of PI3K inhibitors: advances in clinical trials and new strategies (review). Pharmacol Res. 2021;173:105900. https://doi.org/10.1016/j.phrs.2021.105900.
Baselga J, Im SA, Iwata H, Cortés J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904–16. https://doi.org/10.1016/S1470-2045(17)30376-5.
Batalini F, Gulhan DC, Mao V, Tran A, Polak M, Xiong N, et al. Mutational signature 3 detected from clinical panel sequencing is associated with responses to olaparib in breast and ovarian cancers. Clin Cancer Res. 2022. https://doi.org/10.1158/1078-0432.CCR-22-0749.
de Gooijer MC, Zhang P, Buil L, Çitirikkaya CH, Thota N, Beijnen JH, et al. Buparlisib is a brain penetrable pan-PI3K inhibitor. Sci Rep. 2018;8(1):10784. https://doi.org/10.1038/s41598-018-29062-w.
Xing J, Yang J, Gu Y, Yi J. Research update on the anticancer effects of buparlisib. Oncol Lett. 2021;21(4):266. https://doi.org/10.3892/ol.2021.12527.
Garrido-Castro AC, Saura C, Barroso-Sousa R, Guo H, Ciruelos E, Bermejo B, et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020;22(1):120. https://doi.org/10.1186/s13058-020-01354-y.
McRee AJ, Marcom PK, Moore DT, Zamboni WC, Kornblum ZA, Hu Z, et al. A phase I trial of the PI3K inhibitor Buparlisib Combined with Capecitabine in patients with metastatic breast Cancer. Clin Breast Cancer. 2018;18(4):289–97. https://doi.org/10.1016/j.clbc.2017.10.014.
Hainsworth JD, Becker KP, Mekhail T, Chowdhary SA, Eakle JF, Wright D, et al. Phase I/II study of bevacizumab with BKM120, an oral PI3K inhibitor, in patients with refractory solid tumors (phase I) and relapsed/refractory glioblastoma (phase II). J Neurooncol. 2019;144(2):303–11. https://doi.org/10.1007/s11060-019-03227-7.
McGowan DR, Skwarski M, Bradley KM, Campo L, Fenwick JD, Gleeson FV, et al. Buparlisib with thoracic radiotherapy and its effect on tumour hypoxia: a phase I study in patients with advanced non-small cell lung carcinoma. Eur J Cancer. 2019;113:87–95. https://doi.org/10.1016/j.ejca.2019.03.015.
Kojima T, Kato K, Hara H, Takahashi S, Muro K, Nishina T, et al. Phase II study of BKM120 in patients with advanced esophageal squamous cell carcinoma (EPOC1303). Esophagus. 2022;19(4):702–10. https://doi.org/10.1007/s10388-022-00928-3.
Heudel PE, Fabbro M, Roemer-Becuwe C, Kaminsky MC, Arnaud A, Joly F, et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: a stratified type I-type II study from the GINECO group. Br J Cancer. 2017;116(3):303–9. https://doi.org/10.1038/bjc.2016.430.
Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2018;19(1):87–100. https://doi.org/10.1016/S1470-2045(17)30688-5.
Soulières D, Faivre S, Mesía R, Remenár É, Li SH, Karpenko A, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017;18(3):323–35. https://doi.org/10.1016/S1470-2045(17)30064-5.
Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, et al. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol Cancer Ther. 2013;12(11):2319–30. https://doi.org/10.1158/1535-7163.MCT-12-0993-T.
Patnaik A, Appleman LJ, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-hodgkin’s lymphomas. Ann Oncol. 2016;27(10):1928–40. https://doi.org/10.1093/annonc/mdw282.
Doi T, Fuse N, Yoshino T, Kojima T, Bando H, Miyamoto H, et al. A phase I study of intravenous PI3K inhibitor copanlisib in japanese patients with advanced or refractory solid tumors. Cancer Chemother Pharmacol. 2017;79(1):89–98. https://doi.org/10.1007/s00280-016-3198-0.
Liu W, Ping L, Xie Y, Sun Y, Du T, Niu Y, et al. A phase I pharmacokinetic study of copanlisib in chinese patients with relapsed indolent non-hodgkin lymphoma. Cancer Chemother Pharmacol. 2022;89(6):825–31. https://doi.org/10.1007/s00280-022-04417-3.
Grunenberg A, Kaiser LM, Woelfle S, Schmelzle B, Viardot A, Möller P, et al. A phase II study of the PI3K inhibitor copanlisib in combination with the anti-CD20 monoclonal antibody rituximab for patients with marginal zone lymphoma: treatment rationale and protocol design of the COUP-1 trial. BMC Cancer. 2021;21(1):749. https://doi.org/10.1186/s12885-021-08464-6.
Dreyling M, Morschhauser F, Bouabdallah K, Bron D, Cunningham D, Assouline SE, et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann Oncol. 2017;28(9):2169–78. https://doi.org/10.1093/annonc/mdx289.
Dreyling M, Santoro A, Mollica L, Leppä S, Follows G, Lenz G, et al. Long-term safety and efficacy of the PI3K inhibitor copanlisib in patients with relapsed or refractory indolent lymphoma: 2-year follow-up of the CHRONOS-1 study. Am J Hematol. 2020;95(4):362–71. https://doi.org/10.1002/ajh.25711.
Matasar MJ, Capra M, Özcan M, Lv F, Li W, Yañez E, et al. Copanlisib plus rituximab versus placebo plus rituximab in patients with relapsed indolent non-hodgkin lymphoma (CHRONOS-3): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(5):678–89. https://doi.org/10.1016/S1470-2045(21)00145-5.
Chauhan AF, Cheson BD. Copanlisib in the treatment of relapsed follicular lymphoma: utility and experience from the clinic. Cancer Manag Res. 2021;13:677–92. https://doi.org/10.2147/CMAR.S201024.
Yap TA, Bjerke L, Clarke PA, Workman P. Drugging PI3K in cancer: refining targets and therapeutic strategies. Curr Opin Pharmacol. 2015;23:98–107. https://doi.org/10.1016/j.coph.2015.05.016.
Bheemanaboina R. Isoform-Selective. PI3K inhibitors for various Diseases. Curr Top Med Chem. 2020;20(12):1074–92. https://doi.org/10.2174/1568026620666200106141717.
Hudson K, Hancox UJ, Trigwell C, McEwen R, Polanska UM, Nikolaou M, et al. Intermittent high-dose scheduling of AZD8835, a novel selective inhibitor of PI3Kα and PI3Kδ, demonstrates treatment strategies for PIK3CA-Dependent breast cancers. Mol Cancer Ther. 2016;15(5):877–89. https://doi.org/10.1158/1535-7163.MCT-15-0687.
Miller BW, Przepiorka D, de Claro RA, Lee K, Nie L, Simpson N, et al. FDA approval: idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin Cancer Res. 2015;21(7):1525–9. https://doi.org/10.1158/1078-0432.CCR-14-2522.
Rodrigues DA, Sagrillo FS, Fraga C. Duvelisib. A 2018 Novel FDA-Approved small molecule inhibiting phosphoinositide 3-Kinases. Pharmaceuticals (Basel). 2019;12(2). https://doi.org/10.3390/ph12020069.
Fuso P, Muratore M, D’Angelo T, Paris I, Carbognin L, Tiberi G, et al. PI3K inhibitors in advanced breast Cancer: the past, the Present, New Challenges and Future Perspectives. Cancers (Basel). 2022;14(9). https://doi.org/10.3390/cancers14092161.
Chang DY, Ma WL, Lu YS. Role of Alpelisib in the treatment of PIK3CA-Mutated breast Cancer: patient selection and clinical perspectives. Ther Clin Risk Manag. 2021;17:193–207. https://doi.org/10.2147/TCRM.S251668.
Fowler NH, Samaniego F, Jurczak W, Ghosh N, Derenzini E, Reeves JA, et al. Umbralisib, a dual PI3Kδ/CK1ε inhibitor in patients with relapsed or refractory indolent lymphoma. J Clin Oncol. 2021;39(15):1609–18. https://doi.org/10.1200/JCO.20.03433.
Wu X, Xu Y, Liang Q, Yang X, Huang J, Wang J, et al. Recent advances in dual PI3K/mTOR inhibitors for Tumour Treatment. Front Pharmacol. 2022;13:875372. https://doi.org/10.3389/fphar.2022.875372.
Rosich L, Montraveta A, Xargay-Torrent S, López-Guerra M, Roldán J, Aymerich M, et al. Dual PI3K/mTOR inhibition is required to effectively impair microenvironment survival signals in mantle cell lymphoma. Oncotarget. 2014;5(16):6788–800. https://doi.org/10.18632/oncotarget.2253.
Hall CP, Reynolds CP, Kang MH. Modulation of glucocorticoid resistance in Pediatric T-cell Acute Lymphoblastic Leukemia by increasing BIM expression with the PI3K/mTOR inhibitor BEZ235. Clin Cancer Res. 2016;22(3):621–32. https://doi.org/10.1158/1078-0432.CCR-15-0114.
Wu YY, Wu HC, Wu JE, Huang KY, Yang SC, Chen SX, et al. The dual PI3K/mTOR inhibitor BEZ235 restricts the growth of lung cancer tumors regardless of EGFR status, as a potent accompanist in combined therapeutic regimens. J Exp Clin Cancer Res. 2019;38(1):282. https://doi.org/10.1186/s13046-019-1282-0.
Rodon J, Pérez-Fidalgo A, Krop IE, Burris H, Guerrero-Zotano A, Britten CD, et al. Phase 1/1b dose escalation and expansion study of BEZ235, a dual PI3K/mTOR inhibitor, in patients with advanced solid tumors including patients with advanced breast cancer. Cancer Chemother Pharmacol. 2018;82(2):285–98. https://doi.org/10.1007/s00280-018-3610-z.
Wise-Draper TM, Moorthy G, Salkeni MA, Karim NA, Thomas HE, Mercer CA, et al. A phase ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with Everolimus in patients with Advanced Solid Malignancies. Target Oncol. 2017;12(3):323–32. https://doi.org/10.1007/s11523-017-0482-9.
Pongas G, Fojo T. BEZ235: when Promising Science meets clinical reality. Oncologist. 2016;21(9):1033–4. https://doi.org/10.1634/theoncologist.2016-0243.
Salazar R, Garcia-Carbonero R, Libutti SK, Hendifar AE, Custodio A, Guimbaud R, et al. Phase II study of BEZ235 versus Everolimus in patients with mammalian target of rapamycin Inhibitor-Naïve Advanced pancreatic neuroendocrine tumors. Oncologist. 2018;23(7):766–6e90. https://doi.org/10.1634/theoncologist.2017-0144.
Lang F, Wunderle L, Badura S, Schleyer E, Brüggemann M, Serve H, et al. A phase I study of a dual PI3-kinase/mTOR inhibitor BEZ235 in adult patients with relapsed or refractory acute leukemia. BMC Pharmacol Toxicol. 2020;21(1):70. https://doi.org/10.1186/s40360-020-00446-x.
Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, et al. Randomized open-label phase II trial of Apitolisib (GDC-0980), a Novel inhibitor of the PI3K/Mammalian target of Rapamycin Pathway, Versus Everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016;34(14):1660–8. https://doi.org/10.1200/JCO.2015.64.8808.
Brown JR, Hamadani M, Hayslip J, Janssens A, Wagner-Johnston N, Ottmann O, et al. Voxtalisib (XL765) in patients with relapsed or refractory non-hodgkin lymphoma or chronic lymphocytic leukaemia: an open-label, phase 2 trial. Lancet Haematol. 2018;5(4):e170–0e180. https://doi.org/10.1016/S2352-3026(18)30030-9.
Zhao H, Chen G, Liang H. Dual PI3K/mTOR inhibitor, XL765, suppresses glioblastoma growth by inducing ER stress-dependent apoptosis. Onco Targets Ther. 2019;12:5415–24. https://doi.org/10.2147/OTT.S210128.
Schram AM, Gandhi L, Mita MM, Damstrup L, Campana F, Hidalgo M, et al. A phase ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br J Cancer. 2018;119(12):1471–6. https://doi.org/10.1038/s41416-018-0322-4.
Wen PY, Omuro A, Ahluwalia MS, Fathallah-Shaykh HM, Mohile N, Lager JJ, et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17(9):1275–83. https://doi.org/10.1093/neuonc/nov083.
Tarantelli C, Gaudio E, Arribas AJ, Kwee I, Hillmann P, Rinaldi A, et al. PQR309 is a Novel Dual PI3K/mTOR inhibitor with Preclinical Antitumor Activity in Lymphomas as a single Agent and in combination therapy. Clin Cancer Res. 2018;24(1):120–9. https://doi.org/10.1158/1078-0432.CCR-17-1041.
Tarantelli C, Lupia A, Stathis A, Bertoni F. Is there a role for dual PI3K/mTOR inhibitors for patients affected with lymphoma. Int J Mol Sci. 2020;21(3). https://doi.org/10.3390/ijms21031060.
Brandt C, Hillmann P, Noack A, Römermann K, Öhler LA, Rageot D, et al. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood-brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology. 2018;140:107–20. https://doi.org/10.1016/j.neuropharm.2018.08.002.
Wen PY, Cloughesy TF, Olivero AG, Morrissey KM, Wilson TR, Lu X, et al. First-in-human phase I study to evaluate the brain-penetrant PI3K/mTOR inhibitor GDC-0084 in patients with Progressive or Recurrent High-Grade Glioma. Clin Cancer Res. 2020;26(8):1820–8. https://doi.org/10.1158/1078-0432.CCR-19-2808.
Zhang Y, Xie C, Li A, Liu X, Xing Y, Shen J, et al. PKI-587 enhances chemosensitivity of oxaliplatin in hepatocellular carcinoma through suppressing DNA damage repair pathway (NHEJ and HR) and PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11(8):5134–49.
Park YL, Kim HP, Cho YW, Min DW, Cheon SK, Lim YJ, et al. Activation of WNT/β-catenin signaling results in resistance to a dual PI3K/mTOR inhibitor in colorectal cancer cells harboring PIK3CA mutations. Int J Cancer. 2019;144(2):389–401. https://doi.org/10.1002/ijc.31662.
Radovich M, Solzak JP, Wang CJ, Hancock BA, Badve S, Althouse SK, et al. Initial phase I Safety Study of Gedatolisib plus Cofetuzumab Pelidotin for patients with metastatic triple-negative breast Cancer. Clin Cancer Res. 2022;28(15):3235–41. https://doi.org/10.1158/1078-0432.CCR-21-3078.
Rinne N, Christie EL, Ardasheva A, Kwok CH, Demchenko N, Low C, et al. Targeting the PI3K/AKT/mTOR pathway in epithelial ovarian cancer, therapeutic treatment options for platinum-resistant ovarian cancer. Cancer Drug Resist. 2021;4(3):573–95. https://doi.org/10.20517/cdr.2021.05.
Xie Y, Liu C, Zhang Y, Li A, Sun C, Li R, et al. PKI-587 enhances radiosensitization of hepatocellular carcinoma by inhibiting the PI3K/AKT/mTOR pathways and DNA damage repair. PLoS ONE. 2021;16(10):e0258817. https://doi.org/10.1371/journal.pone.0258817.
Langdon SP, Kay C, Um IH, Dodds M, Muir M, Sellar G, et al. Evaluation of the dual mTOR/PI3K inhibitors Gedatolisib (PF-05212384) and PF-04691502 against ovarian cancer xenograft models. Sci Rep. 2019;9(1):18742. https://doi.org/10.1038/s41598-019-55096-9.
Feng Y, Jiang Y, Hao F. GSK2126458 has the potential to inhibit the proliferation of pancreatic cancer uncovered by bioinformatics analysis and pharmacological experiments. J Transl Med. 2021;19(1):373. https://doi.org/10.1186/s12967-021-03050-7.
Qin AC, Li Y, Zhou LN, Xing CG, Lu XS. Dual PI3K-BRD4 inhibitor SF1126 inhibits Colorectal Cancer Cell Growth in Vitro and in vivo. Cell Physiol Biochem. 2019;52(4):758–68. https://doi.org/10.33594/000000053.
Kim MY, Kruger AJ, Jeong JY, Kim J, Shin PK, Kim SY, et al. Combination therapy with a PI3K/mTOR dual inhibitor and Chloroquine enhances synergistic apoptotic cell death in Epstein-Barr Virus-Infected gastric Cancer cells. Mol Cells. 2019;42(6):448–59. https://doi.org/10.14348/molcells.2019.2395.
Simioni C, Cani A, Martelli AM, Zauli G, Alameen AA, Ultimo S, et al. The novel dual PI3K/mTOR inhibitor NVP-BGT226 displays cytotoxic activity in both normoxic and hypoxic hepatocarcinoma cells. Oncotarget. 2015;6(19):17147–60. https://doi.org/10.18632/oncotarget.3940.
Khalafi S, Lockhart AC, Livingstone AS, El-Rifai W. Targeted Molecular Therapies in the treatment of esophageal adenocarcinoma, are we there yet. Cancers (Basel). 2020;12(11). https://doi.org/10.3390/cancers12113077.
Khan KH, Yap TA, Yan L, Cunningham D. Targeting the PI3K-AKT-mTOR signaling network in cancer. Chin J Cancer. 2013;32(5):253–65. https://doi.org/10.5732/cjc.013.10057.
LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34(31):3803–15. https://doi.org/10.1200/JCO.2014.59.0018.
Yudushkin I. Control of akt activity and substrate phosphorylation in cells. IUBMB Life. 2020;72(6):1115–25. https://doi.org/10.1002/iub.2264.
Liao Y, Hung MC. Physiological regulation of akt activity and stability. Am J Transl Res. 2010;2(1):19–42.
Chan TO, Zhang J, Rodeck U, Pascal JM, Armen RS, Spring M, et al. Resistance of akt kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc Natl Acad Sci U S A. 2011;108(46):E1120–7. https://doi.org/10.1073/pnas.1109879108.
Meuillet EJ. Novel inhibitors of AKT: assessment of a different approach targeting the pleckstrin homology domain. Curr Med Chem. 2011;18(18):2727–42. https://doi.org/10.2174/092986711796011292.
Smyth LM, Tamura K, Oliveira M, Ciruelos EM, Mayer IA, Sablin MP, et al. Capivasertib, an AKT kinase inhibitor, as Monotherapy or in combination with fulvestrant in patients with AKT1 E17K-Mutant, ER-Positive metastatic breast Cancer. Clin Cancer Res. 2020;26(15):3947–57. https://doi.org/10.1158/1078-0432.CCR-19-3953.
Banerji U, Dean EJ, Pérez-Fidalgo JA, Batist G, Bedard PL, You B, et al. A phase I open-label study to identify a dosing regimen of the Pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-Mutated breast and gynecologic cancers. Clin Cancer Res. 2018;24(9):2050–9. https://doi.org/10.1158/1078-0432.CCR-17-2260.
Schmid P, Abraham J, Chan S, Wheatley D, Brunt AM, Nemsadze G, et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel as First-Line therapy for metastatic triple-negative breast Cancer: the PAKT Trial. J Clin Oncol. 2020;38(5):423–33. https://doi.org/10.1200/JCO.19.00368.
Howell SJ, Casbard A, Carucci M, Ingarfield K, Butler R, Morgan S, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. 2022;23(7):851–64. https://doi.org/10.1016/S1470-2045(22)00284-4.
Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, et al. Targeting activated akt with GDC-0068, a novel selective akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013;19(7):1760–72. https://doi.org/10.1158/1078-0432.CCR-12-3072.
Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021;398(10295):131–42. https://doi.org/10.1016/S0140-6736(21)00580-8.
Shariati M, Meric-Bernstam F. Targeting AKT for cancer therapy. Expert Opin Investig Drugs. 2019;28(11):977–88. https://doi.org/10.1080/13543784.2019.1676726.
Du L, Yau C, Brown-Swigart L, Gould R, Krings G, Hirst GL, et al. Predicted sensitivity to endocrine therapy for stage II-III hormone receptor-positive and HER2-negative (HR+/HER2-) breast cancer before chemo-endocrine therapy. Ann Oncol. 2021;32(5):642–51. https://doi.org/10.1016/j.annonc.2021.02.011.
Myers AP, Konstantinopoulos PA, Barry WT, Luo W, Broaddus RR, Makker V, et al. Phase II, 2-stage, 2-arm, PIK3CA mutation stratified trial of MK-2206 in recurrent endometrial cancer. Int J Cancer. 2020;147(2):413–22. https://doi.org/10.1002/ijc.32783.
Stover EH, Xiong N, Myers AP, Tayob N, Engvold V, Polak M, et al. A phase II study of MK-2206, an AKT inhibitor, in uterine serous carcinoma. Gynecol Oncol Rep. 2022;40:100974. https://doi.org/10.1016/j.gore.2022.100974.
Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;168(6):960–76. https://doi.org/10.1016/j.cell.2017.02.004.
Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1(1–4):27–36. https://doi.org/10.1007/s12154-008-0003-5.
Paghdal KV, Schwartz RA. Sirolimus (rapamycin): from the soil of Easter Island to a bright future. J Am Acad Dermatol. 2007;57(6):1046–50. https://doi.org/10.1016/j.jaad.2007.05.021.
Law BK. Rapamycin: an anti-cancer immunosuppressant. Crit Rev Oncol Hematol. 2005;56(1):47–60. https://doi.org/10.1016/j.critrevonc.2004.09.009.
Evangelisti C, Cenni V, Lattanzi G. Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria-related disorders. Br J Clin Pharmacol. 2016;82(5):1229–44. https://doi.org/10.1111/bcp.12928.
Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer. 2012;31(1):8–18. https://doi.org/10.5732/cjc.011.10281.
Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109(8):3509–12. https://doi.org/10.1182/blood-2006-06-030833.
Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, et al. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 2008;68(8):2934–43. https://doi.org/10.1158/0008-5472.CAN-07-6487.
Alammar H, Nassani R, Alshehri MM, Aljohani AA, Alrfaei BM. Deficiency in the treatment description of mTOR inhibitor resistance in Medulloblastoma, a systematic review. Int J Mol Sci. 2021;23(1). https://doi.org/10.3390/ijms23010464.
Campbell MT, Millikan RE, Altinmakas E, Xiao L, Wen SJ, Siefker-Radtke AO, et al. Phase I trial of sunitinib and temsirolimus in metastatic renal cell carcinoma. Clin Genitourin Cancer. 2015;13(3):218–24. https://doi.org/10.1016/j.clgc.2014.10.004.
Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8(11):975–84. https://doi.org/10.1016/S1470-2045(07)70285-1.
Rini BI, Bellmunt J, Clancy J, Wang K, Niethammer AG, Hariharan S, et al. Randomized phase III trial of temsirolimus and bevacizumab versus interferon alfa and bevacizumab in metastatic renal cell carcinoma: INTORACT trial. J Clin Oncol. 2014;32(8):752–9. https://doi.org/10.1200/JCO.2013.50.5305.
Kelley RK, Joseph NM, Nimeiri HS, Hwang J, Kulik LM, Ngo Z, et al. Phase II trial of the combination of Temsirolimus and Sorafenib in Advanced Hepatocellular Carcinoma with Tumor Mutation Profiling. Liver Cancer. 2021;10(6):561–71. https://doi.org/10.1159/000518297.
Chan HY, Grossman AB, Bukowski RM. Everolimus in the treatment of renal cell carcinoma and neuroendocrine tumors. Adv Ther. 2010;27(8):495–511. https://doi.org/10.1007/s12325-010-0045-2.
Lim T, Lee J, Lee DJ, Lee HY, Han B, Baek KK, et al. Phase I trial of capecitabine plus everolimus (RAD001) in patients with previously treated metastatic gastric cancer. Cancer Chemother Pharmacol. 2011;68(1):255–62. https://doi.org/10.1007/s00280-011-1653-5.
Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31. https://doi.org/10.1186/s13578-020-00396-1.
Shi JJ, Chen SM, Guo CL, Li YX, Ding J, Meng LH. The mTOR inhibitor AZD8055 overcomes tamoxifen resistance in breast cancer cells by down-regulating HSPB8. Acta Pharmacol Sin. 2018;39(8):1338–46. https://doi.org/10.1038/aps.2017.181.
Pi R, Yang Y, Hu X, Li H, Shi H, Liu Y, et al. Dual mTORC1/2 inhibitor AZD2014 diminishes myeloid-derived suppressor cells accumulation in ovarian cancer and delays tumor growth. Cancer Lett. 2021;523:72–81. https://doi.org/10.1016/j.canlet.2021.09.017.
Li S, Sheng J, Liu Z, Fan Y, Zhang C, Lv T, et al. Potent antitumour of the mTORC1/2 dual inhibitor AZD2014 in docetaxel-sensitive and docetaxel-resistant castration-resistant prostate cancer cells. J Cell Mol Med. 2021;25(5):2436–49. https://doi.org/10.1111/jcmm.16155.
Sammons S, Kornblum NS, Blackwell KL. Fulvestrant-based combination therapy for second-line treatment of hormone receptor-positive advanced breast Cancer. Target Oncol. 2019;14(1):1–12. https://doi.org/10.1007/s11523-018-0587-9.
Morscher RJ, Brard C, Berlanga P, Marshall LV, André N, Rubino J, et al. First-in-child phase I/II study of the dual mTORC1/2 inhibitor vistusertib (AZD2014) as monotherapy and in combination with topotecan-temozolomide in children with advanced malignancies: arms E and F of the AcSé-ESMART trial. Eur J Cancer. 2021;157:268–77. https://doi.org/10.1016/j.ejca.2021.08.010.
Eyre TA, Hildyard C, Hamblin A, Ali AS, Houlton A, Hopkins L, et al. A phase II study to assess the safety and efficacy of the dual mTORC1/2 inhibitor vistusertib in relapsed, refractory DLBCL. Hematol Oncol. 2019;37(4):352–9. https://doi.org/10.1002/hon.2662.
Costa LJ, Maddocks K, Epperla N, Reddy NM, Karmali R, Umyarova E, et al. Diffuse large B-cell lymphoma with primary treatment failure: ultra-high risk features and benchmarking for experimental therapies. Am J Hematol. 2017;92(2):161–70. https://doi.org/10.1002/ajh.24615.
Magaway C, Kim E, Jacinto E. Targeting mTOR and metabolism in Cancer: Lessons and Innovations. Cells. 2019;8(12). https://doi.org/10.3390/cells8121584.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84. https://doi.org/10.1016/j.bbamcr.2006.10.001.
Saini KS, Loi S, de Azambuja E, Metzger-Filho O, Saini ML, Ignatiadis M, et al. Targeting the PI3K/AKT/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat Rev. 2013;39(8):935–46. https://doi.org/10.1016/j.ctrv.2013.03.009.
Yoshitake R, Saeki K, Eto S, Shinada M, Nakano R, Sugiya H, et al. Aberrant expression of the COX2/PGE2 axis is induced by activation of the RAF/MEK/ERK pathway in BRAFV595E canine urothelial carcinoma. Sci Rep. 2020;10(1):7826. https://doi.org/10.1038/s41598-020-64832-5.
Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT. Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18(2):189–218. https://doi.org/10.1038/sj.leu.2403241.
Li DW, Liu JP, Mao YW, Xiang H, Wang J, Ma WY, et al. Calcium-activated RAF/MEK/ERK signaling pathway mediates p53-dependent apoptosis and is abrogated by alpha B-crystallin through inhibition of RAS activation. Mol Biol Cell. 2005;16(9):4437–53. https://doi.org/10.1091/mbc.e05-01-0010.
Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–93. https://doi.org/10.1038/sj.leu.2402945.
Vasjari L, Bresan S, Biskup C, Pai G, Rubio I. Ras signals principally via Erk in G1 but cooperates with PI3K/Akt for cyclin D induction and S-phase entry. Cell Cycle. 2019;18(2):204–25. https://doi.org/10.1080/15384101.2018.1560205.
Doma E, Rupp C, Baccarini M. EGFR-ras-raf signaling in epidermal stem cells: roles in hair follicle development, regeneration, tissue remodeling and epidermal cancers. Int J Mol Sci. 2013;14(10):19361–84. https://doi.org/10.3390/ijms141019361.
Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, et al. Ras activation of the raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127–55. https://doi.org/10.1210/rp.56.1.127.
Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery. Int J Cancer. 2006;119(10):2261–71. https://doi.org/10.1002/ijc.22144.
Dienstmann R, Tabernero J. BRAF as a target for cancer therapy. Anticancer Agents Med Chem. 2011;11(3):285–95. https://doi.org/10.2174/187152011795347469.
Strumberg D, Seeber S. Raf kinase inhibitors in oncology. Onkologie. 2005;28(2):101–7. https://doi.org/10.1159/000083373.
Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5(4):350–6. https://doi.org/10.1016/j.coph.2005.04.007.
Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5(11):875–85. https://doi.org/10.1038/nrm1498.
Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6(4):313–9. https://doi.org/10.1016/j.ccr.2004.09.022.
Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87(4):629–38. https://doi.org/10.1016/s0092-8674(00)81383-5.
Yuryev A, Ono M, Goff SA, Macaluso F, Wennogle LP. Isoform-specific localization of A-RAF in mitochondria. Mol Cell Biol. 2000;20(13):4870–8. https://doi.org/10.1128/MCB.20.13.4870-4878.2000.
Hong SK, Wu PK, Park JI. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell Signal. 2018;42:11–20. https://doi.org/10.1016/j.cellsig.2017.10.001.
Eblen ST. Extracellular-regulated kinases: signaling from ras to ERK Substrates to control Biological Outcomes. Adv Cancer Res. 2018;138:99–142. https://doi.org/10.1016/bs.acr.2018.02.004.
Degirmenci U, Wang M, Hu J. Targeting aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells. 2020;9(1). https://doi.org/10.3390/cells9010198.
Emuss V, Garnett M, Mason C, Marais R. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res. 2005;65(21):9719–26. https://doi.org/10.1158/0008-5472.CAN-05-1683.
Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14(7):455–67. https://doi.org/10.1038/nrc3760.
Greaves WO, Verma S, Patel KP, Davies MA, Barkoh BA, Galbincea JM, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013;15(2):220–6. https://doi.org/10.1016/j.jmoldx.2012.10.002.
Negrao MV, Raymond VM, Lanman RB, Robichaux JP, He J, Nilsson MB, et al. Molecular Landscape of BRAF-Mutant NSCLC reveals an Association between Clonality and driver mutations and identifies targetable Non-V600 driver mutations. J Thorac Oncol. 2020;15(10):1611–23. https://doi.org/10.1016/j.jtho.2020.05.021.
Darp R, Vittoria MA, Ganem NJ, Ceol CJ. Oncogenic BRAF induces whole-genome doubling through suppression of cytokinesis. Nat Commun. 2022;13(1):4109. https://doi.org/10.1038/s41467-022-31899-9.
McCubrey JA, Steelman LS, Franklin RA, Abrams SL, Chappell WH, Wong EW, et al. Targeting the RAF/MEK/ERK, PI3K/AKT and p53 pathways in hematopoietic drug resistance. Adv Enzyme Regul. 2007;47:64–103. https://doi.org/10.1016/j.advenzreg.2006.12.013.
Deak D, Gorcea-Andronic N, Sas V, Teodorescu P, Constantinescu C, Iluta S, et al. A narrative review of central nervous system involvement in acute leukemias. Ann Transl Med. 2021;9(1):68. https://doi.org/10.21037/atm-20-3140.
Pikman Y, Stieglitz E. Targeting the ras pathway in pediatric hematologic malignancies. Curr Opin Pediatr. 2021;33(1):49–58. https://doi.org/10.1097/MOP.0000000000000981.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106(48):20411–6. https://doi.org/10.1073/pnas.0905833106.
Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4(1):61–8. https://doi.org/10.1158/2159-8290.CD-13-0631.
Yuan J, Ng WH, Tian Z, Yap J, Baccarini M, Chen Z, et al. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci Signal. 2018;11(554). https://doi.org/10.1126/scisignal.aar6795.
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006;46:249–79. https://doi.org/10.1016/j.advenzreg.2006.01.004.
Wu PK, Becker A, Park JI. Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. Int J Mol Sci. 2020;21(15). https://doi.org/10.3390/ijms21155436.
Cseh B, Doma E, Baccarini M. “RAF” neighborhood: protein-protein interaction in the Raf/Mek/Erk pathway. FEBS Lett. 2014;588(15):2398–406. https://doi.org/10.1016/j.febslet.2014.06.025.
Yuan J, Dong X, Yap J, Hu J. The MAPK and AMPK signalings: interplay and implication in targeted cancer therapy. J Hematol Oncol. 2020;13(1):113. https://doi.org/10.1186/s13045-020-00949-4.
Giménez N, Martínez-Trillos A, Montraveta A, Lopez-Guerra M, Rosich L, Nadeu F, et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica. 2019;104(3):576–86. https://doi.org/10.3324/haematol.2018.196931.
Ben Mousa A. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J Gastroenterol. 2008;14(1):40–2. https://doi.org/10.4103/1319-3767.37808.
Heo J, Breitbach CJ, Moon A, Kim CW, Patt R, Kim MK, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19(6):1170–9. https://doi.org/10.1038/mt.2011.39.
Colagrande S, Regini F, Taliani GG, Nardi C, Inghilesi AL. Advanced hepatocellular carcinoma and sorafenib: diagnosis, indications, clinical and radiological follow-up. World J Hepatol. 2015;7(8):1041–53. https://doi.org/10.4254/wjh.v7.i8.1041.
Chen Z, Xie H, Hu M, Huang T, Hu Y, Sang N, et al. Recent progress in treatment of hepatocellular carcinoma. Am J Cancer Res. 2020;10(9):2993–3036.
Gyawali B, Shimokata T, Ando M, Honda K, Ando Y. Risk of serious adverse events and fatal adverse events with sorafenib in patients with solid cancer: a meta-analysis of phase 3 randomized controlled trials†. Ann Oncol. 2017;28(2):246–53. https://doi.org/10.1093/annonc/mdw549.
Hou W, Xia H, Zhou S, Fan Z, Xu H, Gong Q, et al. The MEK inhibitors enhance the efficacy of sorafenib against hepatocellular carcinoma cells through reducing p-ERK rebound. Transl Cancer Res. 2019;8(4):1224–32. https://doi.org/10.21037/tcr.2019.07.11.
Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38(5):614–22. https://doi.org/10.1038/aps.2017.5.
Kim A, Cohen MS. The discovery of vemurafenib for the treatment of BRAF-mutated metastatic melanoma. Expert Opin Drug Discov. 2016;11(9):907–16. https://doi.org/10.1080/17460441.2016.1201057.
Nucera C, Nehs MA, Nagarkatti SS, Sadow PM, Mekel M, Fischer AH, et al. Targeting BRAFV600E with PLX4720 displays potent antimigratory and anti-invasive activity in preclinical models of human thyroid cancer. Oncologist. 2011;16(3):296–309. https://doi.org/10.1634/theoncologist.2010-0317.
Smalley KS. PLX-4032, a small-molecule B-Raf inhibitor for the potential treatment of malignant melanoma. Curr Opin Investig Drugs. 2010;11(6):699–706.
Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, et al. PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell Melanoma Res. 2010;23(6):820–7. https://doi.org/10.1111/j.1755-148X.2010.00763.x.
Ascierto PA, Simeone E, Giannarelli D, Grimaldi AM, Romano A, Mozzillo N. Sequencing of BRAF inhibitors and ipilimumab in patients with metastatic melanoma: a possible algorithm for clinical use. J Transl Med. 2012;10:107. https://doi.org/10.1186/1479-5876-10-107.
Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105(8):3041–6. https://doi.org/10.1073/pnas.0711741105.
Nehs MA, Nucera C, Nagarkatti SS, Sadow PM, Morales-Garcia D, Hodin RA, et al. Late intervention with anti-BRAF(V600E) therapy induces tumor regression in an orthotopic mouse model of human anaplastic thyroid cancer. Endocrinology. 2012;153(2):985–94. https://doi.org/10.1210/en.2011-1519.
Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13(8):782–9. https://doi.org/10.1016/S1470-2045(12)70269-3.
Cheng Y, Tian H. Current Development Status of MEK inhibitors. Molecules. 2017;22(10). https://doi.org/10.3390/molecules22101551.
Zhu X, Cao Y, Liu W, Ju X, Zhao X, Jiang L, et al. Stereotactic body radiotherapy plus pembrolizumab and trametinib versus stereotactic body radiotherapy plus gemcitabine for locally recurrent pancreatic cancer after surgical resection: an open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2021;22(8):1093–102. https://doi.org/10.1016/S1470-2045(21)00286-2.
Holt SV, Logié A, Odedra R, Heier A, Heaton SP, Alferez D, et al. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012;106(5):858–66. https://doi.org/10.1038/bjc.2012.8.
Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAFV600E/K-mutated melanoma. Clin Cancer Res. 2013;19(8):2257–64. https://doi.org/10.1158/1078-0432.CCR-12-3476.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30. https://doi.org/10.1038/nature08902.
Papale A, Morella IM, Indrigo MT, Bernardi RE, Marrone L, Marchisella F, et al. Impairment of cocaine-mediated behaviours in mice by clinically relevant Ras-ERK inhibitors. Elife. 2016;5. https://doi.org/10.7554/eLife.17111.
Yap JL, Worlikar S, MacKerell AD Jr, Shapiro P, Fletcher S. Small-molecule inhibitors of the ERK signaling pathway: towards novel anticancer therapeutics. ChemMedChem. 2011;6(1):38–48. https://doi.org/10.1002/cmdc.201000354.
Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2(3):135–64. https://doi.org/10.18632/oncotarget.240.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012;3(10):1068–111. https://doi.org/10.18632/oncotarget.659.
Ku BM, Jho EH, Bae YH, Sun JM, Ahn JS, Park K, et al. BYL719, a selective inhibitor of phosphoinositide 3-Kinase α, enhances the effect of selumetinib (AZD6244, ARRY-142886) in KRAS-mutant non-small cell lung cancer. Invest New Drugs. 2015;33(1):12–21. https://doi.org/10.1007/s10637-014-0163-9.
Wang Z, Zhou J, Fan J, Qiu SJ, Yu Y, Huang XW, et al. Effect of rapamycin alone and in combination with sorafenib in an orthotopic model of human hepatocellular carcinoma. Clin Cancer Res. 2008;14(16):5124–30. https://doi.org/10.1158/1078-0432.CCR-07-4774.
Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin Cancer Res. 2011;17(20):6482–9. https://doi.org/10.1158/1078-0432.CCR-11-0933.
Papadimitrakopoulou V. Development of PI3K/AKT/mTOR pathway inhibitors and their application in personalized therapy for non-small-cell lung cancer. J Thorac Oncol. 2012;7(8):1315–26. https://doi.org/10.1097/JTO.0b013e31825493eb.
Ewald F, Nörz D, Grottke A, Hofmann BT, Nashan B, Jücker M. Dual inhibition of PI3K-AKT-mTOR- and RAF-MEK-ERK-signaling is synergistic in cholangiocarcinoma and reverses acquired resistance to MEK-inhibitors. Invest New Drugs. 2014;32(6):1144–54. https://doi.org/10.1007/s10637-014-0149-7.
Kuger S, Flentje M, Djuzenova CS. Simultaneous perturbation of the MAPK and the PI3K/mTOR pathways does not lead to increased radiosensitization. Radiat Oncol. 2015;10:214. https://doi.org/10.1186/s13014-015-0514-5.
Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the akt(PKB) signaling pathway. Genes Dev. 1998;12(4):502–13. https://doi.org/10.1101/gad.12.4.502.
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–74. https://doi.org/10.1172/JCI34739.
Mundt F, Rajput S, Li S, Ruggles KV, Mooradian AD, Mertins P, et al. Mass Spectrometry-Based Proteomics reveals potential roles of NEK9 and MAP2K4 in resistance to PI3K inhibition in Triple-Negative breast cancers. Cancer Res. 2018;78(10):2732–46. https://doi.org/10.1158/0008-5472.CAN-17-1990.
Paraiso KH, Xiang Y, Rebecca VW, Abel EV, Chen YA, Munko AC, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71(7):2750–60. https://doi.org/10.1158/0008-5472.CAN-10-2954.
Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE. 2011;6(12):e28973. https://doi.org/10.1371/journal.pone.0028973.
Acknowledgements
The figures in the article were created by Biorender.
Funding
None.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Data collection and analysis were performed by Qingfang Li and Huashan Shi. The first draft of the manuscript was written by Qingfang Li and Ting Luo. Qingfang Li, Huashan Shi, and Zhihui Li revised the article. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Q., Li, Z., Luo, T. et al. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol Biomed 3, 47 (2022). https://doi.org/10.1186/s43556-022-00110-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-022-00110-2