





Abstract
Periostin (POSTN) may serve as a biomarker for Type-2 mediated eosinophilic airway inflammation in asthma. We hypothesised that a Type-2 cytokine, interleukin (IL)-13, induces airway epithelial expression of POSTN, which in turn contributes to epithelial changes observed in asthma.
We studied the effect of IL-13 on POSTN expression in BEAS-2B and air–liquid interface differentiated primary bronchial epithelial cells (PBECs). Additionally, the effects of recombinant human POSTN on epithelial-to-mesenchymal transition (EMT) markers and mucin genes were assessed. POSTN single cell gene expression and protein levels were analysed in bronchial biopsies and induced sputum from asthma patients and healthy controls.
IL-13 increased POSTN expression in both cell types and this was accompanied by EMT-related features in BEAS-2B. In air–liquid interface differentiated PBECs, IL-13 increased POSTN basolateral and apical release. Apical administration of POSTN increased the expression of MMP-9, MUC5B and MUC5AC. In bronchial biopsies, POSTN expression was mainly confined to basal epithelial cells, ionocytes, endothelial cells and fibroblasts, showing higher expression in basal epithelial cells from asthma patients versus those from controls. A higher level of POSTN protein expression in epithelial and subepithelial layers was confirmed in bronchial biopsies from asthma patients when compared to healthy controls. Although sputum POSTN levels were not higher in asthma, levels correlated with eosinophil numbers and with the coughing-up of mucus.
POSTN expression is increased by IL-13 in bronchial epithelial cells and is higher in bronchial biopsies from asthma patients. This may have important consequences, as administration of POSTN increases epithelial expression of mucin genes, supporting the relationship of POSTN with Type-2 mediated asthma and mucus secretion.
Abstract
High levels of IL-13 in Type-2 driven asthma play a crucial role in the development of the abnormal epithelial phenotype by induction of periostin (POSTN) secretion. POSTN induces mucin production in airway epithelial cells. https://bit.ly/3hg7ETu
Introduction
Many susceptibility genes for the development of asthma are expressed in the airway epithelium [1–3]. Airway epithelial barrier dysfunction is thought to play a central role in allergic sensitisation and the development of asthma [4, 5]. There are abnormalities in the epithelial barrier in asthma, including goblet cell metaplasia and loss of epithelial cell–cell adhesion molecule E-cadherin (CDH1) [5]. Loss of epithelial barrier function may have important consequences, not only increasing the access of allergens to the submucosa, but also propagating airway immunity, remodelling and hyper-responsiveness [4, 5]. The exact mechanisms through which aberrant expression of epithelial genes is translated into functionally altered responses to aero-allergens in asthma are still unknown.
Airway epithelial cells (AECs) from asthma patients display an intrinsic inefficiency in forming a functional barrier [6, 7]. It has been observed that loss of epithelial barrier function in asthma is accompanied by increased production of the Type-2 driving factors thymic stromal lymphopoietin (TSLP) and interleukin (IL)-33 in vitro [7], and increased expression of periostin (POSTN) in the airways in vivo [8]. Importantly, POSTN is a proposed systemic biomarker of IL-13 driven airway eosinophilia in asthma [9]. However, reduced POSTN expression upon treatment with the monoclonal IL-13 antibody lebrikizumab is not accompanied by reduced eosinophil numbers, limiting its use as a biomarker [10]. POSTN is an extracellular matrix-derived bioactive peptide (a matrikine) expressed by various lung resident cells and secreted by the airway epithelium in response to the Type-2 cytokines IL-4 and IL-13, as well as the profibrotic transforming growth factor-β (TGF-β). Induction of POSTN expression is associated with epithelial-to-mesenchymal transition (EMT), a process of aberrant epithelial repair that may be involved in loss of epithelial barrier function and airway remodelling in asthma [5]. Both IL-4 and IL-13 downregulate CDH1 expression in lung epithelial cells [11] disrupting epithelial barrier function [11, 12]. IL-13 has been shown to induce expression of CDH1 repressor and EMT marker, zinc finger protein (SNAI1), in intestinal epithelial cells [13]. Additionally, the asthma epithelium is reported to be more prone to undergoing EMT in response to TGF-β compared to the healthy epithelium [14]. Our previous data indicates that during TGF-β induced EMT, increased POSTN expression is accompanied by increased fibronectin (FN) and matrix metalloproteinase-2 (MMP-2) and MMP-9 expression in the bronchial epithelial cell line BEAS-2B [15]. Overexpression of POSTN in BEAS-2B cells promotes epithelial expression of MMP-2, MMP-9 and alpha-1 Type-I collagen (COL1A1) in a TGF-β dependent manner [16]. Furthermore, it has been shown to mediate the effects of TGF-β in fibroblasts [17], indicating that POSTN is not merely an EMT marker but may also play an active role in EMT. We hypothesise that IL-13 induced production of POSTN plays a crucial role in the abnormal phenotype of the asthma epithelium. We have assessed the expression of POSTN following exposure to IL-13 and have studied the effect of POSTN on expression of EMT markers, as well as mucins, in AECs. Additionally, we have assessed POSTN expression in bronchial biopsies (single-cell RNA-sequencing (scRNA-seq) and protein levels) and sputum from well-characterised asthma patients and healthy controls.
Methods
Subjects
We included bronchial biopsies and sputum samples from patients with asthma and from healthy controls with normal lung function, defined as forced expiratory volume in 1 s (FEV1)/forced vital capacity (FVC) >lower limit of normal (LLN), FEV1 >80% predicted, an absence of bronchial hyper-responsiveness (BHR) to methacholine (provocative concentration causing a 20% fall in FEV1 (PC20) methacholine >16 mg·mL−1) and no allergies (see table 1 for subject characteristics). The medical ethics committee of University Medical Center Groningen (UMCG) approved the study and all subjects gave written informed consent.
Subject characteristics
Primary AECs were obtained by protease digestion of tracheal bronchial tissue from normal donor lungs received at the UMCG. The study protocol was consistent with the research code of the UMCG (www.umcg.nl/EN/Research/Researchers/General/ResearchCode/Paginas/default.aspx) and national ethical and professional guidelines (the Code of Conduct of the Dutch Federation of Biomedical Scientific Societies; www.federa.org). Bronchial biopsies were subjected to immunohistochemical analysis for POSTN as described in the supplementary material.
Sputum induction
Sputum was induced by inhalation of nebulised hypertonic saline (5%) for three consecutive periods of 5 min. Whole sputum samples were processed as described previously [19].
Culture of human bronchial epithelial cells
Bronchial epithelial BEAS-2B cells and AECs were cultured and treated. Quantitative PCR and enzyme-linked immunosorbent assay (ELISA) were performed as described in the supplementary material.
Single-cell RNA-sequencing
Bronchial biopsies were collected through bronchoscopy and processed for scRNA-seq with the Chromium platform (10x Genomics, Pleasanton, CA, USA) using the 3′ protocol (version 2) according to the manufacturer's instructions. The details of this study have been described elsewhere [20]. Briefly, the data underwent quality control by use of SOUP-X correction, with removal of doublets as well as low quality cells [20]. We used Seurat, version 3 for analysis of POSTN gene expression in the dataset, using cluster annotation as previously described [20]. We combined the epithelial and non-epithelial datasets to perform principal component analysis (PCA), using the resulting principal components to calculate the uniform manifold approximation and projection (UMAP).
Statistical analysis
Data were analysed using the nonparametric, two-tailed Wilcoxon-signed rank test for paired observations in primary cells and the t-test for paired observations in the cell line, unless otherwise stated. Correlations were determined using a linear regression analysis.
Results
IL-13 induces airway epithelial POSTN expression and induction of EMT markers in BEAS-2B cells
We first assessed whether IL-13 induced POSTN expression and if this was accompanied by downregulation of CDH1 and induction of EMT markers. We used Type-2 cytokine IL-5 as a negative control and TGF-β as a positive control. Treatment of BEAS-2B cells with both TGF-β and IL-13 for 48 h significantly upregulated POSTN gene expression (figure 1a). Furthermore, both TGF-β and IL-13 (but not IL-5) induced a change to a spindle-shaped morphology in BEAS-2B cells at 24 h (figure 1b) and downregulation of CDH1 gene expression at 24 h (figure 1c). In contrast to our previous findings, the TGF-β induced increase in mRNA expression of EMT-marker FN did not reach significance [15], although TGF-β significantly increased mRNA expression of α-smooth muscle actin (SMA). FN and SMA were not induced by IL-13 or IL-5 (figure 1c). MMP-9 expression was below the detection limit of the assay in BEAS-2B cells.

Interleukin (IL)-13 induces changes in human bronchial epithelial cell morphology and epithelial plasticity with an increase in epithelial-to-mesenchymal transition (EMT) markers. BEAS-2B cells were seeded in 24-well plates in duplicate, grown to confluence in RPMI/10% fetal calf serum (FCS) and serum-deprived overnight. Subsequently, cells were exposed to vehicle control (medium), IL-13 (10 ng·mL−1), transforming growth factor-β (TGF-β) (5 ng·mL−1) as a positive control, or IL-5 (10 ng·mL−1) as a negative control. Techniques were then as follows: a) cells were harvested after 48 h for RNA isolation and periostin (POSTN) mRNA expression was measured by quantitative PCR. Expression of POSTN was related to the expression of the β2-microglobulin (B2M) and peptidylprolyl isomerase A (PPIA) housekeeping genes, and expressed as 2−ΔΔCt compared to baseline; b) morphology was studied by phase-contrast microscopy at 24 h; c) cells were harvested for RNA isolation after 24 h or 48 h and expression of E-cadherin (CDH1), fibronectin (FN) and α-smooth muscle actin (SMA) was related to the expression of the B2M and PPIA housekeeping genes, and expressed as 2−ΔΔCt compared to baseline. Data are presented as mean±sem (n=4–5). Ct: cycle threshold; sem: standard error of the mean. *: p<0.05; **: p<0.01; ***: p<0.001 (between the indicated value and baseline, as analysed by t-test for paired observations).
Next, we studied the effects of TGF-β, IL-13 and IL-5 on POSTN expression in air–liquid interface differentiated AECs. Interestingly, 48-h treatment with either TGF-β or IL-13 (but not IL-5) significantly increased POSTN expression in these cells, with the strongest increase, approximately 20-fold, observed after IL-13 treatment (figure 2a). POSTN protein levels were measured in cell-free supernatants at baseline and upon TGF-β and IL-13 stimulation. POSTN was detected with a higher concentration at baseline in the apical compartment. POSTN levels in both compartments were increased by IL-13 treatment, while no significant effect was observed for TGF-β (figure 2b).
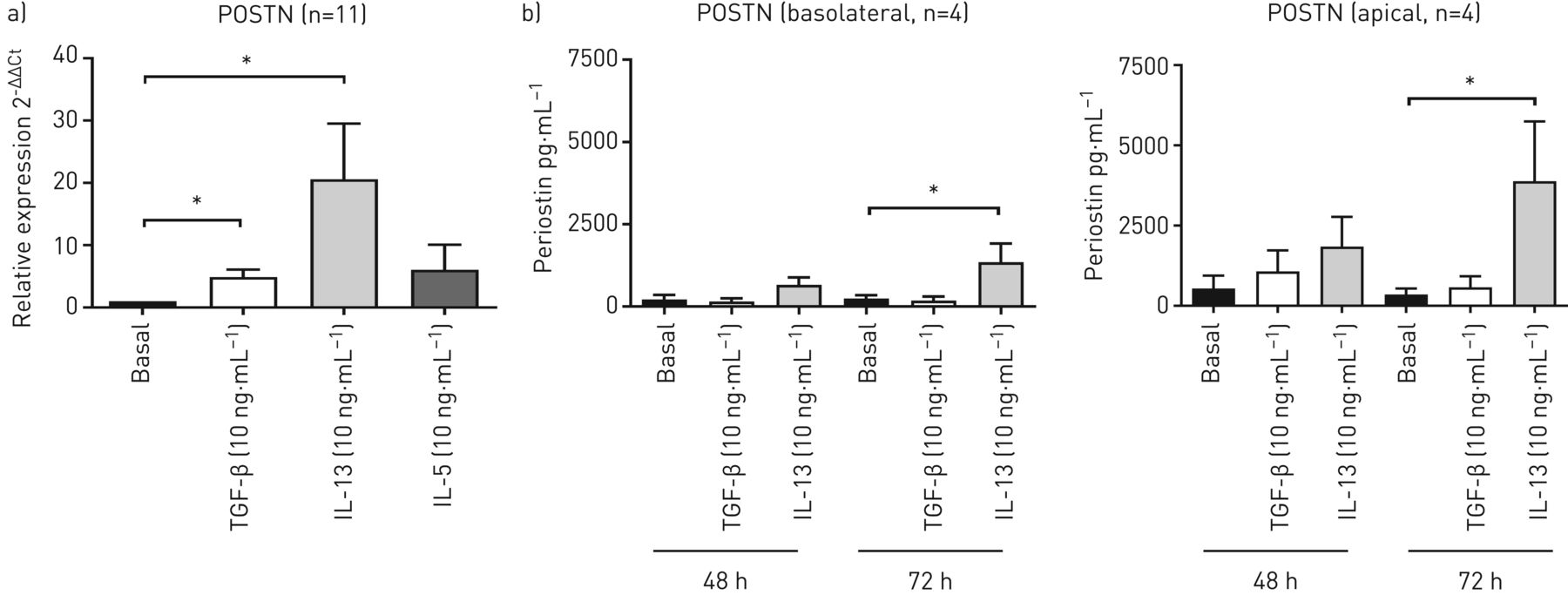
Interleukin (IL)-13 induces periostin (POSTN) mRNA expression and release in human primary bronchial epithelial cells (PBECs). PBECs were differentiated at an air–liquid interface for 4 weeks, exposed to hormonally-deprived medium for 24 h (basolateral and apical side) and treated with or without transforming growth factor-β (TGF-β) (10 ng·mL−1) or IL-13 (10 ng·mL−1) from the apical side for 48 h or 72 h. Techniques were then as follows: a) cells were harvested after 48 h for RNA isolation and POSTN mRNA expression was measured by quantitative PCR. Expression of POSTN was related to the expression of the β2-microglobulin (B2M) and peptidylprolyl isomerase A (PPIA) housekeeping genes, and expressed as 2−ΔΔCt compared to baseline (n=11); b) at 48 h and 72 h, basolateral and apical supernatants were collected and POSTN was measured in cell-free supernatant (n=4). Data are presented as mean±sem. Ct: cycle threshold; sem: standard error of the mean. *: p<0.05 (between the indicated values as analysed by a) the Wilcoxon signed rank test or b) one-way ANOVA for repeated measurements with Bonferroni's multiple comparison test).
POSTN induces expression of MMP-9, MUC5B and MUC5AC in differentiated primary epithelial cells
To examine whether the IL-13 induced release of POSTN could be responsible for changes in the epithelial phenotype observed in asthma, we studied the effect of recombinant human POSTN on the expression of CDH1 and EMT markers SNAI1, MMP-9, FN, COL1A1 and SMA in air–liquid interface differentiated AECs at 48 h. TGF-β (10 ng·mL−1) was again used as a positive control based on previous findings on the induction of EMT by TGF-β in human bronchial epithelial cells [14].
POSTN was applied from the apical side as IL-13 induced the strongest release of POSTN into the apical compartment (figure 2b). Apical stimulation with TGF-β was used as a positive control. Similar to TGF-β, POSTN significantly increased MMP-9 expression, with a trend towards increased FN expression (figure 3a). TGF-β (but not POSTN) tended to increase COL1A1 expression, while neither TGF-β nor POSTN significantly affected the expression of CDH1, SNAI1, COL1A1 (figure 3a) or SMA (data not shown). TGF-β has previously been reported to reduce CDH1 protein expression in air–liquid interface differentiated AECs after 72 h [14]. However, no significant difference was observed in CDH1 expression, while TGF-β (but not POSTN) significantly increased COL1A1 expression at this time point (figure 3b). With MMP-9 being the only marker significantly affected by POSTN, we assessed MMP-9 protein levels in culture supernatants and observed a trend towards increased MMP-9 secretion with both TGF-β (basolaterally and apically) and POSTN (apically) at 72 h (supplementary figure S1).

Effects of periostin (POSTN) on epithelial-to-mesenchymal transition (EMT) markers and barrier function in human primary bronchial epithelial cells (PBECs). PBECs were differentiated at an air–liquid interface for 4 weeks, exposed to hormonally-deprived medium for 24 h and treated with or without POSTN (1 ng·mL−1) or transforming growth factor-β (TGF-β) (10 ng·mL−1) as a positive control, both from the apical side. Techniques were then as follows: a) cells were harvested for RNA isolation after 48 h and expression of E-cadherin (CDH1), zinc finger protein (SNAI1), matrix metalloproteinase-9 (MMP-9), fibronevtin (FN), alpha-1 Type-I collagen (COL1A1) and α-smooth muscle actin (SMA) was related to the expression of the β2-microglobulin (B2M) and peptidylprolyl isomerase A (PPIA) housekeeping genes, and expressed as 2−ΔΔCt compared to baseline; b) cells were harvested for RNA isolation after 72 h and expression of CDH1 and COL1A1 was related to the expression of the B2M and PPIA housekeeping genes, and expressed as 2−ΔΔCt compared to baseline. Data are presented as mean±sem (n=8). Ct: cycle threshold; sem: standard error of the mean. *: p<0.05; **: p<0.01; ***: p<0.001 (between the indicated value and baseline, as analysed by the Wilcoxon signed rank test).
Since both IL-13 and MMP-9 have been implicated in mucus production and goblet cell hyperplasia, while reduced allergen-induced goblet cell metaplasia has been observed in POSTN-deficient mice [21], we determined the effect of apical POSTN exposure on mucins MUC5B and MUC5AC in air–liquid interface differentiated cells. We observed a significant increase in both MUC5B and MUC5AC mRNA expression (figure 4a). We did not detect MUC5AC protein in the apical compartment after 72 h of medium exposure of the air–liquid interface cultured cells, while MUC5B protein levels were significantly higher in the presence of POSTN despite the high variation between donors (figure 4b).

Effect of periostin (POSTN) on MUC5B and MUC5AC expression in human primary bronchial epithelial cells (PBECs). PBECs were differentiated at an air–liquid interface for 4 weeks, exposed to hormonally-deprived medium for 24 h and treated with or without POSTN (1 ng·mL−1) from the apical side. Techniques were then as follows: a) cells were harvested for RNA isolation after 72 h and expression of MUC5B (n=11) and MUC5AC (n=6) was related to the expression of the β2-microglobulin (B2M) and peptidylprolyl isomerase A (PPIA) housekeeping genes, and expressed as 2−ΔΔCt compared to the control (basal); b) cell-free supernatants were harvested after 72 h and MUC5B levels were determined by enzyme-linked immunosorbent assay (ELISA) (n=9). Data are presented as mean±sem. Ct: cycle threshold; sem: standard error of the mean. *: p<0.05 (between the indicated values, as analysed by the Wilcoxon signed rank test).
Single cell analysis of POSTN expression in bronchial biopsies reveals expression in suprabasal epithelial cells and increased expression in asthma
We recently generated a scRNA-seq dataset from airway wall biopsies of healthy individuals and asthma patients [20]. In this dataset, we observed the presence of increased numbers of goblet cells and mucous ciliated cells (ciliated cells expressing MUC5AC and other goblet cell genes) in the airway epithelium of asthma patients [20]. Using this dataset to explore POSTN expression in the different subsets of epithelial cells, we observed that club and ciliated cells hardly express POSTN, while substantial POSTN expression is observed in basal, activated basal (also referred to as suprabasal [22]), mucous ciliated and goblet cells, as well as in ionocytes (a rare bronchial epithelial cell type that has recently been described) (figures 5a–5c) [23]. In addition to epithelial cells, expression was also observed in endothelial cells and fibroblasts. When comparing expression levels between asthma patients and healthy subjects, POSTN gene expression in healthy controls was observed to be confined to the basal cell cluster, representing the most undifferentiated, resting basal cells. However, in asthma patients, POSTN expression was maintained and even increased in suprabasal, activated basal and cycling basal cells (figure 5b). POSTN expression in goblet cells and mucous ciliated cells was also only observed in asthma patients (figure 5b). Since this dataset consists of only six healthy donors and six asthma donors, we do not have the power to statistically test whether the POSTN expression level is different between the two disease states for each epithelial cell subset. However, we have included the per-donor expression levels of POSTN for the different epithelial cell subsets in supplementary figure S2.

Single cell RNA expression of periostin (POSTN) and the interleukin (IL)-13 response gene expression signature in healthy controls and asthma patients. Bronchial biopsies were obtained from six healthy controls and six asthmatic subjects. Single-cell RNA-sequencing (scRNA-seq) was performed and the information obtained is displayed as follows: a) uniform manifold approximation and projection (UMAP) plot of the identified clusters of cells and their annotations; b) distribution of the normalised expression of POSTN split between asthmatic and control subjects in the expressing clusters; c) normalised expression of POSTN split between control subjects and asthmatic subjects (first and second plots, respectively) in the expressing cell types; d) expression of IL-13 response genes split between asthmatic and control subjects in all combined clusters (left) and the difference in the expression of IL-13 response genes between asthmatic and control subjects in different basal cell types/states (right). DC: dendritic cell.
We have previously reported the presence of pathogenic, IL-13 expressing Type-2 T-helper (Th2) cells in airway wall biopsies from these asthma patients [20] and have observed increased expression of an IL-4/IL-13 induced gene signature (based on in vitro studies using primary AECs [24]) in the bronchial epithelial cells of asthma patients [20]. Therefore, we next asked whether the IL-13 induced gene signature [24] in the asthmatic airway epithelium [20] overlapped with POSTN gene expression patterns. The IL-13 induced gene signature was indeed increased in epithelial cells in asthma patients (figure 5d), with consistently increased expression in basal cell subsets as well as goblet and mucous ciliated cells (figure 5d).
Increased expression of POSTN in bronchial biopsies of asthma patients and association of sputum levels of POSTN with sputum eosinophils and symptoms of mucus hypersecretion
Given the small number of asthma patients and healthy controls in the scRNA-seq dataset, we next asked whether the increased expression of POSTN was also evident at the protein level in bronchial biopsies from a larger cohort of asthma patients and controls. In accordance with the scRNA-seq data, we observed that the intensity of POSTN in the biopsies from asthma patients was significantly stronger than in those from healthy controls (figures 6a and 6b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of periostin (POSTN) protein in biopsies and sputum from asthma patients and healthy controls. Panels are as follows: a) exemplar immunohistochemistry results for POSTN in bronchial biopsies (26 asthma patients and 22 healthy control subjects); b) staining intensity for POSTN in bronchial biopsies (positive pixels per total tissue area (whole biopsy)) quantified using computer-assisted imaging; c) levels of POSTN (pg·mL−1) in induced sputum (73 asthma patients and 87 healthy control subjects) measured by enzyme-linked immunosorbent assay (ELISA). Mean levels±sem are indicated; d) eosinophil counts in sputum, correlation between absolute eosinophil counts and log-transformed levels of POSTN in sputum, and correlation between the score for coughing-up sputum (by questionnaire) and log-transformed levels of POSTN in sputum. *: p<0.05; ****: p<0.0001 (between the indicated values, as analysed by the Mann–Whitney U-test). Correlations were analysed by linear regression and Pearson's correlation for normally distributed data (eosinophils) or Spearman's correlation for nonparametric data (coughing-up sputum). A p-value of less than 0.05 was considered statistically significant. Open symbols indicate smokers (some asthma group (n=13) and control group (n=15) smokers had POSTN levels in sputum below the detection limit and are thus not indicated).
Finally, we observed that POSTN was detectable in sputum of asthma patients and healthy control subjects. While we did not observe a significant difference in POSTN levels between the two groups (figure 6c), sputum POSTN levels in asthma patients positively correlated to sputum eosinophil numbers (figure 6d). Moreover, we observed that sputum POSTN levels in asthma patients were significantly correlated with coughing-up mucus (figure 6d).
Discussion
We hypothesised that high levels of IL-13 in Type-2 driven asthma may result in increased airway POSTN levels. Here, we show that IL-13 induces airway epithelial expression of POSTN and its release from both the apical and basolateral side. In bronchial biopsies, scRNA-seq analysis revealed that POSTN is expressed in basal epithelial cells, ionocytes, endothelial cells and fibroblasts, and is maintained in differentiated epithelial cells from asthma patients compared to healthy controls. Consistent with a role for IL-13 in epithelial POSTN expression, the IL-13 induced gene signature observed in the asthmatic AECs [20] overlapped with the POSTN gene expression patterns. At the protein level, POSTN was expressed in both airway epithelial and subepithelial layers, with higher expression in asthma patients. Exaggerated POSTN release may have important consequences for asthma pathophysiology, as administration of POSTN increased the expression of MUC5B and MUC5AC in differentiated AECs. Although POSTN levels in sputum of asthma patients were not significantly higher compared to those in healthy controls, the levels correlated with eosinophil numbers in asthma, supporting the relationship of POSTN with Type-2 mediated asthma and mucus hypersecretion.
We observed that IL-13 induces a strong increase in POSTN release by differentiated AECs, particularly into the apical compartment. This is in contrast to previous observations in undifferentiated primary epithelial cells, where POSTN could not be detected in apical washes, with or without 4-day IL-13 treatment [16]. Thus, POSTN may not be released by the bronchial epithelial cells under culture conditions that maintain them in a basal phenotype. Accordingly, Suzaki et al. [25] demonstrated that 14-day differentiation of AECs into goblet cells using IL-13 strongly increased POSTN secretion from both the basolateral and apical side. This suggests that more differentiated cells, such as the suprabasal, club, ciliated or goblet cells, were the main source of POSTN in this model. Whether goblet cells in particular were responsible for the apical release of POSTN in our study needs further investigation. Our scRNA-seq analysis revealed that POSTN was expressed mainly by basal epithelial cells in dissociated airway biopsies, with maintenance of expression in suprabasal, basal activated and cycling epithelial cells from asthma patients. Furthermore, POSTN was expressed in goblet and mucous ciliated cells exclusively in asthma patients. The overlapping expression patterns between the IL-13 induced gene signature and POSTN in epithelial cells from asthma patients further supports the notion that POSTN might well be an IL-13 induced gene in vivo, although a role for IL-4 cannot be excluded given the strong overlap between IL-13 and IL-4 induced gene expression patterns in primary human AECs [24].
It is tempting to speculate that the increased POSTN expression in asthma may have consequences, leading to higher basolateral as well as apical POSTN secretion and potentially also higher mucus production in the airways of asthmatic patients. As epithelial barrier integrity is compromised in asthma, POSTN may be less restricted to the basolateral side and our results indicate that apically secreted POSTN especially acts to induce transcriptional upregulation of mucin genes. We observed that administration of recombinant human POSTN from the apical side increases the expression of the mucin genes MUC5B and MUC5AC and also increases MUC5B protein levels. In line with a role for apical POSTN release in inducing mucus hypersecretion, we observed that sputum levels of POSTN correlate with symptoms of mucus hypersecretion in asthma patients. Mucus plugs in asthma patients were reported to contain both MUC5AC and MUC5B-containing domains [26]. In vitro, IL-13 induced the formation of heterogeneous mucus gels and impaired mucociliary transport. This was related to the tethering of MUC5AC-containing mucus gel domains to mucus-producing cells in the epithelium [26]. In our study, we did not observe an increase in mucin gene expression when cells were stimulated with either IL-13 or TGF-β. A limitation of our study, however, is that we only administered these cytokines from the apical side to be able to compare them to the apical addition of POSTN.
Our findings that POSTN increased mucin expression correspond with previous data from Bentley et al. [21], who reported that POSTN-deficiency in a mouse model of allergic airway disease resulted in reduced house dust mite (HDM) induced goblet cell hyperplasia and reduced airway inflammation and hyper-responsiveness. However, these findings are in contrast with those of Sehra et al. [27], which suggest an opposing role for POSTN in ovalbumin (OVA)-induced allergic airways disease, with increased MUC5AC expression upon allergen challenge in POSTN-deficient mice. Discrepancies between these two studies may be due to the different models that were used and the intranasal HDM-sensitisation model used by Bentley et al. [21] may be more relevant for human disease.
In addition to mucin genes, we observed that POSTN increases MMP-9 mRNA in differentiated AECs. Increased MMP-9 expression could be involved in the observed POSTN-induced increase in mucin expression and epithelial remodelling, as MMP-9 has been implicated in epidermal growth factor receptor (EGFR)-induced MUC5AC production and goblet cell metaplasia [28, 29], thus affecting innate immune defense of the epithelial barrier. MMP-9 is a marker of EMT but we did not find evidence for POSTN regulation of epithelial plasticity/EMT. Nevertheless, it will be of interest to focus future studies on the potential role of POSTN in airway hyper-responsiveness, since airway smooth muscle cell contractile responses may be regulated by MMP-9 [30]. Furthermore, MMP-9 may induce secretion of chemokines involved in the attraction of eosinophils, leading to peribronchial fibrosis and peribronchial numbers of eosinophils [31]. The mechanisms underlying the observed POSTN-induced upregulation of mucin genes and MMP-9 need further investigation. Of note, POSTN acts on αMβ2/CD11b [32] and may thus directly induce eosinophil migration, which may subsequently act to further increase mucus secretion as eosinophilic cationic protein is a well-known mediator of mucus production. It is also of interest to mention that high levels of POSTN were detected in epithelial columnar cell-high sputum of asthma patients with eosinophilia [33]. This may reflect an eosinophil and/or Type-2 immune signalling function of epithelial cells that secrete high levels of POSTN.
Our observation that POSTN levels in sputum were associated with eosinophil levels in asthmatic patients is in line with a previous study on a cohort of poorly-controlled asthma patients [34], showing that sputum POSTN levels were significantly higher in eosinophilic asthma (defined as sputum eosinophils ≥3%) compared to noneosinophilic asthma. Similarly, Bobolea et al. [35] observed that sputum levels of POSTN were higher in asthma patients with fixed airflow limitation compared to variable airflow limitation and in patients with eosinophilic (>2%) compared to mixed granulocytic asthma. The lack of a significant difference in sputum levels of POSTN between asthma patients and healthy controls in our study, with POSTN being undetectable in many of the asthma patients, can be explained by the fact that our patients had relatively mild asthma and were able to refrain from medication for at least 1 month prior to sample collection.
In addition to promoting mucus production, POSTN is known to have a structural role interacting with other extracellular matrix proteins to promote matrix production and subepithelial fibrosis, especially when secreted in the basolateral direction [16]. Our data do not support a role for POSTN in epithelial collagen synthesis, as reported previously for stable overexpression of POSTN [16], although we did not investigate the effects of basolateral secretion of POSTN, which is more likely to play a role in matrix remodelling.
All together, our data support the notion that IL-13 induced secretion of POSTN by AECs mediates mucus hypersecretion and acts to perpetuate eosinophilic inflammation, aggravating the symptoms of asthma. As such, targeting POSTN may potentially be a part of a novel, multipronged therapeutic approach in asthma.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-01286-2020.Supplement
Shareable PDF
Supplementary Material
This one-page PDF can be shared freely online.
Shareable PDF ERJ-01286-2020.Shareable
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: J.K. Burgess has nothing to disclose.
Conflict of interest: M.R. Jonker has nothing to disclose.
Conflict of interest: M. Berg has nothing to disclose.
Conflict of interest: N.T.H. ten Hacken has nothing to disclose.
Conflict of interest: K.B. Meyer has nothing to disclose.
Conflict of interest: M. van den Berge reports research grants paid to their institution from AstraZeneca, Novartis and Genentech, outside the submitted work.
Conflict of interest: M.C. Nawijn reports grants from Lung Foundation Netherlands (grant numbers 5.1.14.020 and 4.1.18.226), GSK Ltd, the European Commission (grant number 874656, discovAIR) and the Chan Zuckerberg Initiative, during the conduct of the study.
Conflict of interest: I.H. Heijink reports grants from Roche, during the conduct of the study; and grants from the Netherlands Lung Foundation and Boehringer Ingelheim, outside the submitted work.
Support statement: This study was supported by research grants from Roche (HLR-14) to I.H. Heijink and from the Netherlands Lung Foundation to M.C. Nawijn (LF5.1.14.020 and LF4.1.18.226). Funding information for this article has been deposited with the Crossref Funder Registry.
- Received April 20, 2020.
- Accepted August 17, 2020.
- ©ERS 2021. For reproduction rights and permissions contact permissions{at}ersnet.org
References










