Abstract
Dimensionally stable anodes (DSA) are widely used in water electrolysis, cathodic protection, wastewater treatment, metal electrowinning and chlor-alkali industry. An important parameter of these anodes is their lifetime under harsh anodic conditions. In this work, the incorporation of TiO2 nanoparticles into a thermally prepared IrO2 anode as a way of improving its service lifetime was investigated. The results show that incorporation of up to 25 wt% TiO2 nanoparticles into the coating formed crack-free structure during the thermal decomposition process and thus enhances the service lifetime of modified electrode dramatically by up to 10x compared to the pure IrO2 anode. Importantly, these nanoparticle additions have minimal effect of the anodes performance toward the oxygen evolution reaction. Further addition of TiO2 nanoparticles reduced the lifetime of the anode due to uneven distribution of the active coating over the substrate leading to electrolyte penetration to, and passivation of, the underlying titanium substrate. It is proposed that adding the right quantity of nanoparticles into the IrO2 layer improved the anodes lifetime by minimising electrolyte penetration to the substrate while increasing the mechanical strength of the layer.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Dimensionally stable anodes (DSA), have been widely used for water electrolysis, electro chlorination, cathodic protection, wastewater treatment, and metal electrowinning.1–5 While these anodes have achieved wide-spread use in industry, they can still suffer from short service lifetimes in the strongly acidic and anodic conditions in which they often operate in. In general, the failure or deactivation of DSA is caused by a combination of substrate passivation and dissolution of the active electrocatalytic layer.6–9 In order to overcome this problem, additional oxides such as TiO2, Ta2O5, or SnO2 are often added to the electrocatalytic oxide layer to improve the corrosion resistance of the active IrO2 and RuO2 materials.10–12 Others have used thin interlayers between the active coating and metallic substrate in order to reduce the passivation of the electrode by preventing penetration of electrolyte through the active oxide layer to the substrate.7,9,13–16
As cracks in the DSA active layer facilitate electrolyte penetration to the underlying substrate,1,9 the observations that the presence of nanoparticles within the active layer can reduce these cracks,16,17 may suggest that adding nanoparticles to DSA can improve service lifetimes. This is partially supported by others who suggest the improved service lifetime of a IrO2 anode when a nanoparticle-like SiO2 interlayer was used was attributed to both the suppression of electrolyte transport to the Ti substrate and improved mechanical strength of the resulting composite layer.15 This proposed strengthening of the nanoparticle composites has been widely investigated in other systems, with many reporting enhanced wear resistance of the when nanoparticles are added into surface coatings.18–20 Others have found that TiO2 nanoparticles added into electrochemically deposited PbO2 films can significantly change the microstructure of the film, and improves service life of the electrode under strong anodic conditions.21
In this research, the incorporation of TiO2 nanoparticles into IrO2 DSA layers is investigated as a means to improve the service lifetime of these electrodes. The morphological and electrochemical properties of these nanoparticle modified IrO2 electrodes are determined to understand and quantify the impact this nanoparticle addition has on the oxygen evolution reaction, the electrochemically active surface area and the service lifetime under accelerated testing conditions.
Experimental
Titanium substrates (15.5 × 17 mm) were pre-treated by etching in 10% hydrochloric acid at room temperature for 1 h, before rinsing with deionized water and drying at room temperature. The precursor solution was prepared by mixing a solution of 0.02 M IrCl3 (99.9%, American Elements) in deionized water with a suspension of 0.02 M TiO2 nanoparticles (Degussa P25 99.9%) in isopropanol. P25 TiO2 nanoparticles is a standard material found in applications (especially photocatalysis) and is characterized as mixture of anatase and rutile (75–84% anatase) with a BET surface area of approximately 49 m2 g−1, and average crystallite sizes of 25–29 nm (anatase phase) and 55–85 nm (rutile phase).22,23 The size of the TiO2 nanoparticles used in this work are larger than those used to stabilize PbO2 films (approx. 5 nm),21 but are smaller than the SiO2 particle-like film used to stabilize IrO2 films (approx. 500 nm).15 The mixture of IrCl3 and TiO2 nanoparticles was ultrasonicated (Kudos 100W Ultrasonic bath) for at least 5 minutes to ensure that the TiO2 nanoparticles were well-dispersed. This suspension was typically stable for at least 2 hours without evidence of settling or separation. To coat the titanium substrates, the suspension was sprayed over the Ti substrates which were heated at 150ᵒC using a small airbrush (Blackridge Professional) with a nozzle size of 0.2 mm. The electrode was coated with 6 layers, with each layer annealed in air at 450ᵒC for 5 min and the final layer annealed for 1 h. Electrodes containing 0, 10, 25 and 40 wt% TiO2 nanoparticles were prepared in this way and in all cases the total oxide loading was determined to be 0.35 ± 0.06 mg cm−2.
The surface morphology of the prepared anodes was investigated using a JEOL JSM 7000F field emission, high-resolution scanning electron microscope (at 15 kV) and EDS analysis performed with the JEOL energy dispersive X-ray analysis system.
The electrochemical behavior of the prepared anodes were assessed in 0.5 M H2SO4 electrolyte using a three-electrode cell system controlled by a Gamry Interface 1000 potentiostat. Electrochemical impedance spectroscopy (EIS) was used to determine the resistance between the working and reference electrode. The EIS was conducted in hybrid mode, with a DC current of 0.6 mA with an AC amplitude of 10 mV between 100 kHz and 0.1 Hz. Accelerated lifetime tests were performed on the TiO2 nanoparticle + IrO2 anodes at 50°C using a large stainless steel cathode (16 cm2) and 2 M sulphuric acid as the electrolyte. In these tests, the anodes were operated at 1 A cm−2 until the cell potential reached 10 V at which point the anodes were deemed to have failed.
Results and Discussion
Scanning electron microscopy revealed that the surface structure and the morphology of fabricated electrodes vary considerable as a function of the TiO2 nanoparticles: IrO2 ratio (Figure 1). As expected, the layer of pure IrO2 exhibits the cracked mud structure commonly observed for DSA electrodes.1,2,17 For the electrode layer containing 10 wt% TiO2 nanoparticles, a similar morphology to the pure IrO2 anode is found, but with much smaller cracks across surface of the electrode. Quite different morphology is observed for the electrodes containing 25 and 40 wt% TiO2 nanoparticles, with these showing pronounced roughness and a particle-like structure. While the surface morphology of electrode layer containing 25 wt% TiO2 nanoparticles is uniform on the ∼10 μm scale (as determined by EDS mapping), the electrode containing 40 wt% TiO2 nanoparticles has an uneven distribution of large agglomerates, with some regions of the substrate poorly by the IrO2-TiO2 composite (marked by a dashed region on the figure). EDS analysis of this electrode revealed that the poorly coated regions had Ti:Ir atomic ratios of 25:2, whereas the coated regions had Ti:Ir atomic ratios of 4:1. This suggests that the poorly coated regions were still covered with IrO2, but as the Ti ratio was very high, it suggests that the coating in these regions were very thin. It must be noted that in all cases, the EDS revealed higher than expected Ti:Ir ratios (compared to the layer composition) as the a significant fraction of the Ti signal would have originated from the underlying Ti substrate due to the penetration depth of EDS.

Figure 1. SEM micrographs of the surface morphology of IrO2 anodes containing (a) 0, (b) 10, (c) 25 and (d) 40 wt% TiO2 nanoparticles. In (d), the dashed region highlights a poorly coated region, where the dotted region highlights an example of an agglomerate.
We propose that the mechanism for these observed changes in morphology due to the addition of nanoparticles is related to differences in thermal expansion coefficient of composite and substrate and the improved drying characteristics of the precursor suspension. It is reported that the differences in thermal expansion coefficient of the layer and the substrate during the thermal decomposition method may be linked to the formation of cracks,9 presumably through induced mechanical stress on the coating. In our case, IrO2 has a smaller thermal expansion coefficient (5 × 10−6 / °C24) than Ti (1 × 10−5 / °C25) which would explain the observed cracking, whereas the thermal expansion coefficient of TiO2 (9 × 10−6 / °C26,27) is closer to Ti, and thus the presence of the TiO2 nanoparticles may reduce some mechanical stress. It is also possible that the composite has stronger mechanical strength as observed in some metal-ceramic composites.28 We also speculate that the initial drying and decomposition of the precursor suspension plays a role in determining the final morphology, with many investigations revealing that the drying of suspensions leads to complex and interesting structures.29,30 It is also likely that the presence of the TiO2 nanoparticles provides the IrCl3 (the IrO2 precursor) nucleation sites from which a network of IrO2 crystallites can grow.
Cyclic voltammetry of the fabricated anodes in 0.5 M H2SO4 solution revealed similar electrochemical behavior to the traditional IrO2 DSA electrodes,31–33 which suggests that the surface electrochemistry is most likely dominated by iridium species34,35 (Figure 2).
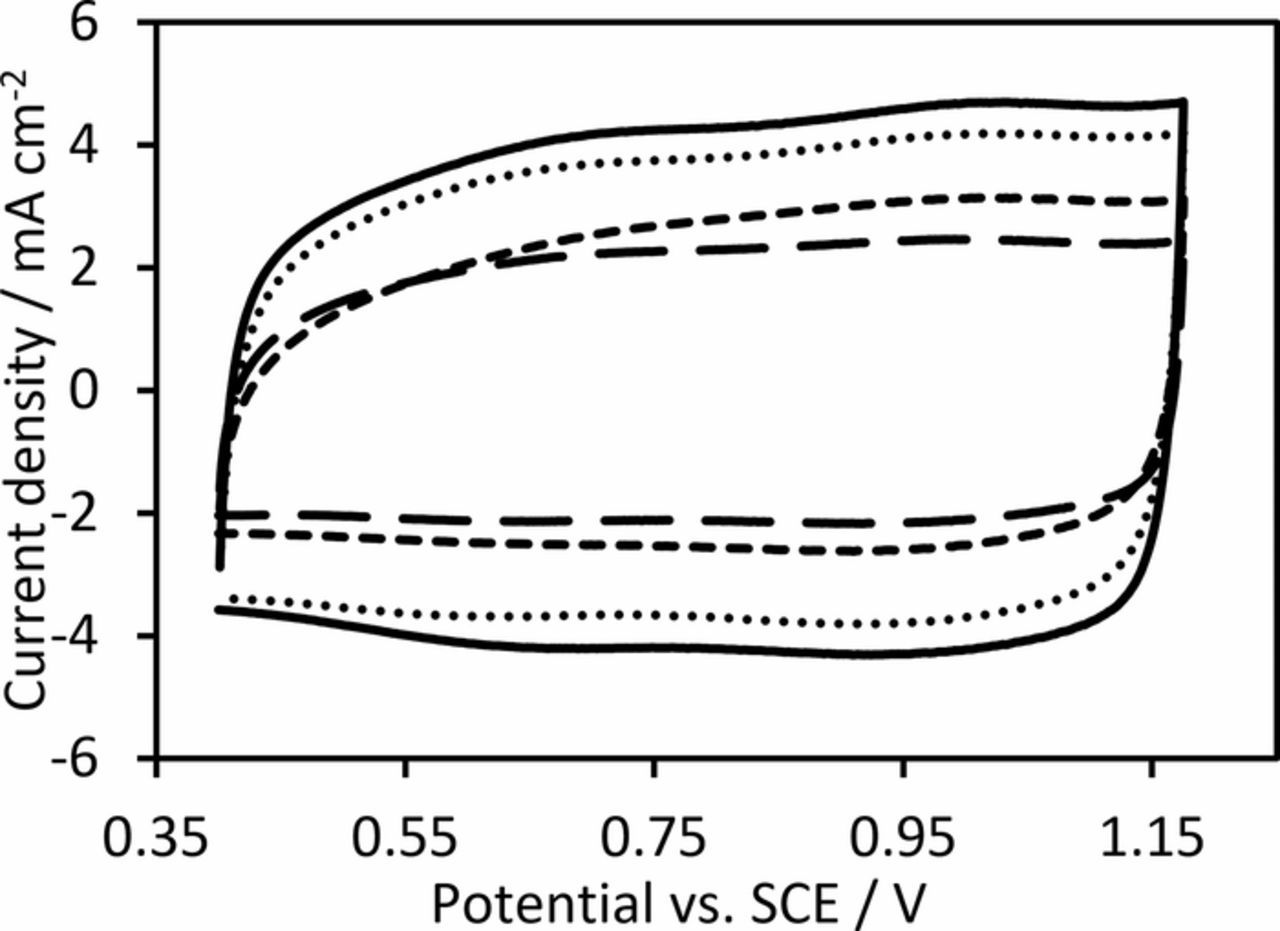
Figure 2. Cyclic voltammetry IrO2 anodes containing (—) 0, (···) 10, (‒ ‒ ‒) 25 and (‒ ‒) 40 wt% TiO2 nanoparticles at 50 mV s−1 in 0.5 M H2SO4.
As expected, the anodic charge (a measure of the electrochemically active surface area1,36,37) was found to decrease with the addition of TiO2 nanoparticles due to the decreasing mass of IrO2 in the active layer (Figure 3). This suggests that the TiO2 nanoparticles do not help improve the utilization of the IrO2 phase within the composite layer, unlike the addition of 10 wt% Sb-doped SnO2 nanoparticles to IrO2 which did increase the overall electrochemically active surface area.17 In this case, by calculating the anodic charge on an IrO2 mass basis, it is found that the specific active area of the IrO2 decreased upon additional of 10 wt% TiO2 nanoparticles before increasing with further TiO2 nanoparticle additions (Figure 3). We propose that the initial decrease in IrO2 utilization is due to the reduced crack size, which would restrict the transport of electrolyte into the active layer.36 Interestingly the anodic charge on an IrO2 mass basis measured at 200 mV s−1 is higher for the anode containing 40 wt% TiO2 nanoparticles compared to the pure IrO2 anode (Figure 3), which suggests that a great proportion of the Ir atoms are easily accessible36 in this anode, presumably due to the particle-like morphology this anode exhibits. However, it is important to note that the TiO2 used in this work has limited electrical conductivity, and it is likely that for thicker films, this will reduce the utilization of the IrO2 due to ohmic losses between the film and the titanium substrate.
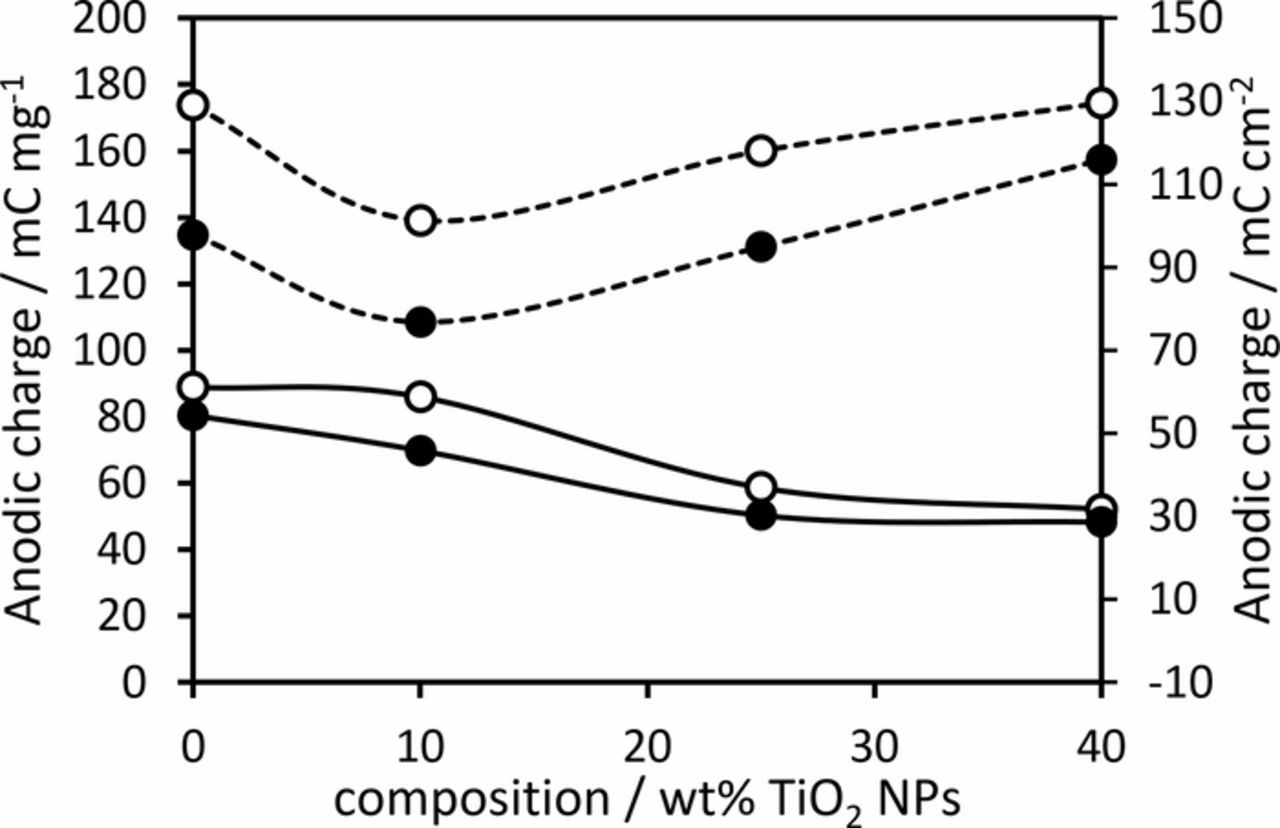
Figure 3. Anodic charge calculated from cyclic voltammetry measured at (○) 50 and (●) 200 mV s−1 in 0.5 M H2SO4. Solid lines = anodic charge normalized on an anode geometric area basis, Dashed lines = anodic charge normalized on an IrO2 mass basis. Lines are guides only.
The electrocatalytic performance of fabricated electrodes toward oxygen evolution reaction was investigated by linear sweep voltammetry in 0.5 M H2SO4 with a scan rate of 1 mV s−1 (Figure 4). This shows that the onset of the oxygen evolution reaction begins at around 1.2 V vs. SCE for all electrodes, with Tafel slopes between 75–85 mV at low overpotentials, before curving upwards to Tafel slopes of 150–190 mV at higher overpotentials. The lower Tafel slope is similar to that report for IrO2 in KOH38 and the 60–90 mV reported on some IrxTi(1-x)O2 anodes in acid.39 These slopes are significantly higher than that normally reported for IrO2 in acidic electrolytes,40–42 which may suggest that the OER is operating via a different mechanism on these anodes. This could be due to the low IrO2 loading on the anodes used in this work (total oxide loading = 0.35 mg cm−2), with some evidence suggesting that low total loadings of the active phase can increase the Tafel slope for OER.39,43,44 Regardless of this, very little difference is found between the anodes, with the difference in potential between the most active anode (containing 0 wt% TiO2 nanoparticles) and least active anode (containing 40 wt% TiO2 nanoparticles) only around 30 mV at useful current densities (>10 mA cm−2). This shows that the OER performance can be largely maintained even with the addition of up to 40% TiO2 nanoparticles, with any small loss in performance likely only due to fewer IrO2 sites within the anode. We expect this loss in performance could be minimized by increasing the anode's total oxide loading as the quantity of TiO2 nanoparticles within the layer increases, to ensure that the IrO2 loading remains approx. constant.
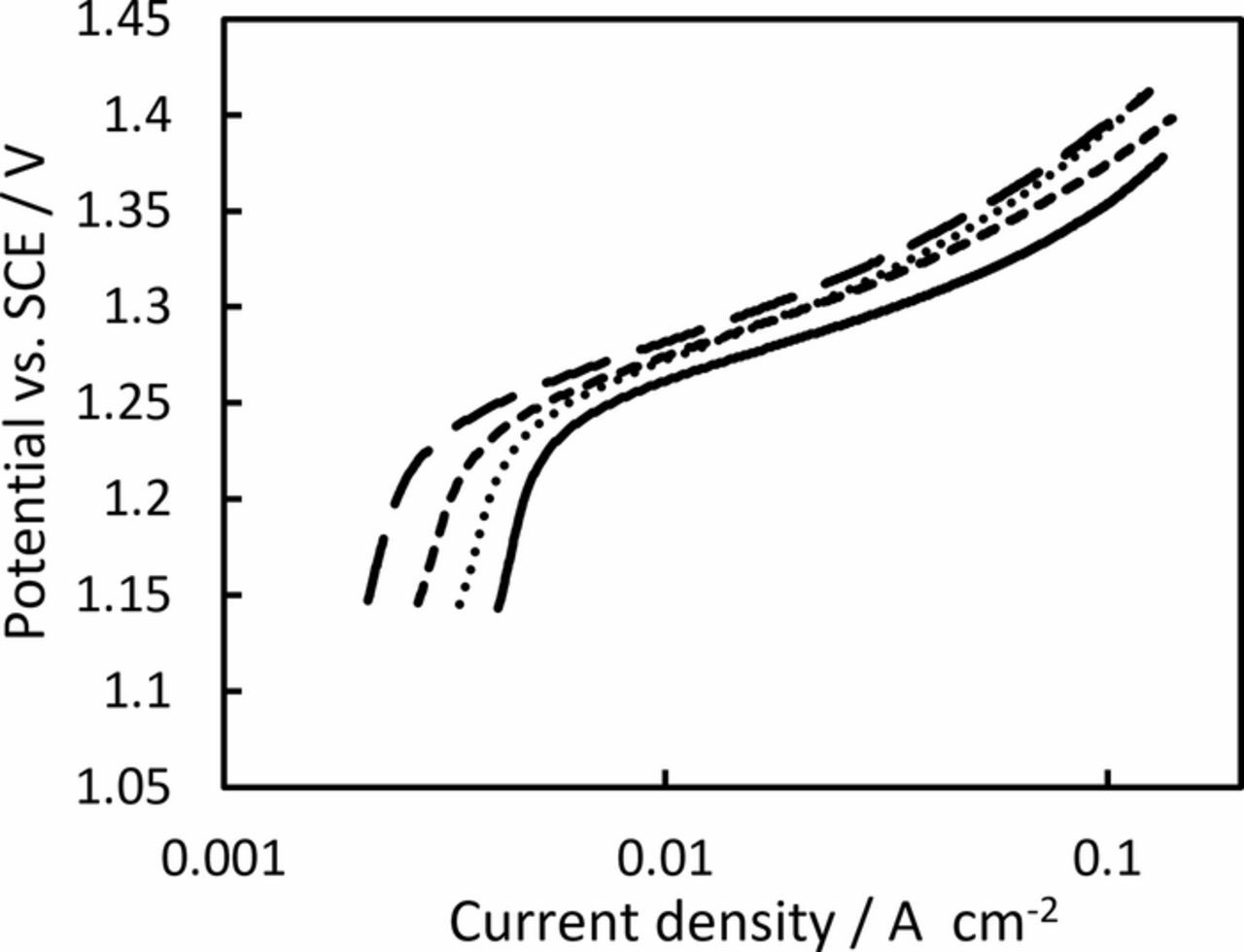
Figure 4. Oxygen evolution reaction polarization curves in 0.5 M H2SO4 for IrO2 anodes containing (—) 0, (···) 10, (‒ ‒ ‒) 25 and (‒ ‒) 40 wt% TiO2 nanoparticles. The data was measured at 1 mV s−1 and was iR corrected post-run using the resistance determined by EIS.
The accelerated life tests of the fabricated anodes showed that while the lifetime of the IrO2 anode containing no TiO2 nanoparticles was only 17 h, the addition of 10 wt% and 25 wt% TiO2 nanoparticles increased this by two and ten times respectively (Figure 5). The lifetime for the anode containing 40 wt% TiO2 nanoparticles was much shorter at 2.7 h. This important result indicates that simply by adding an optimum amount of inert nanoparticles into a DSA layer can significantly increase the service life of these anodes. We interpret the short lifetime of the anode containing 40 wt% TiO2 nanoparticles as being related to the poor coverage of the film over the titanium substrate which would have led to rapid passivation and possibly mechanical failure of the particle-like coating. Indeed, SEM analysis after this electrode had failed showed large regions of anode were no longer coated, suggesting that at least part of the failure mechanism was due to erosion of the coating. Given that the density of IrO2 (11.67 g cm−3) is much higher than TiO2 (4.25 g cm−3)4,5 for the anode containing 40 wt% TiO2 nanoparticles, the 60 wt% IrO2 in the layer only corresponds to around 35 vol% IrO2 which might not be enough to act as an effective binder for the TiO2 nanoparticles.
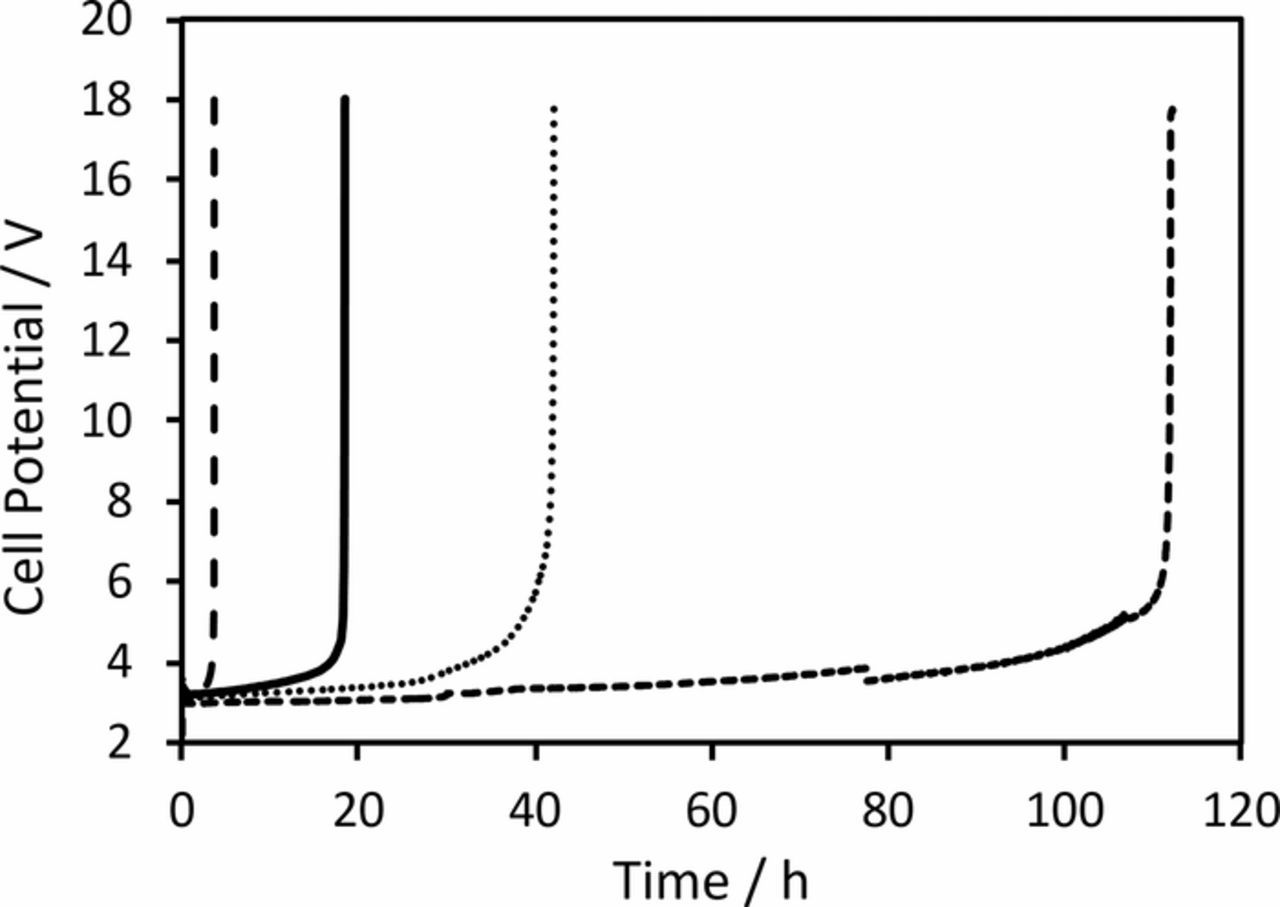
Figure 5. Accelerated lifetime evaluation of IrO2 anodes containing (—) 0, (···) 10, (‒ ‒ ‒) 25 and (‒ ‒) 40 wt% TiO2 nanoparticles at 1 A cm−2 in 2 M H2SO4 at 50°C.
The deactivation of DSA electrodes is generally assumed to be due to erosion, corrosion and/or passivation of the underlying substrate.6,46–48 Erosion occurs when the all or part of the active layer detaches from electrode substrate due to poor adhesion of the coating or mechanical stress, resulting from bubble formation at the cracks and pores of the coating. This differs to corrosion, where the active species are anodically oxidized to a soluble or volatile species during the OER.49 Passivation is caused by the formation of insulating layer on the (typically) titanium substrate and is widely known as final stage of DSA deactivation, and often accelerates the failure of the anodes following the partial detachment or corrosion of the coating.6,50 In this work, as the TiO2 nanoparticles are unlikely to influence the corrosion rate of the active IrO2 phase, the improved lifetime of the anodes containing 10 and 25 wt% TiO2 nanoparticles must rise from improved mechanical strength and/or reduced electrolyte penetration through the layer leading to reduced substrate passivation. As the addition of 10 wt% TiO2 nanoparticles resulted in noticeably smaller cracks in the layer compared to the pure IrO2 anode, it is reasonable to propose that this anode has an improved lifetime due to less electrolyte penetration and substrate passivation. For the anode containing 25 wt% TiO2 nanoparticles, no cracked-mud structure was found and instead the layer exhibited a particle-like morphology. While this layer is porous, the coverage of IrO2 over the substrate was even, which suggests that electrolyte penetration to the substrate may be impeded. We also propose that the composite layer may have improved mechanical strength (similar effect to that found in other nanoparticle composites18–20) which reduces the erosion rate under strong oxygen evolution reaction rates.
Conclusions
This study investigated the effects of addition of TiO2 nanoparticles on service lifetime and electrochemical behavior of IrO2 anodes. The accelerated service lifetime test show that the addition of TiO2 nanoparticles into the oxide matrix enhances the durability of the anodes, with 10 wt% and 25 wt% TiO2 nanoparticle additions increasing the service lifetime of the electrodes by 2x and 10x respectively. When 40 wt% TiO2 nanoparticles were added, the anodes had much lower lifetimes due in part to poor coverage of the coating on the titanium substrate. Importantly, the simplicity of embedding the oxide nanoparticles into the thermally prepared IrO2 layers should be compatible with standard industrial DSA production methods and maybe further optimisable to improve both service lifetime and electrochemically active surface area.
Acknowledgments
The authors thank Nathan Burns for the preparation of the anodes and conducting the accelerated lifetime tests reported in this work.
ORCID
Aaron T. Marshall 0000-0002-3530-7251