Abstract
Batteries for grid storage applications must be inexpensive, safe, reliable, as well as have a high energy density. Here, we utilize the high capacity of sulfur (S) (1675 mAh g−1, based on the idealized redox couple of S2−/S) in order to demonstrate for the first time, a reversible high capacity solid-state S-based cathode for alkaline batteries. To maintain S in the solid-state, it is bound to copper (Cu), initially in its fully reduced state as the sulfide. Upon charging, the sulfide is oxidized to a polysulfide species which is captured and maintained in the solid-state by the Cu ions. This solid-state sulfide/polysulfide cathode was analyzed versus a zinc (Zn) anode which gives a nominal >1.2 V cell voltage based on the sulfide/polysulfide redox cathode chemistry. It was found that in order for the S cathode to have the best cycle life in the solid-state it must not only be bound to Cu ions but bound to Cu ions in the +1 valence state, forming Cu2S as a discharge product. Zn/Cu2S batteries cycled between 1.45 V and 0.4 V vs. Zn displayed capacities of ∼1500 mAh g−1 (based on mass of S) or ∼300 mAh g−1 (based on mass of Cu2S) and high areal (>23 mAh cm−2) and energy densities (>135 Wh L−1), but suffered from moderate cycle lifes (<250 cycles). The failure mechanism of this electrode was found to be disproportionation of the charged S species into irreversible sulfite releasing the bound Cu ions. The Cu ions become free to perform Cu specific redox reactions which slowly changes the battery redox chemistry from that of S to that of Cu with a S additive. Batteries utilizing the Cu2S cathode and a 50% depth of charge (DOC) cathode cycling protocol, with 5 wt% Na2S added to the electrolyte, retained a cathode capacity of 838 mAh g−1 (based on the mass of S) or 169 mA h g−1 (based on mass of Cu2S) after 450 cycles with >99.7% coulombic efficiency. These Zn/Cu2S batteries provided a grid storage relevant energy density of >42 W h L−1 (at 65 wt% Cu2S loading), despite only using a 3% depth of discharge (DOD) for the Zn anode. This work opens the way to a new class of energy dense grid storage batteries based on high capacity solid-state S-based cathodes.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Due to the continually increasing penetration of renewable energy onto the electric grid, batteries for grid reliability are needed and must have the following metrics: low cost, safe, non-toxic, high cycle-ability, and have high energy density.1 Sulfur (S) is considered a high capacity material for battery applications [based on the idealized redox couple of S2−/S (1675 mAh g−1)], with suitable terrestrial abundance and overall economics.2 It is currently being developed for use as the cathode in lithium-sulfur (Li-S) batteries which are anticipated to be of lower cost and higher energy density (>500 W h kg−1) than current lithium-ion technology (50–300 W h kg−1);2–4 however, for grid electrical energy storage applications this chemistry is still considered too costly (∼$200 per kWh),4 as well as potentially unsafe for widespread use, due to the flammability of the organic electrolyte in these cells. Technical considerations that must be overcome for S-based electrochemistry in these batteries include: polysulfide shuttling leading to self-discharge and the low conductivity of sulfur/sulfides.5
Recently, Li/S batteries utilizing non-flammable water-in-salt electrolyte (WiSE) have been demonstrated, with high capacity and cycle life, largely due to the insolubility of sulfur-based species in this electrolyte.6 While the use of WiSE also addresses safety concerns for Li-ion batteries, a cost reduction in the fluorinated electrolytes, exemplified by bis(trifluoroimide) salts, would be needed.6 Aqueous S batteries consisting of a dissolved S redox active sulfide/polysulfide electrolyte are also under development. In these batteries, S-based cathodes are paired with anodes (such as Zn) in separate compartments due to polysulfides' high solubility and shuttle mechanism which would result in self-discharge of the cell, if sulfide "crossover" were to occur.6–9 This high solubility is needed for these fuel-cell type cathodes as their energy density is directly proportional to the active materials solubility. These cells have been shown to achieve high cycle lives (>200 cycles), with lower S active material capacities of ≤120 mA h g−1.8
Alkaline aqueous Zn/S batteries where the anode consists of a Zn composite electrode and the cathode is a dissolved S redox active sulfide/polysulfide electrolyte have been historically researched as primary cells.10 These cells have been shown to have high capacity and high energy density; however, the separator crossover of the highly soluble sulfide and reaction with the Zn anode during discharge forms an insulating ZnS layer rendering the Zn anode inert to recharge.10,11 In an attempt to develop a rechargeable Zn/S battery, a solid-state alkali metal conducting ceramic separator has recently been used to compartmentalize the Zn dissolution/precipitation anode reaction from the soluble sulfide/polysulfide cathode reaction.12 Although, a reversible battery with a high cathode active material capacity (>800 mAh g−1 of S) at nearly 100% coulombic efficiency was obtained from the low-cost electrode materials, the battery suffers from low rate capability due to the nominal 1 mm thickness of the ceramic separator. While thinner separators with lower resistance have been achieved via mechanical milling,13 the thinner ceramic separators become brittle and generally harder to manage/utilize. Additionally, the energy intensive synthesis of the ceramic significantly increases the overall cost and is currently far too high for broad use in the grid energy storage market.
It has been recently proposed that in alkaline electrolytes that sulfide (e.g. S2−) exists in the form of the bisulfide ion, HS−, with no actual free S2− ions.14 Accordingly, HS− is known to have the following redox chemistry in alkaline electrolyte:15
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0002.gif)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0003.gif)
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0004.gif)
Whereby, polysulfides of various atomic chain length are reduced to bisulfide and hydroxide ions. Additionally, Zn as an anode in alkaline batteries has the well-established electrochemical oxidation reaction:16
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0005.gif)
Wherein, Zn oxidizes to the hydroxide complexed Zn2+ species, zincate [Zn(OH)2 −4], that can precipitate through dehydration to ZnO. Therefore, a battery combining these electrodes would result in the following overall reaction:
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0006.gif)
A table of the different polysulfide products and their theoretical performance metrics (gravimetric capacity and voltage vs. Zn) are provided in Table I. A ≥1 V potential vs. Zn and ≥800 mA h g−1 of S specific capacity would result in an energy density on the order of ≥200 Wh L−1 (with 65% active cathode material loading, ∼ 3% Zn anode DOD and similar dimensions as other alkaline grid storage batteries).17 This is more than adequate for grid storage applications, would compete well with the best lead acid and alkaline batteries currently being considered/used for grid storage (∼ ≤ 100 Wh L−1) and is similar in energy density (150–250 WhL−1) as Li-ion batteries based on LiMnO2 or LiFePO4.18 For example, due to the inexpensiveness of the materials and ease of manufacturing, alkaline grid storage batteries based on Zn/MnO2 are being commercialized based on an even lower capacity limited depth of discharge (DOD) technology with energy densities of ∼10–40 Wh L−1 at overall costs of ∼$150–250 kWh−1.17 Higher energy dense Zn/MnO2 batteries are also currently under development19,20 with the goal of reaching $50 kWh−1 at the cell level.18,21 Hence, interest in high energy dense alkaline battery system based on inexpensive abundant elements is of interest.21
Table I. Theoretical performance metrics of polysulfide reductions in alkaline solutions.
| Chemical | Polysulfide | # of e− | Potential vs. | Specific Capacity | Specific Capacity |
|---|---|---|---|---|---|
| Reduction Equation | Species | per S atom | Zinc (V) | (mA h g−1 S) | (mA h g−1 Cu2S) |
| 1 | S2 −5 | 1.60 | 1.202 | 1340 | 268 |
| 2 | S2 −4 | 1.50 | 1.232 | 1256 | 251 |
| 3 | S2 −3 | 1.33 | 1.296 | 1117 | 223 |
| 4 | S2 −2 | 1.00 | 1.497 | 838 | 168 |
Herein, we report on the first use of a solid-state sulfide/polysulfide electrochemical couple(s) in alkaline electrolyte (8.5 M KOH).22 Specifically, copper sulfide (Cu2S) is demonstrated as a solid-state sulfide/polysulfide electrode for use in alkaline Zn/Cu2S batteries, with nominal cell voltages of >1.2 V. Theoretical capacities of the Cu2S cathode based on the mass of S or Cu2S and the redox (# of e−s per S atom) of the Sn2−/HS− couple can be found in Table I. S is kept in the solid-state by utilizing Cu ions as polysulfide/sulfide complexing agents. Cu's ability to complex with soluble polysulfides limits their solubility in alkaline solutions and prevents and/or postpones the deposition of insulating ZnS on the Zn anode.
Zn/Cu2S batteries utilizing a 50% depth of charge (DOC) cathode cycling protocol, with 5 wt% Na2S added to the electrolyte, retained a cathode capacity of 838 mAh g−1 (based on the mass of S) or 169 mA h g−1 (based on mass of Cu2S at 65 wt% loading) after 450 cycles with >99.7% coulombic efficiency. The Zn/Cu2S batteries provided a grid storage relevant areal capacity of 8.3 mA h cm−2 (with 65% active material) and energy density of >42 W h L−1 over the 450 cycles, despite only using a 3% depth of discharge (DOD) for the Zn anode. Standard Celgard separators were sufficient for battery operation, suggesting a good binding between the Cu and S, limiting solubility, as both Cu and S are known to react negatively with Zn after crossover. The role of Cu valence on the sulfide electrochemistry and the effect of disproportionation reactions of the charged S species is also further elucidated.
Experimental
Chemicals and materials
All chemicals were purchased and used as received. Cu2S, CuS, Cu, CuO, Cu2O, graphite, KOH, Na2S and S were purchased from Sigma-Aldrich, Cu(OH)2 was purchased from Alfa Aesar, PEG 400 was purchased from EMD Chemical and tartaric acid was purchased from Fisher Scientific. SDBS was purchased from Acros Organics. Polytetrafluoroethylene (PTFE) DISP 30 was obtained from Chemours. Expanded Cu mesh (5CU5) and expanded nickel mesh (5NI5) was purchased from Dexmet Corporation. Cellophane 350P00 was obtained from Innovia Films Inc. Celgard 3501 was purchased from Celgard, LLC. Polypropylene 'Flex-A-Top' containers used as battery casings were purchased from LA Container Inc. Cellulose fibre tissue wicking separators were purchased from Kimberly-Clark.
Zinc electrodes
Zn electrodes were made by mixing 83.1 wt% Zn, 9.8 wt% ZnO, 2.2 wt% SDBS, and 5 wt% PTFE. Isopropanol (IPA) was added to help uniformly mix the materials into a malleable putty. The putty was then rolled to a thickness of ∼0.8 mm. The composite Zn putty was then put into an oven @ 60°C for 1 h to bake off the IPA. The material was then cut into ¾" × 1" rectangles which comprised the anode. The anode was then pressed onto Cu mesh with a Ni tab at 414 mPa (anode thickness after pressing: ∼0.6 mm).
Electrodes composed of Cu and/or S compounds
Cathodes comprised of Cu and/or S compounds were made by mixing 65 wt% Cu and/or S compound, 30 wt% graphite, and 5 wt% PTFE. IPA was added to help uniformly mix these materials into a malleable putty. In the case of elemental Cu and S, these were mixed in a 2:1 molar ratio. The Cu and/or S composite putty was then rolled to a thickness of ∼0.40 mm. The material was then placed into a 60°C oven for 1 h. The material was then cut into 1 × 1 cm squares to make electrodes. The electrode was then pressed into Ni mesh with a Ni tab at 414 mPa (cathode thickness after pressing: ∼0.27 mm).
Electrolyte
KOH flakes were added to DI water to obtain an 8.5 M KOH solution. 4000 ppm tartaric acid and 3000 ppm PEG 400 were added to this solution to limit the hydrogen evolution reaction on the Zn anode.
Battery assembly
Zn electrodes were wrapped in one layer of Celgard 3501 (25 μm thick) followed by 4 layers (each 25 μm thick) of cellulose fibre tissue wicking material. The Cu- and/or S-based electrodes were wrapped in 3 layers of Cellophane 350P00 (each 25 μm thick) followed by 4 layers of cellulosic fibre tissue wicking material. Electrodes were placed facing each other into a battery casing. A thick shim was placed into the case to keep separators from unravelling. Three mL of the electrolyte above was then added to the case. Batteries were then soaked in this state for at least 16 h. Following this soaking, another thick plastic shim was added to provide the needed compression for these cells. The total thickness of each cell is ∼0.12 cm.
Battery cycling
Full depth of charge/discharge cells were cycled between 0.4 and 1.45 V vs. Zn. Cells were cycled at a 0.2 C rate based on either the Cu/Cu2+ or S2−/S active redox couple in Cu-only or S-only cathodes and the Cu/Cu2+ redox couple when mixed. For example, the rate for Cu2S based on the Cu/Cu2+ redox couple is 0.2 C; however, the rate is 0.4 C based on the S2−/S redox couple as there is half as many S atoms as Cu atoms in Cu2S.
50% DOD Zn/Cu2S batteries were discharged at a 0.4 C rate based on the S2−/S redox couple for 1.25 h with a cutoff voltage of 0.4 V vs. Zn. They were subsequently charged at the same rate to a 1.45 V end charge voltage.
50% DOC Zn/Cu2S batteries were charged at a 0.4 C rate based on the S2−/S redox couple for 1.25 h with a cutoff voltage of 1.45 V vs. Zn. They were subsequently discharged at the same rate to a 0.4 V end discharge voltage.
Volumetric energy density calculations
Volumetric energy densities were calculated similar to recent literature reports.17,19,20 The energy in units of Wh was obtained by a Maccor battery tester. This was divided by the volume of the cell in units of liters. The volume of the cell was determined by considering the total volume of both electrodes (after compression) and all separators. The volume of the current collectors was negligible as those were essentially pressed into the electrodes upon compression. The volume of electrolyte was also not considered as per literature,17,19,20 but was fixed at 3 mL per cell. Further improvements in volume electrolyte and cell build will be conducted in the future once cycle life has been optimized.
Raman spectroscopy
Raman spectra were obtained using a WiTec Alpha300M instrument. The laser source was a 532 nm Nd:YAG laser. The power was set to 0.5 mW. Spectra were obtained using 10 accumulations of 10 sec.
Drop-cast particle films
For cyclic voltammetry analysis, drop-cast particle films were used. To prepare, particle inks were made that consisted of 25 mg of particle powders in a solution of 7.5 mL DI H2O, 2.0 mL IPA, and 0.5 mL of a 5 wt% Nafion solution. The solution was sonicated for 15 min to obtain a suspension. 5 μL of the suspension was then drop cast on a glassy carbon disc electrode (area = 0.707 mm2). After drying, the particles were immersed in 8.5 M KOH and then immediately electrochemically cycled.
Results and Discussion
It is well-established that S and Cu individually both dissolve at high concentrations in highly alkaline environments with Cu forming Cu(OH)42− and/or Cu(OH)3− (pKsp = 20.36 for CuO and pKsp = 19.34 for Cu(OH)2)23 and S forming polysulfide(s), Sx2−, and bisulfide, HS−, ions (pKsp = 10.52 for S).14,23–25 This is demonstrated by the deep color they produce upon immersion into alkaline solutions, yellow-orange for S and blue for Cu, as seen in Figure 1a. Thus, their high solubility in strong base limits their individual ability to form stable solid-state cathodes in alkaline batteries. However, S and Cu have a strong affinity for each other and form insoluble copper sulfides/polysulfides (pKsp = 36.26 for CuS and pKsp = 48.61 for Cu2S)23 when combined in alkaline, as demonstrated in the precipitate formation that occurs when combining two concentrated solutions of each species in Figure 1a. This is the premise of the current research; whereas, copper sulfides should largely exist as solid-state S redox cathodes in alkaline batteries.
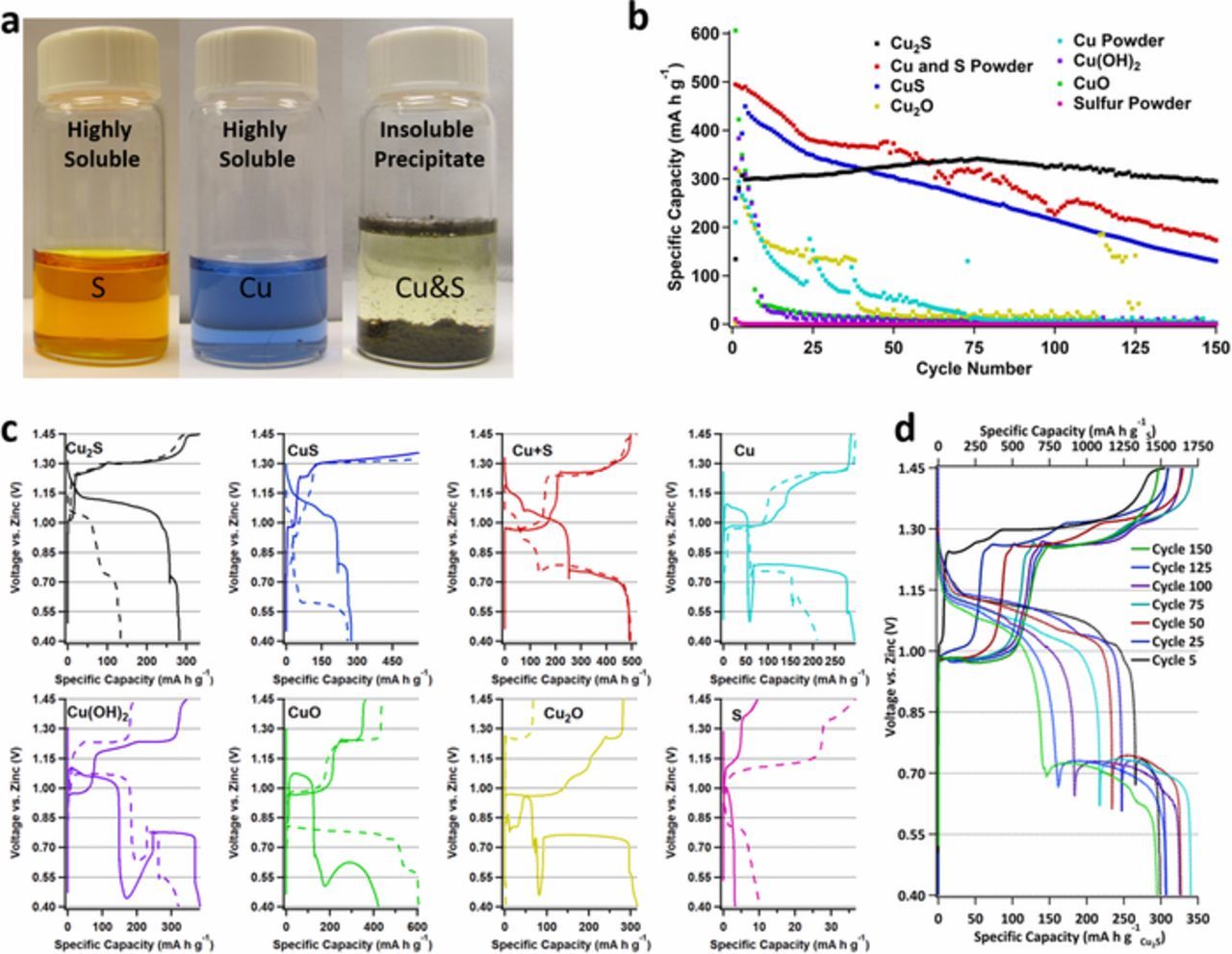
Figure 1. a. 5 wt% solution of sulfur in 8.5 M KOH, saturated solution of copper in 8.5 M KOH, and their mixed product in 8.5 M KOH. b. Specific capacity (based on mass of active material) versus cycle number for a variety of Cu and S materials as the active materials in the cathode versus a Zn anode. c. The first (dashed line) and second (solid line) galvanostatic charge/discharge curves for the materials represented in b (note: each cycle begins with discharge). d. Cu2S cathode charge and discharge profiles at cycle 5, 25, 50, 75, 100, 125, and 150.
To understand the electrochemistry of Cu and S compounds individually and combined under alkaline conditions, a variety of Cu and S compounds as the active material in cathodes versus Zn anodes were galvanostatically cycled (rate: 0.2 C based on Cu/Cu2+ redox species in Cu-only or mixed Cu/S cathodes and 0.4 C rates based on the S2−/S active redox species in S-only cathodes) for 150 cycles between the potentials of 1.45 and 0.40 V vs. Zn. The results of these experiments can be found in Figure 1b. Electrodes containing Cu in the form of Cu metal, CuO, and Cu2O powders all fail within 50 cycles, with failure defined as when the electrodes reach ≤15% of their theoretical capacity. Additionally, the S-only electrode fails within two cycles, with most of the elemental S dissolving before any electrochemistry can even take place, as evidenced by electrolyte color. However, Cu containing S electrodes [(Cu2S, CuS and (Cuo+So)] have enhanced cycle lives with all of them lasting at least 150 cycles. The best performing of these was Cu2S which retained 100% of its capacity over the 150 cycles. In fact, Zn/Cu2S batteries retained a cathode capacity of ∼1500 mAh g−1 (based on the mass of S) or ∼300 mA h g−1 (based on mass of Cu2S at 65 wt% loading) after 150 cycles with >99.5% coulombic efficiency. These Zn/Cu2S batteries provided an impressive areal capacity of >23 mA h cm−2 (with 65% active material) and energy density of >135 Wh L−1 (see supporting information Figure S1), justifying further investigation.
To help deduce the reason why Cu2S is the best performing material, an examination of each compound's first two charge/discharge profiles is presented. These charge/discharge profiles are found in Figure 1c. The results indicate that only Cu2S, CuS, and S have high oxidation plateaus on their first charge above 1.30 V. This high voltage charge plateau is attributed to the oxidation of sulfide/bisulfide to polysulfide by chemical reactions 1 through 3 above. In the case of CuS, the oxidation above 1.30 V continues until the charge capacity reaches the theoretical two electron capacity of S cut off [561 mA h g−1 based on the total mass of CuS (66.5 wt% Cu and 33.5 wt% S)]. This indicates that the sulfide is being fully oxidized to S and that Cu also participates in the CuS electrochemistry by oxidizing during the charge producing higher than theoretical capacities. However, the complete oxidation to S is not advantageous as S is very unstable (soluble, i.e. as per Fig. 1a) in strong alkaline environments with a disproportionation reaction occurring represented by the following equation:26
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0007.gif)
This disproportionation of S plays a dominant role in the instability of CuS, which is characterized by the coulombic inefficient subsequent discharge that reduced less than 43% of the sulfide that was oxidized, e.g. achieving only ∼ 290 mA h g−1 based on mass of CuS. Additionally, the plateau above 1.30 V quickly diminishes upon cycling CuS and is completely gone by the 4th charge cycle (see supporting information Figure S2). As the oxidation above 1.30 V is attributed to sulfide oxidation, its disappearance indicates that the CuS electrode (with electrochemistry initially dominated by sulfide/polysulfide) transitions to a Cu based battery, whereby the sulfide/polysulfide now functions as an additive. In fact, cycle 4 for CuS (Figure S2) is essentially identical in shape to that of Cuo+So (Figure 1C (Cu+S)), consistent with this notion. This has the effect of reducing the cycle-ability of CuS electrode to arrive at that similar of a Cuo+So battery, as evidenced by similar Specific Capacity vs. Cycle Number profiles exhibited in Figure 1b.
Additional charge cycle data for Cu2S that correlates to Figure 1b is provided in Figure 1d. It is clear that Cu2S is the only compound tested that performs S redox chemistry over all 150 cycles (existence of high voltage plateau upon charge >1.30 V vs. Zn for all cycles) and that the discharge profile (under the cycling conditions used here) progresses over time from a predominantly high voltage plateau to one of lower voltage and dramatically fails once the high voltage plateau is diminished giving a total cycle life of ∼250 cycles. The details of this transition will be discussed but first we focus on the role of copper valence on sulfur electrochemistry.
The crystal structure of Cu2S is in the low chalcolite monoclinic form (see XRD data in figure S3) with the space group of Pc which consists of a unit cell with 96 Cu2S units with no S-S bonds.27 This absence of S-S bonds, as apparent in the absence of any S-S vibrational modes in the Raman spectra (see supporting information Figure S4), reduces the ability to form long chain polysulfides upon cycling Cu2S. It is proposed that Cu in the +1 oxidation state is needed to form stable bonds with S. For example, both Cu2S and CuS, the latter of which stoichiometrically should contain Cu in the +2 oxidation state, are found in the literature by XPS to only have Cu+ character, thus demonstrating that Cu+ is the preferred oxidation state of Cu for S binding and/or polysulfide capture.28 Similarly, this concept has recently been demonstrated in organic electrolyte Li/S batteries; where, Cu was shown to capture polysulfides in the solid-state form of CuSn and Cu2Sn thus preventing the well-known polysulfide shuttle reactions that reduce the cycle ability of Li/S batteries.5
To further demonstrate the importance of the Cu +1 valence state in supporting the sulfide redox in alkaline electrolyte, Cu2S electrodes were discharged more deeply to 0.05 V at various cycle number intervals (e.g. every cycle, every 10th cycle, every 25th cycle, or every 50th cycle). This experiment was performed since linear sweep voltammetry (LSV) on drop casted films of Cu2S showed that during the initial negative sweep a major reduction occurs at a potential of 0.15 V vs. Zn which is below the end of discharge potential of 0.40 V used during the standard Cu2S cycling shown in Fig. 1b and Fig. 1d. This result can be seen in Figure S6. This cathodic peak is assigned to the reduction of the Cu+ to Cu0 as copper is in the +1 valence state in Cu2S. The cycle-ability of the cells discharged at various cycle numbers to 0.05V can be seen in Figure S6a. Discharge curves before and after full discharge are seen in Figure S6b. The results indicate that once Cu+ is reduced to metal, its presence and binding to S is not found in the subsequent charge indicated by the absence of the charge plateau above 1.30 V and thus the battery's subsequent behavior is no longer indicative of Cu2S but CuS for the remainder of cycling. Hence the battery's cycle life is adversely affected. Thus, it is apparent that the Cu valence state of +1 is needed for a sustained cycle-ability of cathode made from Cu and S species in alkaline electrolyte.
To analyze the charge/discharge mechanism of Cu2S cathodes, Raman spectroscopy was employed as it is a method well suited for distinguishing the differences in Cu metal oxide species as well as sulfides and polysulfides.29,30 Figure 2a represents a typical galvanostatic charge/discharge curve for a Cu2S electrode with the numbers representing important points of discussion along the curve. For Raman analysis, Zn/Cu2S batteries were cycled 5 times and then stopped at the designated point along the charge/discharge curve. From there, the cell was taken apart and the Cu2S cathode was analyzed by Raman spectroscopy, the spectra of which are shown in Figure 2b. Point one represents the end of the 1st charge step/reaction and the following discussion begins with the charge sequence from this point.
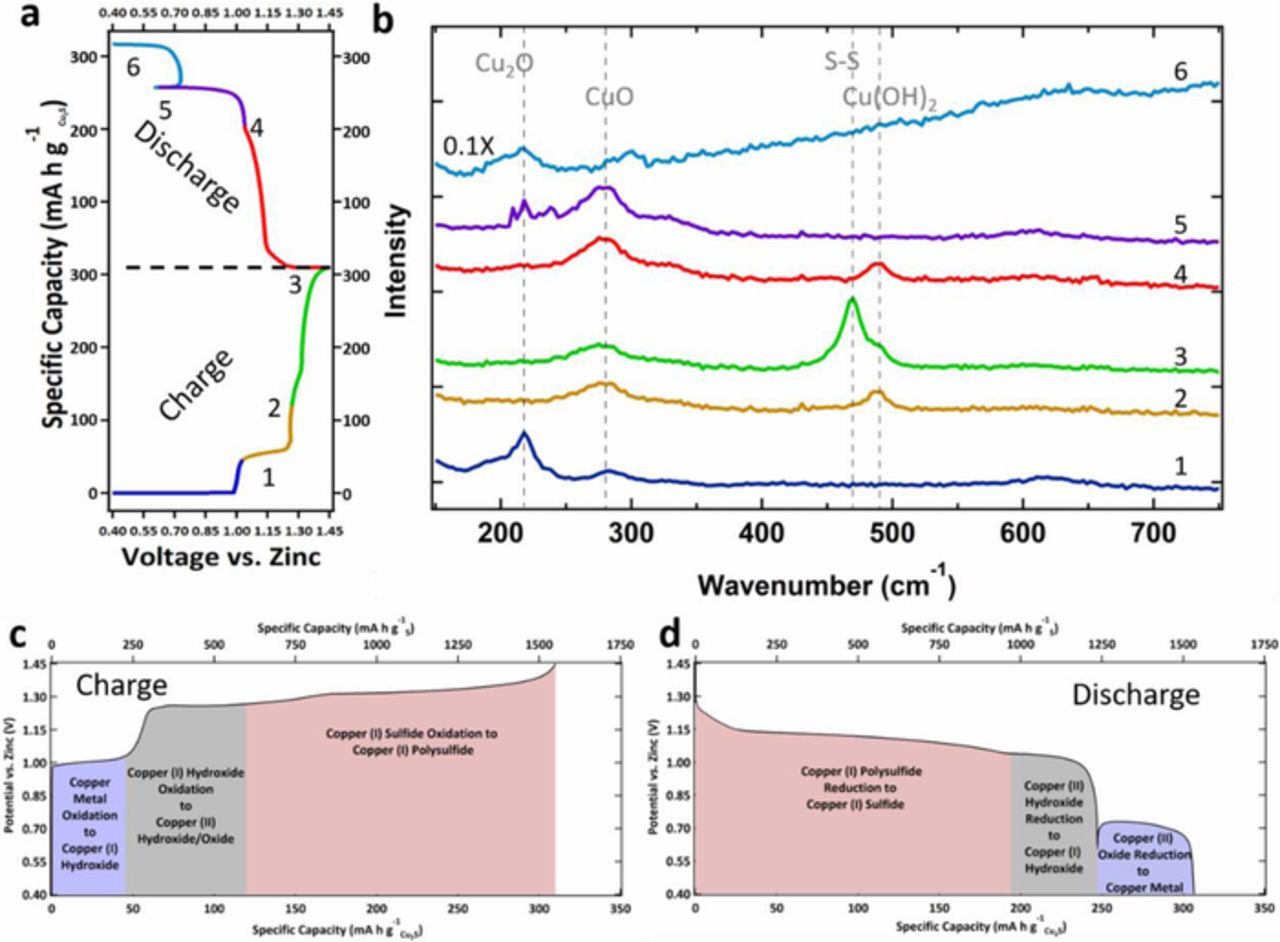
Figure 2. a. Representative charge/discharge curve with numeric figures indicating positions along the curve where Raman spectra was taken. b. Ex Situ Raman spectra of electrodes taken along their charge/discharge profile at the different numeric points indicated in a. c. Typical charge curve (25th cycle) for a Cu2S cathode indicating the phase transitions inferred by the Raman spectroscopy in b. d. Typical discharge curve (25th cycle) for a Cu2S cathode indicating the phase transitions inferred by the Raman spectroscopy in b.
Upon charging, the Raman data indicates that the Cu metal oxidizes to Cu(OH) at step 1 due to the appearance of the Cu2O [dehydration product of Cu(OH)] mode at 218 cm−1 in the Raman spectra. This oxidation is represented by the following reaction:
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0008.gif)
This 1 e− oxidation of Cu is well characterized in the literature with cyclic voltammetry indicating a 59 mV peak potential difference per decade of hydroxide concentration indicative of a 1 e− transfer.31,32 It should be noted that some CuO may be present here and throughout the Raman spectra experiments due to its role as the thermodynamic sink for Cu in alkaline solutions. Upon further charge, the Raman spectra of point two shows a disappearance of the Cu(OH) mode, an appearance of the Cu(OH)2 mode at 490 cm−1, and an enhanced CuO peak represented by the Raman mode at 281 cm−1 which indicates that oxidation to Cu(OH)2 and CuO has taken place through the following reactions:
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0009.gif)
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0010.gif)
The dehydration of Cu(OH)2 to form CuO could also be present.
![Equation ([11])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0011.gif)
Further oxidation to 1.45 V vs. Zn results in the appearance of the S-S bonding mode at 470 cm−1 in the Raman spectra, at step 3. This is indicative of the formation of polysulfides by the following reaction:
![Equation ([12])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0012.gif)
This S redox chemistry is supported by electrochemically analyzing a solution of Na2S in 8.5 M KOH; whereas, a nickel foil was scanned positively in the solution producing an anodic wave at −0.1 V vs. Hg/HgO (equivalent to ∼ 1.3 V vs. Zn) as seen in supporting information Figure S7a. Electrolysis of this same solution at 0.05 V vs. Hg/HgO (equivalent to 1.45 V vs. Zn) resulted in a change in color of this solution (Figure S7b) as well as a change in its UV-Vis absorbance (Figure S7c) with new polysulfide absorbance bands, 3S2 −2, S2 −3, and S52 −, appearing after electrolysis, indicating that the HS− was oxidized to polysulfide species through Reactions 1–3 above. The absorbance band for S2 −4 is noticeably missing most likely due to known disproportionation reactions:5
![Equation ([13])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0013.gif)
![Equation ([14])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0014.gif)
Reduction of the electrode follows in the reverse order with the first discharge plateau which is represented by a sloping discharge profile indicative of S galvanostatic curves, as similarly observed in sulfur cathodes in Li/S batteries. Here, the S-S Raman mode at 470 cm−1 disappears at the end of this discharge step as indicted in point 4. However, after the discharge of the second plateau, point 5 in Figure 2a, the Cu2O [again the dehydrated form of Cu(OH)] mode appears at 218 cm−1 and the Cu(OH)2 mode at 490 cm−1 disappears indicating that Cu(OH)2 is reduced to Cu(OH) through the following reaction:
![Equation ([15])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0015.gif)
This is additionally supported by LSV of drop-casted Cu(OH)2 particles on a glassy carbon electrode which shows a cathodic wave with an onset of ∼1.0 V vs. Zn (see supporting Figure S5) indicating Cu(OH)2 reduction at this relatively high potential. Further reduction to 0.4 V vs. Zn shows a huge increase in fluorescence indicative of zero valent metal formation (note: spectrum 6 was reduced by a multiple of 0.1 x). Therefore, it is proposed that CuO is reduced to Cu at step 6 metal through the following reaction:
![Equation ([16])](https://content.cld.iop.org/journals/1945-7111/166/4/A687/revision1/d0016.gif)
Additionally, it appears that Cu2O/CuOH do not reduce to Cu metal as evidenced by the absence of a cathodic wave above 0.4 V vs. Zn for a Cu2O drop-casted nanoparticle film as well as the second cathodic wave of the Cu(OH)2 films as indicated in supporting Figure S5. This indicates that the reduction at 0.70 V is not due to a Cu+ to Cu0 mechanism. Additionally, this is evident by the absence of a reduction plateau for Cu2O's first discharge to 0.4 V vs. Zn as seen in Fig. 1c.
For further analysis of the battery chemistry over cycle number, the voltage profile evolution over cycle lifetime was examined as represented in Figure 3a. The three main discharge reactions were inventoried over 150 cycles. These are plotted in Figure 3b. From this data, it was found that the cathode capacity slowly converts from that mostly derived from S electrochemistry to that derived mostly from Cu. Since the Cu redox transfers >1 e- per Cu atom, the capacity of the electrode increases initially over the first 75 cycles as S which transfers only 1 e- per Cu atom slowly releases Cu for the Cu electrochemistry. Note: this increase in capacity is eventually met with a concomitant decrease in energy density as the voltage is lower for the Cu redox. Nonetheless, this change in capacity can be quantitatively seen, if one assumes that for every Cu atom released from the S matrix, it can form either Cu(OH)2 which then undergoes a further 1 e− reduction, or CuO which undergoes a further 2e− reduction. With any increase in capacity for Cu(OH)2 there will be a 1:1 decrease in Cu2S capacity and for any increase in the capacity for CuO there will be a ½ decrease in the capacity of Cu2S since CuO transfers twice the electrons of Cu2S per copper atom. Thus, to conserve Cu's role as the inert element when bound to S as Cu2S and its role in the electrochemistry when bound to O as either Cu(OH)2 or CuO; the sum of the capacity of Cu2S, Cu(OH)2, and ½ CuO must be maintained for each cycle. As seen in Fig. 3b, under the cycling conditions used here, this summation of the redox reactions holds up until the 75th charge/discharge cycle. However, at the 75th cycle, the capacity begins to decrease due to the instability of the Cu cycle-ability by itself as indicated previously by early cycle failure for Cu only compounds in Figure 1b. Additionally, it can be observed in Figures 3b that the decrease in the S reaction was linear versus cycle number over all 150 cycles under the cycling conditions used for this experiment. The cell has a dramatic capacity fade after 150 cycles and drops below 50% of the theoretical S/S2− capacity around 250 cycles thus indicating that the sulfur redox chemistry is crucial to long term cycle life.
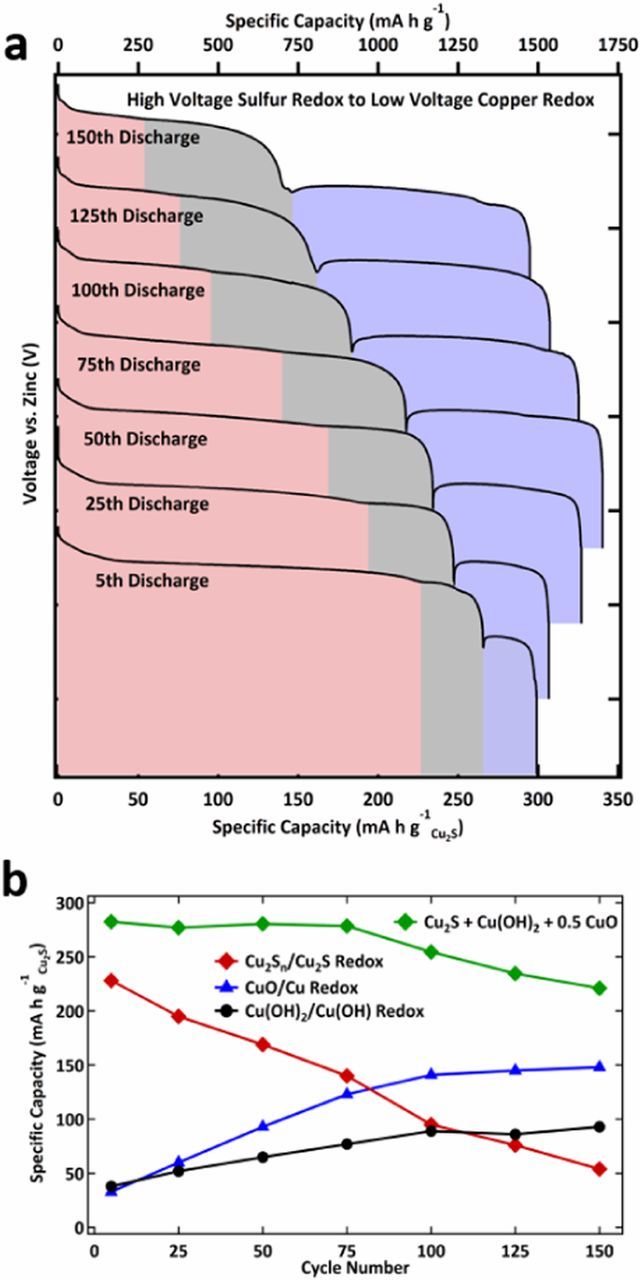
Figure 3. a. Discharge curves of Cu2S at a 0.4 C rate (note: based on S2−/S redox; however, the rate is 0.2 C based on Cu/Cu2+ redox) from cycle 5 to the 150th cycle with the three redox reactions represented by shading; b. Specific capacity (based on the mass of Cu2S) of the three redox reactions varying with cycle number obtained from the data in b.
To examine whether this linear degradation in S-based capacity is a construct of electrochemistry or whether the degradation is a result of chemical transformations that occur in the highly alkaline electrolyte, an experiment was performed where Zn/Cu2S batteries were paused for 36 hours every 15th cycle on either charge or discharge. Specific capacity versus cycle number and specific capacity versus experimental time for these cells are provided in Figs. 4a and 4b. The cell paused after the charge step to 1.45 V vs. Zn precipitously loses capacity after every paused step and overall shorter cell lifetimes were obtained for the cell upon resting on charge. It is apparent from these experimental time curves that the longer the electrodes are in the oxidized state (@ 1.45V vs. Zn), the greater the propensity for the sulfur species that is formed to disproportionate. As for the cell that is paused after discharge to 0.4 V vs. Zn, this cell also has a lower cycle life than the cell which is neither paused on charge or discharge. However, the cell paused on discharge demonstrates similar capacity versus time behavior as the control cell. Thus, it appears the continued degradation of the S species is a chemical (time dependent) and not electrochemical (cycle dependent) phenomenon and clearly this degradation happens in the charged state, under these cycling conditions.
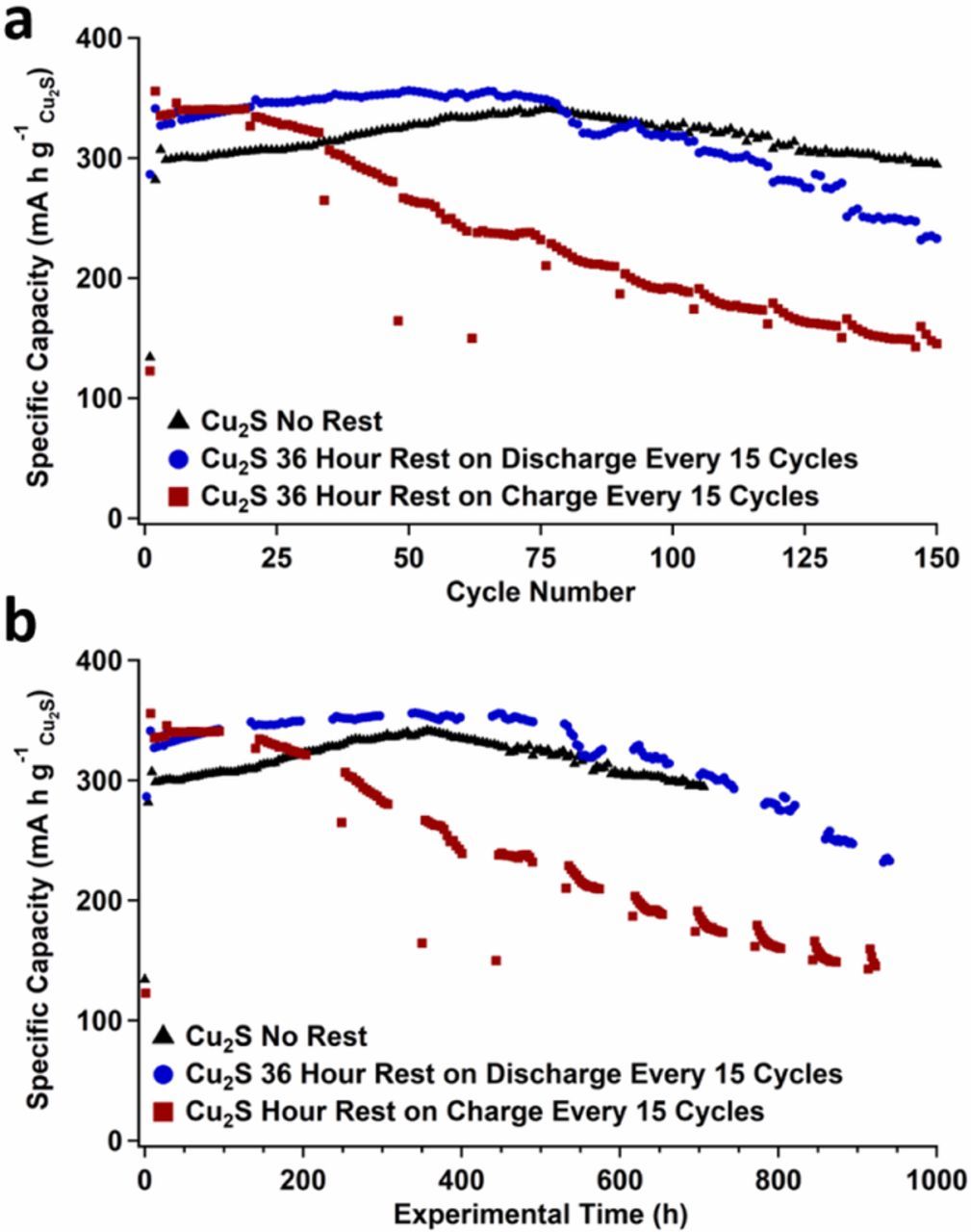
Figure 4. a. Specific capacity versus cycle number of a Zn/Cu2S battery with no rest and resting every 15 cycles for 36 hours upon discharge (to 0.4 V vs. Zn) or upon charge (to 1.45V vs. Zn). b. Specific capacity versus experimental time of a Zn/Cu2S battery with no rest and resting every 15 cycles for 36 hours upon discharge or upon charge.
Utilizing the above results as guidance, cells were cycled based on limited 50% DOD and/or DOC based on 2 e− S cycling (S2−/S) as seen in Fig. 5a. Cells were considered failed when their capacity dropped below 150 mA h g−1 based on the mass of Cu2S (∼250 cycles for full DOD/DOC cells). The cells limited on charge were able to last 20% longer on average which is attributed to the restricted generation of the fully oxidized sulfur species which can disproportionate by means of Equation 7. Additionally, as seen in Figure 5b, cells based on limited DOC have a considerably higher energy efficiency. This is most likely due to a decrease in the polarization of the limited DOC when compared to the limited DOD cell due (1) to full discharge of any CuO species to conductive copper metal and (2) the limiting of the full charge product of insulating polysulfides both of which improve the conductivity of the limited DOC electrode. This can also be attributed to why the energy efficiency of the full depth of charge/discharge electrode is in between the DOD and DOC energy efficiencies as both the insulating polysulfide species and the conductive Cu metal are formed upon charge and discharge, respectively, for the full DOD electrode. Additionally, Na2S was used as an additive to the electrolyte at 5 wt% to increase the presence of sulfide during the charging and discharging of these cells to minimize or mitigate loss of sulfur species due again to the chemical sulfur disproportionation. This hypothesis is supported by the increase in coulombic efficiency of the cathodes cycled in Na2S as seen in Figure 5c. (Note: addition of Na2S reduces the ionic conductivity of the electrolyte which increases the polarization of the oxidation and reduction reactions thus decreasing the energy efficiency of these cathodes as seen in Figure 5b.) As hypothesized, the decrease in the disproportionation to sulfide results in a lower self-discharge of the oxidized S charge product allowing more to be available for the subsequent electrochemical discharge and thus increasing the coulombic efficiency of the electrodes cycled in the 5 wt% Na2S electrolytes.
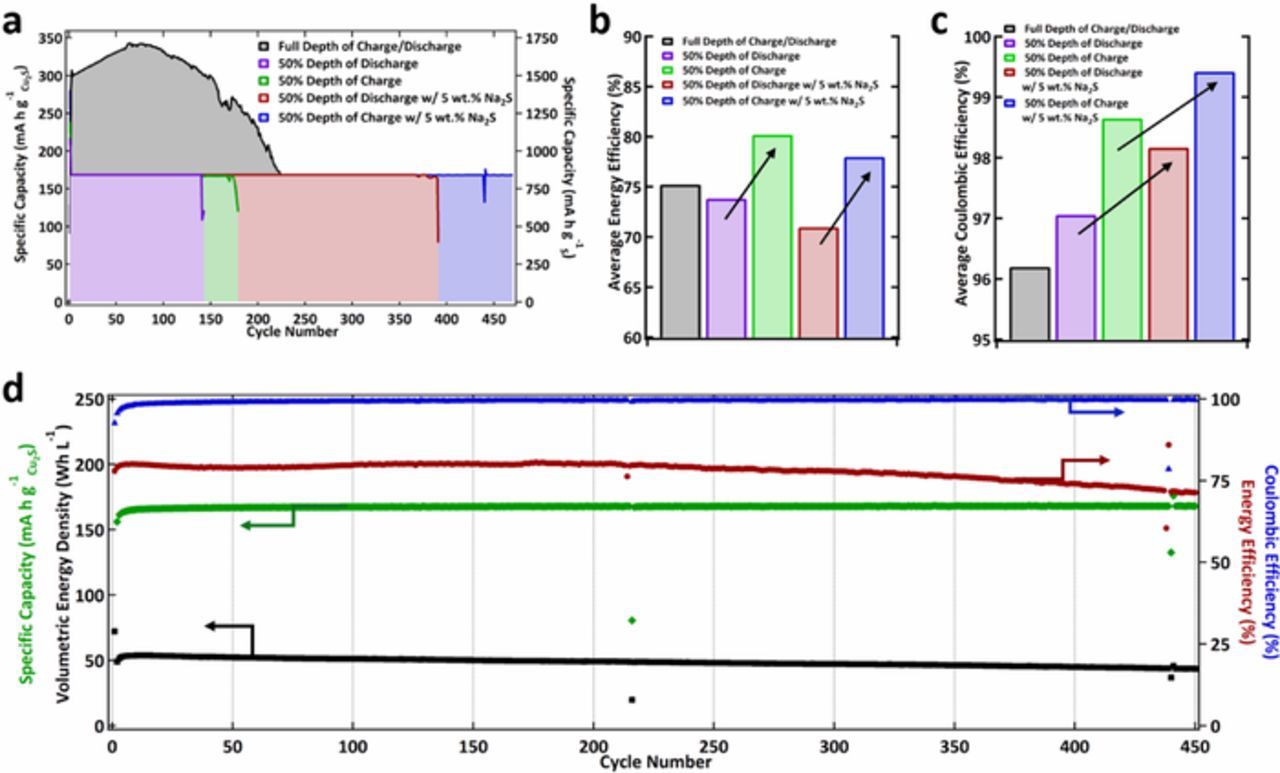
Figure 5. a. Cu2S cathode cycling at various charge discharge protocols: full depth of charge (black), 50% depth of discharge with no additive (purple), 50% depth of charge with no additive (green), 50% depth of discharge with 5 wt% Na2S in the electrolyte (red), and 50% depth of charge with 5 wt% Na2S in the electrolyte (blue). b. Average energy efficiency of cells cycled in a. c. Average coulombic efficiency of cells cycled in a. d. Performance metrics of Cu2S electrode cycled at 50% depth of charge with 5 wt% Na2S added to the electrolyte over 450 cycles.
The performance metrics of the Zn/Cu2S battery at 50% DOC with 5 wt% Na2S added to the electrolyte versus cycle number are found Figure 5d. Here, the specific capacity of the cathodes is retained at 838 mAh g−1 (based on the mass of sulfur) or 169 mA h g−1 (based on the mass of Cu2S) and an areal cathode capacity of 8.3 mA h cm−2 (with 65% active material) is retained even after 450 cycles. The volumetric energy density of the battery over 450 cycles (based on the volume of both pressed electrodes and separators: ∼0.12 cm3) is greater than 42 Wh L−1, despite only using ∼3% DOD of the Zn anode (see Figure S8 for materials performance of a cell utilizing ∼15% DOD of the Zn anode). These values are attractive and could render this chemistry suitable for grid storage applications once suitable cycle life has been obtained. For example, cells based on Zn/MnO2 that utilize a limited DOD (typically less than 10%) MnO2 cathode and a similarly low Zn utilization (∼ 0.9–1.8% depending on MnO2 DOD chosen) have shown promise for grid storage, with similar cells achieving ∼40 Wh L−1 with cycle life numbers of roughly 500, 20 Wh L−1 with cycle life numbers of ∼ 3000, or 10 Wh L−1 with cycle life numbers >4000.17 Additionally, the coulombic and energy efficiency for Zn/Cu2S starts out low and then stabilizes at a level over 99.7% for the former and retains >71% for the latter after 450 cycles. This discovery of a reversible high capacity solid-state S-based cathode for alkaline batteries provides a promising new avenue of exploration for a potentially low cost and safe electrical grid storage technology.22 Further optimization of sulfide/polysulfide stabilization is underway in order to provide increased cycle life and these advanced solid-state cathodes will be reported in due course.
Conclusions
A new battery chemistry is demonstrated that consists of a solid-state Cu2S cathode paired with a Zn anode in alkaline electrolyte. The nominal voltage of this battery is >1.202 V with a empirical capacity (see Table I) of ≥168 mA h g−1 based on the mass of Cu2S (and ≥838 mA h g−1 based on the mass of S). It was found that in order for the S cathode to have the best cycle life in the solid-state it must not only be bound to Cu ions but bound to Cu ions in the +1 valence state, forming Cu2S as a discharge product. Zn/Cu2S batteries cycled between 1.45 V and 0.4 V vs. Zn displayed capacities of ∼1500 mAh g−1 (based on mass of S) or ∼300 mAh g−1 (based on mass of Cu2S) and high areal (>23 mAh cm−2) and energy densities (>135 Wh L−1), but suffered from short cycle life (<250 cycles where end of cycle life is when the capacity reaches <15% of the theoretical capacity). The charge/discharge mechanism of these cells was examined by Raman spectroscopy which revealed the capacity to arise from a combination of S and Cu electrochemistry. Additionally, the mechanism was shown to be dependent on the starting phase of these materials with Cu2S being the best performing phase. The failure mechanism of these cells was found to be a slow conversion from S redox to Cu redox chemistry with a subsequent rapid decline in capacity upon the full conversion from the S to Cu redox chemistry. This conversion was shown to be a chemical rather than electrochemical mechanism that occurred in the oxidized state. Batteries utilizing this cell chemistry at 50% depth of charge (DOC), with 5 wt% Na2S added to the electrolyte, retained a cathode capacity of 838 mAh g−1 (based on the mass of sulfur) or 169 mA h g−1 (based on the mass of Cu2S) after 450 cycles while also demonstrating >99.7% coulombic efficiency and >71% energy efficiency after the 450 cycles. Zn/Cu2S batteries with areal capacities of (8.3 mA h cm−2, with 65% active material) provided for a grid relevant energy density of >42 Wh L−1 after 450 cycles. These results represent the first example of a rechargeable solid-state sulfur cathode in an alkaline battery and augurs for future work into increasing cycle life of the cell chemistry.
Acknowledgments
This work was supported by the Laboratory Directed Research and Development program at Sandia National Laboratories. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. Additional support was also provided by Dr. Imre Gyuk, Energy Storage Program Manager, Office of Electricity Delivery and Energy Reliability. The views expressed in the article do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
ORCID
Timothy N. Lambert 0000-0002-2359-2876