
Abstract
AlCu solid-solution alloys were dealloyed by immersion in alkaline solution to produce a high surface density of copper nanoparticles, simulating the region around a cathodic intermetallic phase in an industrial AlCuMg alloy. The cathodic behavior of the copper-enriched surface in neutral chloride or sulfate solutions showed an abnormally low limiting current density for oxygen reduction, less than 10% of the value shown by a planar copper electrode, but if the solution was substituted by a borate buffer, the limiting current increased sharply. In the unbuffered solution there is corrosion of the aluminum by cathodically generated alkali, and the surface pH rises to the value where the net or apparent cathodic limiting current density maintains the surface pH at about 9 [for a polarization potential around −800 mV (SCE)]. This process greatly reduces the available cathodic current to drive remote pits or crevice corrosion sites. The implications for corrosion in real environments of varying buffer capacity are discussed. © 2002 The Electrochemical Society. All rights reserved.
Export citation and abstract BibTeX RIS
A number of authors have shown that the alkalinity developed at cathodic intermetallic particles on corroding aluminum alloys in aerated solutions can dissolve the adjacent alloy matrix, creating grooves or pit-like clusters.1 2 Later on, by a process not well understood, these cavities may switch to an acid-pitting mechanism. These two distinct stages were clearly distinguished in recent work by Park et al. ,3 but other authors refer to the alkaline attack itself as pitting4 5 or treat the problem as one of galvanic corrosion between particle and matrix.6 In fact, since alkaline anodic dissolution of aluminum shows potential-independent behavior indicative of diffusion control,7 the local galvanic effect per se is irrelevant, and no significant potential differences can exist over such small distances in a conducting electrolyte, except in the case of rapid pitting which is not the case here. The alkaline attack has been imaged in transmission electron microscopy (TEM) studies of thin foils exposed to water.8
The corrosion that occurs around intermetallic particles under open-circuit conditions, with oxygen reduction as the cathodic reaction, is essentially the same thing as cathodic corrosion occurring under more extreme conditions of cathodic polarization and normally involving hydrogen evolution as the source of alkalinity. The descriptive term "cathodic corrosion" means that corrosion occurs faster with cathodic current than under simple immersion. This was known before WWII and clearly described in the first edition of Uhlig's Corrosion Handbook.9 Cathodic corrosion can be a problem in systems containing aluminum in contact with magnesium, and has recently been topical in the electrodeposition of paint for automotive applications.10 11 It also increases pit densities in ac or pulsed treatments such as electrograining; the surface is made bare by cathodic corrosion, then experiences a sudden anodic step above the pitting potential and produces a huge number of pits.12 There is obviously a connection between this sequence and the tendency for acid pits to initiate in alkaline grooves around intermetallics.
In a copper-bearing system such as the AlCuMg alloy 2024-T3, the naturally aged matrix contains significant solid-solution copper, at least 1%. This copper will enrich on the surface as nanoparticles during alkaline dissolution around cathodic sites.13 Acting as microelectrodes, these particles should be able to supply a limiting cathodic current equivalent to a continuous copper surface, provided that they are much closer together than the diffusion layer thickness.14 With time it is possible that the cathodic activity will spread or shift from the intermetallic particles to the enriched solid solution. A remote crevice or pit site may thus be able to draw current from an expanding area of the alloy surface, and thus corrosion may become more severe with time. However it is apparent that if the enriched solid solution is composed of small copper particles on top of an AlCu substrate, alkalinity will be generated around these particles and might activate the substrate leading to detachment of the particles, local anodic currents, and other complexities. The present work is part of a study that aims to quantify the time evolution of the cathodic behavior of AlCu and AlCuMg surfaces, taking into account the important role of cathodic corrosion.
Experimental
Materials.—
Al-1% Cu and Al-4% Cu alloy sheets were obtained from Arizona State University (Professor Karl Sieradzki), having been prepared from high-purity starting materials. Their compositions were as follows in mass %: Al-1% Cu: 0.001% Si, 0.98% Cu, 0.001% Zn, and 0.002% Ti, and Al-4% Cu: 0.001% Si, 0.001% Fe, 3.96% Cu, 0.001% Zn, and 0.002% Ti. They were supplied in the solution-annealed and quenched condition.
Samples for electrochemistry were prepared by grinding with 800 grit silicon carbide paper lubricated with diethylene glycol, then ultrasonically cleaned for 10 min in pure ethanol and dried with cold air. Lacquer was used to expose an area of 0.5 cm2 for testing. Mass loss experiments used larger (30 cm2) flag-shaped samples prepared in the same way.
Samples of pure copper were also used as controls.
Surface copper enrichment.—
Samples of the AlCu alloys were immersed in alkaline solution (pH 11, made of deionized (DI) water and NaOH pellets) for various times to enrich the surfaces with metallic copper nanoparticles. After each immersion the sample was briefly rinsed with DI water, dried with cold air, then lacquered to expose an area of 0.5 cm2, and immersed in the required solution (0.1 M  0.1 M
0.1 M  with NaOH added to pH 7, or 0.1 M
with NaOH added to pH 7, or 0.1 M  ), prepared with deionized water. The specimens were lacquered after precorrosion because the Cu enrichment was not homogeneous; hydrogen evolution was more prominent at the edges of the alloy sheet, and the exposed area after lacquering was in the middle of the sample. A three-electrode electrochemical cell (open beaker) was used. All the solutions were naturally aerated apart from when they were oxygenated with pure oxygen gas. The reference electrode was generally
), prepared with deionized water. The specimens were lacquered after precorrosion because the Cu enrichment was not homogeneous; hydrogen evolution was more prominent at the edges of the alloy sheet, and the exposed area after lacquering was in the middle of the sample. A three-electrode electrochemical cell (open beaker) was used. All the solutions were naturally aerated apart from when they were oxygenated with pure oxygen gas. The reference electrode was generally 
Electrochemical procedures.—
Immediately after immersion in the sulfate solution, a potential equivalent to −800 mV (SCE) was applied and the current was measured (cathodic polarization curves starting at the open-circuit potential had been used to confirm that this potential was in the limiting cathodic current region, and a similar potential had been measured during crevice corrosion of 2024-T3 alloy15). Following a set immersion period, the sample was lifted out of the beaker and immersed immediately, after being quickly rinsed with DI water, in another beaker with identical electrochemical connections containing either (i) the same solution (to show that the transfer did not perturb the process) or (ii) a pH 7 buffered solution made from 0.1 M  with NaOH added or (iii) an oxygen saturated 0.1 M
with NaOH added or (iii) an oxygen saturated 0.1 M  solution.
solution.
Reverse transfers were also done in various sequences.
The open-circuit potentials of the two alloys were measured as a function of pH, with and without prior copper enrichment in the pH 11 solution, by titrating 1 M NaOH solution into 0.1 M  pH 8.2-8.3 originally,
pH 8.2-8.3 originally,  10.33. The open-circuit potential was measured at each step in pH after allowing time for stabilization.
10.33. The open-circuit potential was measured at each step in pH after allowing time for stabilization.
Mass loss.—
ASTM procedure G 1-90 (nitric acid immersion with periodic mass measurements) was used to measure the mass loss of the 30 cm2 samples subject to cathodic corrosion at −800 mV (SCE).
Results and Discussion
Evidence of low cathodic limiting current on AlCu alloys.—
Figure 1 shows current-time curves in the cathodic-limiting current region [−800 mV (SCE)] for small samples of the Al-1% Cu alloy in aerated 0.1 M  Fresh and copper-enriched (immersed 5 min in pH 11 solution) samples are compared with pure copper. The precorroded sample was initially anodic, but settled in the limiting cathodic current region after ∼200 s. The fresh specimen settled in the limiting cathodic current region after several thousand seconds. The cathodic-limiting current density on the copper-enriched sample is much lower than that of the copper. Different amounts of precorrosion were tried, but always the limiting current was low. It was hypothesized that alkalinity generated by the cathodic reduction of oxygen was activating the AlCu matrix so that the measured current was a net current and did not represent the true rate of the cathodic reaction. The alloy surface was visibly altered by cathodic polarization, consistent with the occurrence of significant corrosion.
Fresh and copper-enriched (immersed 5 min in pH 11 solution) samples are compared with pure copper. The precorroded sample was initially anodic, but settled in the limiting cathodic current region after ∼200 s. The fresh specimen settled in the limiting cathodic current region after several thousand seconds. The cathodic-limiting current density on the copper-enriched sample is much lower than that of the copper. Different amounts of precorrosion were tried, but always the limiting current was low. It was hypothesized that alkalinity generated by the cathodic reduction of oxygen was activating the AlCu matrix so that the measured current was a net current and did not represent the true rate of the cathodic reaction. The alloy surface was visibly altered by cathodic polarization, consistent with the occurrence of significant corrosion.
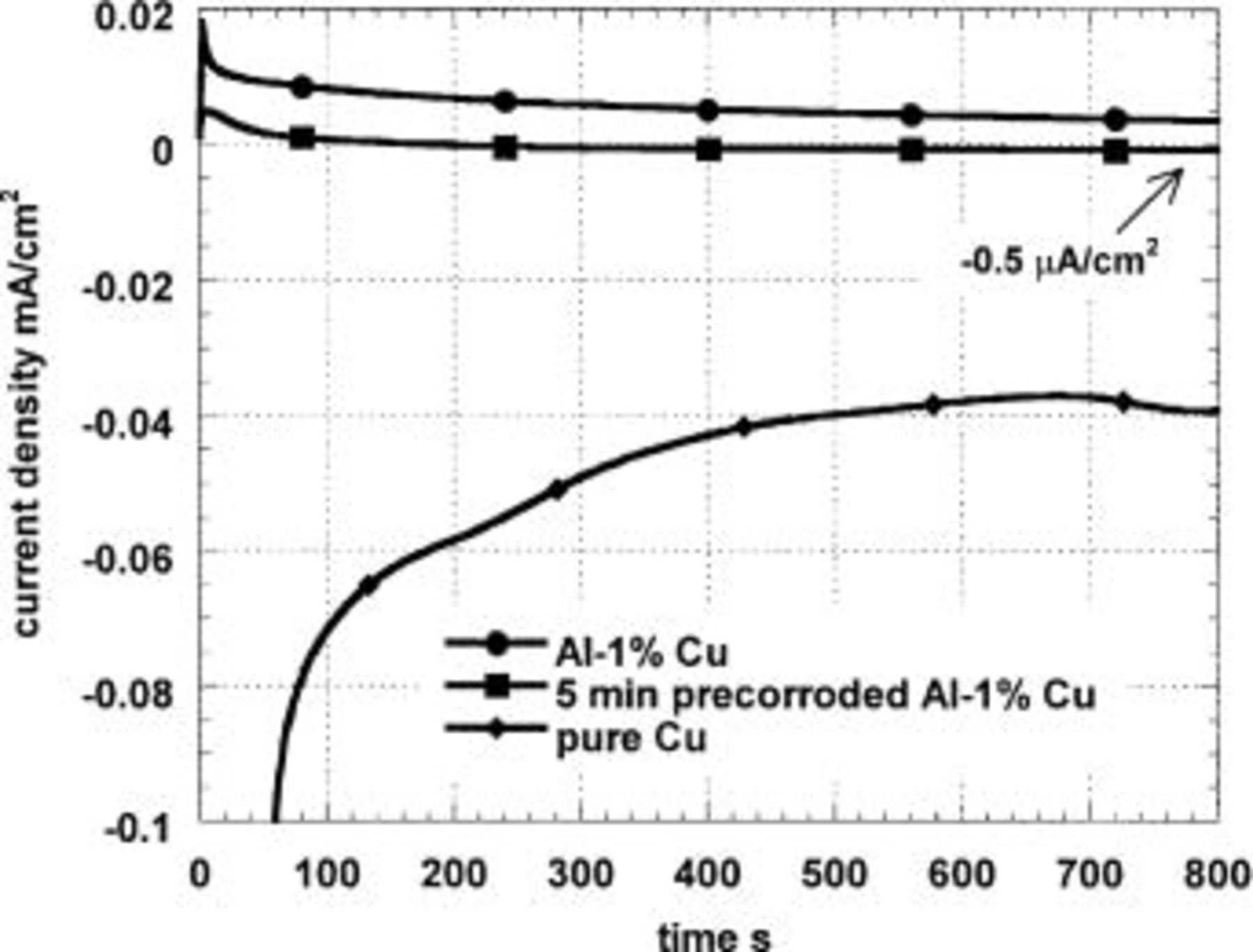
Figure 1. Cathodic polarization at −800 mV (SCE) in naturally aerated 0.1 M  of three specimens, fresh Al-1% Cu, Al-1% Cu precorroded for 5 min at pH 11, and pure Cu.
of three specimens, fresh Al-1% Cu, Al-1% Cu precorroded for 5 min at pH 11, and pure Cu.
The slow transient observed on pure Cu in Fig. 1 with the final current density of 40 μA/cm2 suggests that with time the  reduction reaction may progress to a 2e mechanism. Higher current densities were measured on platinum. This is contrary to some literature,16 but, as we obtained 40 μA/cm2 on copper with different kinds of deionized and distilled water, it does not seem to be a simple impurity issue. Fast potential cycling gives a higher limiting current density. Possibly this is a new and valid result; anyway it does not affect the conclusions of this paper. A possible mechanism of the transition from 4e to 2e is underpotential monolayer oxidation of the copper.
reduction reaction may progress to a 2e mechanism. Higher current densities were measured on platinum. This is contrary to some literature,16 but, as we obtained 40 μA/cm2 on copper with different kinds of deionized and distilled water, it does not seem to be a simple impurity issue. Fast potential cycling gives a higher limiting current density. Possibly this is a new and valid result; anyway it does not affect the conclusions of this paper. A possible mechanism of the transition from 4e to 2e is underpotential monolayer oxidation of the copper.
Transfer experiments on Al-1% Cu from aerated sulfate solution to borate solution.—
Figure 2 shows the results of a sequence of transfers between the sulfate solution and the borate buffer solution using small samples. The control transfer A to B gave no change in the stable limiting current density  and at the end of the experiment (E)
and at the end of the experiment (E)  again reverted to the same value of ca. 0.5 μA/cm2. However in the borate buffer solution
again reverted to the same value of ca. 0.5 μA/cm2. However in the borate buffer solution  was much higher, approaching 20 μA/cm2 in the steady state and 35 μA/cm2 transiently. This result confirms that by preventing the increase of surface pH, the AlCu surface can be made to exhibit as a net current a good fraction of its hydrodynamic limiting current for oxygen reduction. The remaining discrepancy with the pure Cu result (40 μA/cm2) is due to a porous deposit of aluminum hydroxide visible on the precorroded AlCu surface especially when dried, which continues to thicken during the cathodic corrosion.
was much higher, approaching 20 μA/cm2 in the steady state and 35 μA/cm2 transiently. This result confirms that by preventing the increase of surface pH, the AlCu surface can be made to exhibit as a net current a good fraction of its hydrodynamic limiting current for oxygen reduction. The remaining discrepancy with the pure Cu result (40 μA/cm2) is due to a porous deposit of aluminum hydroxide visible on the precorroded AlCu surface especially when dried, which continues to thicken during the cathodic corrosion.
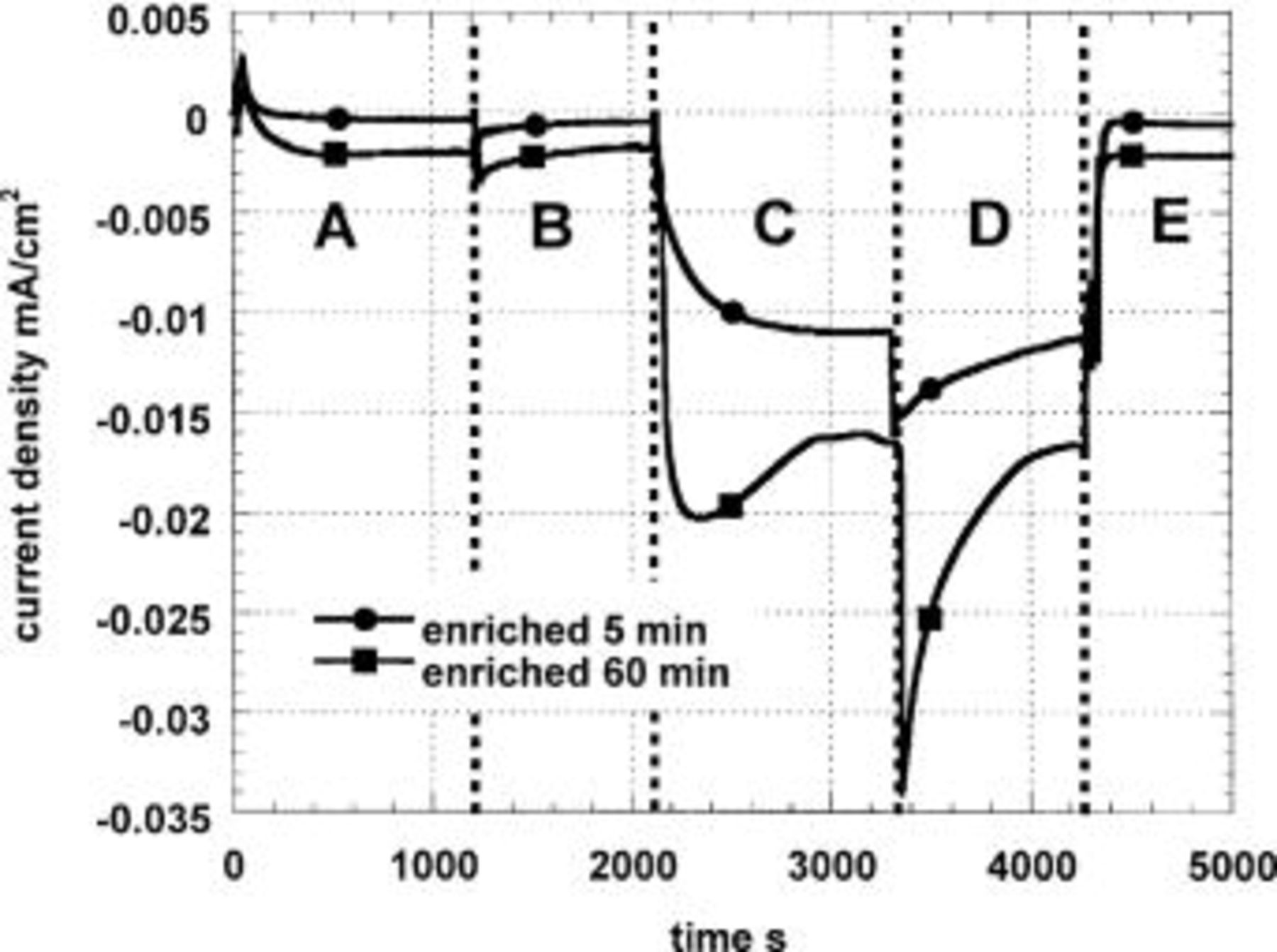
Figure 2. Cathodic polarization in naturally aerated solutions at −800 mV (SCE) of Al-1% Cu previously immersed 5 and 60 min in a pH 11 solution. In zones A, B, and E, the electrolyte is 0.1 M  and in C and D, 0.1 M
and in C and D, 0.1 M  adjusted to pH 7.
adjusted to pH 7.
Figure 2 shows a long transient on first transferring the sample to the buffer solution, but a fast one on making the reverse transfer to the sulfate solution. It appears that aluminum that has undergone alkaline dissolution has a memory of its high surface pH and takes some time to adjust to a neutral environment. We speculate that the surface is covered by an alumina gel that only slowly releases its  In view of this long transient, it is very likely that the limiting current in borate is only a rough estimate of that which would be strictly comparable with the sulfate solution.
In view of this long transient, it is very likely that the limiting current in borate is only a rough estimate of that which would be strictly comparable with the sulfate solution.
Transfer experiments on Al-4% Cu from aerated sulfate solution to borate solution.—
Figure 3 shows that for the 4% Al alloy, the limiting current is significantly higher in the unbuffered solution than for Al-1Cu, the effect of switching to the buffered solution is less, and the effect of the precorrosion time is not significant even though the surface appearances after 5 or 60 min of precorrosion were radically different (after 5 min, the surface was mildly copper colored, but after 60 min it had a deep bronze color). Microscopic examination showed that this alloy had patches of enriched copper that appeared to be continuous metallic films. While electron microscopy would probably show these to be particulate or porous, their existence does suggest that in certain regions of the surface there is so much copper that it may act as a microcathode without interference from dissolution of the underlying alloy. Only about one such site per  area would be required to give a microelectrode array that would function almost like a planar copper electrode (modified by any overlying hydrous alumina deposit).
area would be required to give a microelectrode array that would function almost like a planar copper electrode (modified by any overlying hydrous alumina deposit).
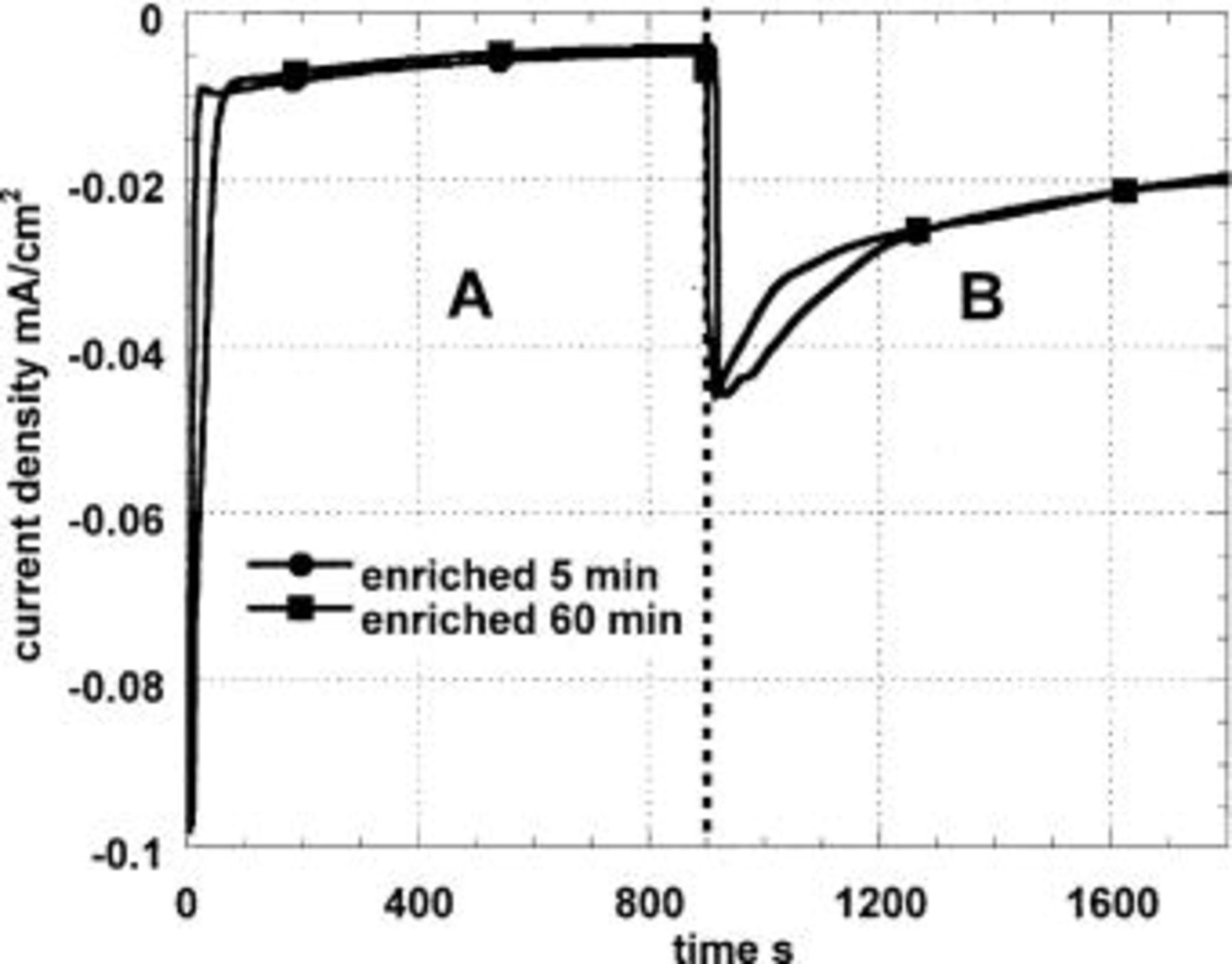
Figure 3. Cathodic polarization in naturally aerated solutions at −800 mV (SCE) of Al-4% Cu previously immersed 5 and 60 min in a pH 11 solution. In zone A, the electrolyte is 0.1 M  and in B, 0.1 M
and in B, 0.1 M  (pH 7).
(pH 7).
Transfer experiments from aerated to oxygen saturated sulfate solutions.—
Figure 4 shows the effect of oxygenation and Cu concentration on the current response during the same kind of cathodic polarization in 0.1 M  The specimens were polarized 900 s in a naturally aerated solution and then in an oxygenated solution. In the case of the Al-1% Cu, the ratio
The specimens were polarized 900 s in a naturally aerated solution and then in an oxygenated solution. In the case of the Al-1% Cu, the ratio  is only 1.5 despite the five times increase in
is only 1.5 despite the five times increase in  confirming that the current is a net current and does not reflect the actual rate of
confirming that the current is a net current and does not reflect the actual rate of  reduction. In the case of the Al-4% Cu, the ratio is almost four, suggesting once again that this alloy has plaques of copper on the surface that are capable of delivering an almost conventional limiting current, albeit this is affected by a hydrous alumina layer.
reduction. In the case of the Al-4% Cu, the ratio is almost four, suggesting once again that this alloy has plaques of copper on the surface that are capable of delivering an almost conventional limiting current, albeit this is affected by a hydrous alumina layer.
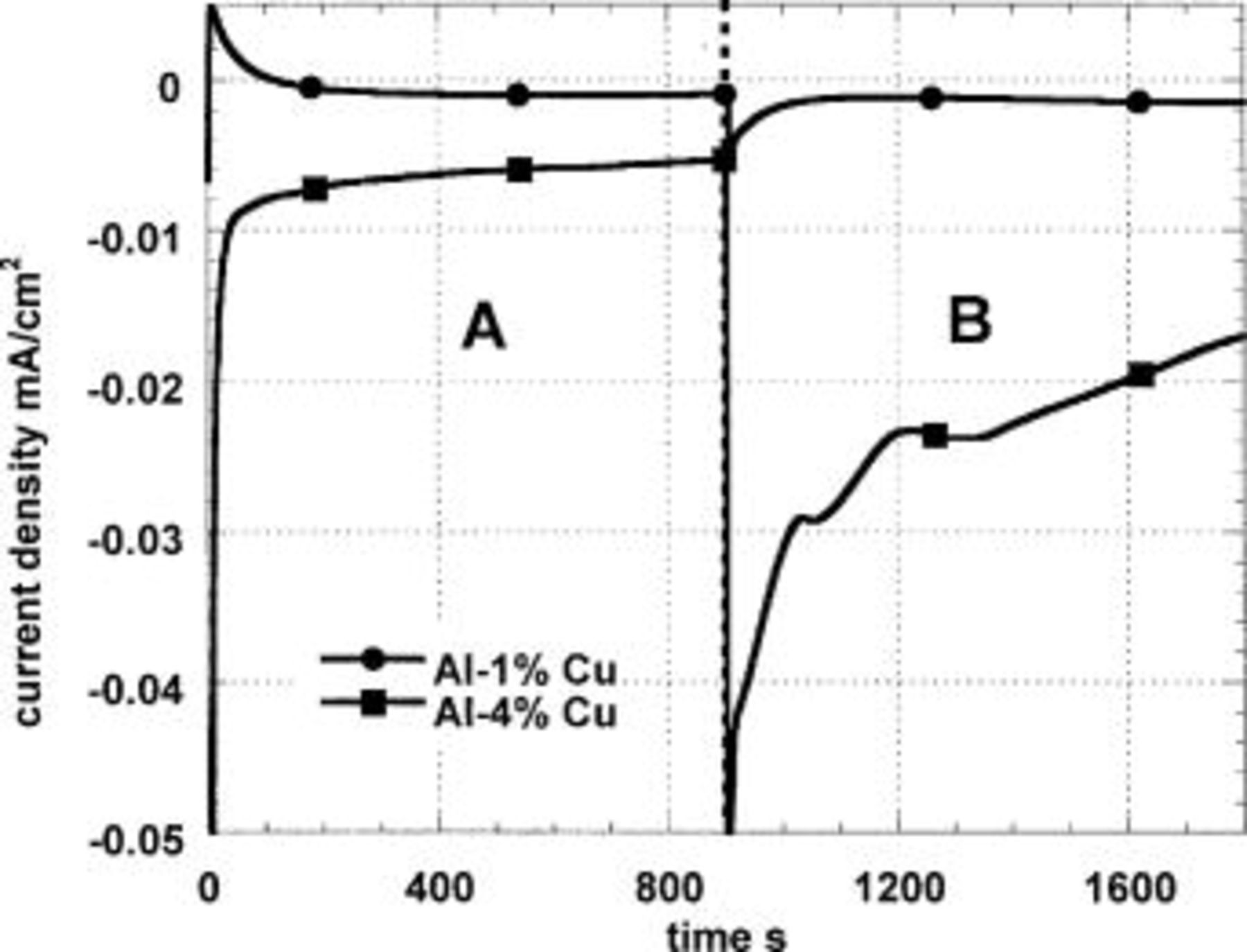
Figure 4. Cathodic polarization at −800 mV (SCE) of Al-1% Cu and Al-4% Cu (both immersed 5 min in a pH 11 solution) in 0.1 M  solution. In zone A, the solution is naturally aerated and in B, oxygenated.
solution. In zone A, the solution is naturally aerated and in B, oxygenated.
Mass loss measurements.—
After 4 days of polarization at −800 mV (SCE) in aerated 0.1 M  multiple mass loss measurements on 30 cm2 samples revealed that Al-1Cu and Al-4Cu had lost
multiple mass loss measurements on 30 cm2 samples revealed that Al-1Cu and Al-4Cu had lost  and
and  of metal, respectively. The corresponding anodic partial charge densities are
of metal, respectively. The corresponding anodic partial charge densities are  and
and  respectively. Figures 5 and 6 show the corresponding net current density responses. Every 24 h, the 0.1 M
respectively. Figures 5 and 6 show the corresponding net current density responses. Every 24 h, the 0.1 M  solutions were replaced for 30 min by 0.1 M
solutions were replaced for 30 min by 0.1 M  (pH 7) solutions to allow quantification of the true cathodic partial current densities. From the values of the cathodic current after 30 min in the buffer solution, we were able, assuming that the cathodic currents were varying linearly during each 24 h period, to calculate and integrate the corresponding partial cathodic charge densities,
(pH 7) solutions to allow quantification of the true cathodic partial current densities. From the values of the cathodic current after 30 min in the buffer solution, we were able, assuming that the cathodic currents were varying linearly during each 24 h period, to calculate and integrate the corresponding partial cathodic charge densities,  and
and  for Al-1Cu and Al-4Cu, respectively. The integrated net cathodic current densities in the sodium sulfate solution were
for Al-1Cu and Al-4Cu, respectively. The integrated net cathodic current densities in the sodium sulfate solution were  and
and  respectively; thus the predicted values of anodic partial charge density are
respectively; thus the predicted values of anodic partial charge density are  and
and  compared with +1.52 and +2.34 C/cm2 estimated from the mass loss. This is in reasonable agreement considering that (i) the mass loss measurement on corroded AlCu is not an exact science when there are heavy surface deposits of alumina, and (ii) the technique of transferring to borate solution very likely overestimates the cathodic partial current that was flowing in the sulfate solution, possibly because an outer gel layer present in the sulfate solution is washed off by the transfer process. It is also likely that recrystallization of surface Cu adatoms is a time-dependent process that influences the results.
compared with +1.52 and +2.34 C/cm2 estimated from the mass loss. This is in reasonable agreement considering that (i) the mass loss measurement on corroded AlCu is not an exact science when there are heavy surface deposits of alumina, and (ii) the technique of transferring to borate solution very likely overestimates the cathodic partial current that was flowing in the sulfate solution, possibly because an outer gel layer present in the sulfate solution is washed off by the transfer process. It is also likely that recrystallization of surface Cu adatoms is a time-dependent process that influences the results.
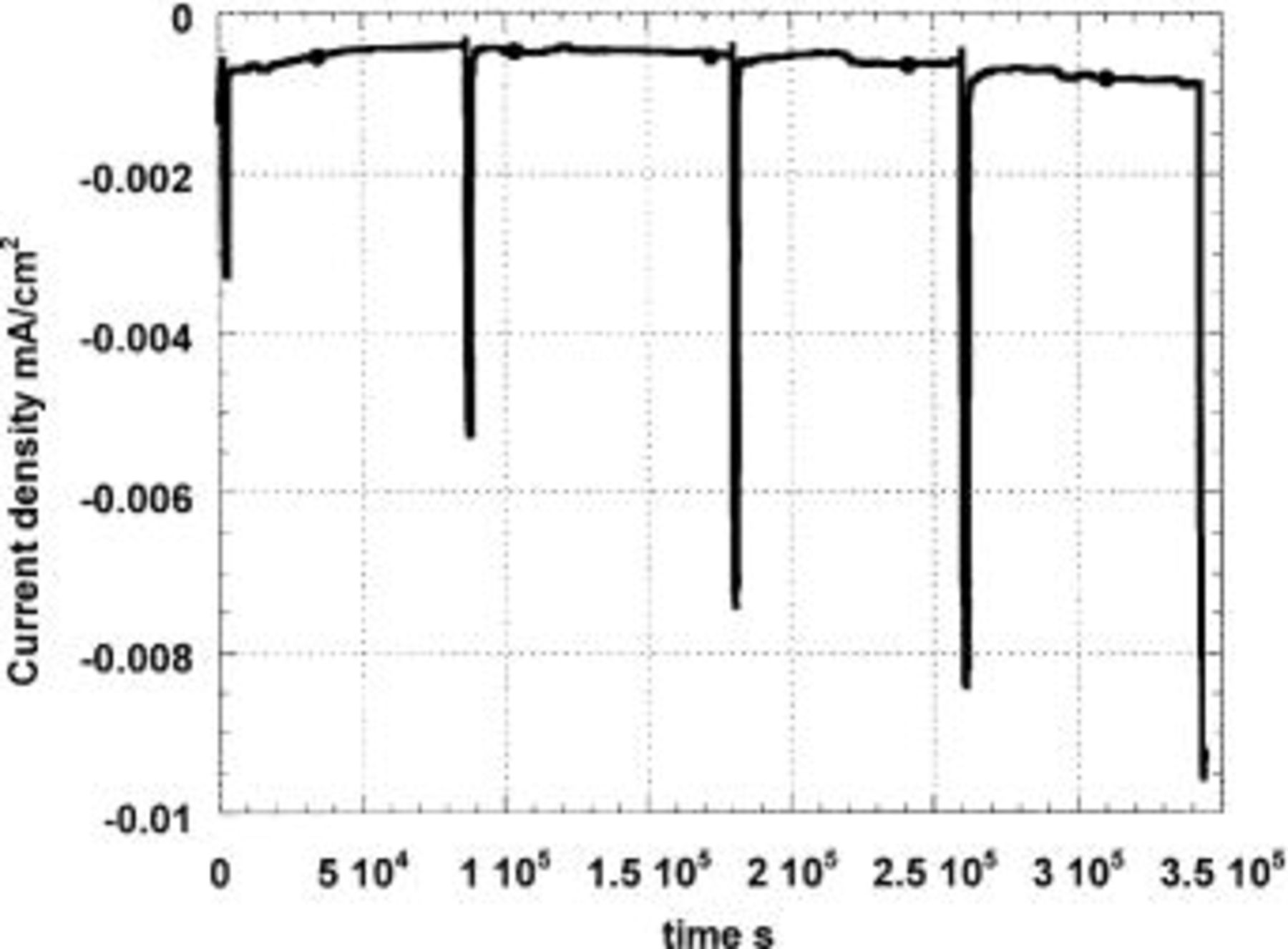
Figure 5. Cathodic polarization in naturally aerated solution at −800 mV (SCE) of Al-1% Cu previously immersed 5 min in a pH 11 solution. The specimen is polarized in 0.1 M  and transferred to 0.1 M
and transferred to 0.1 M  (pH 7) every 24 h for 30 min.
(pH 7) every 24 h for 30 min.
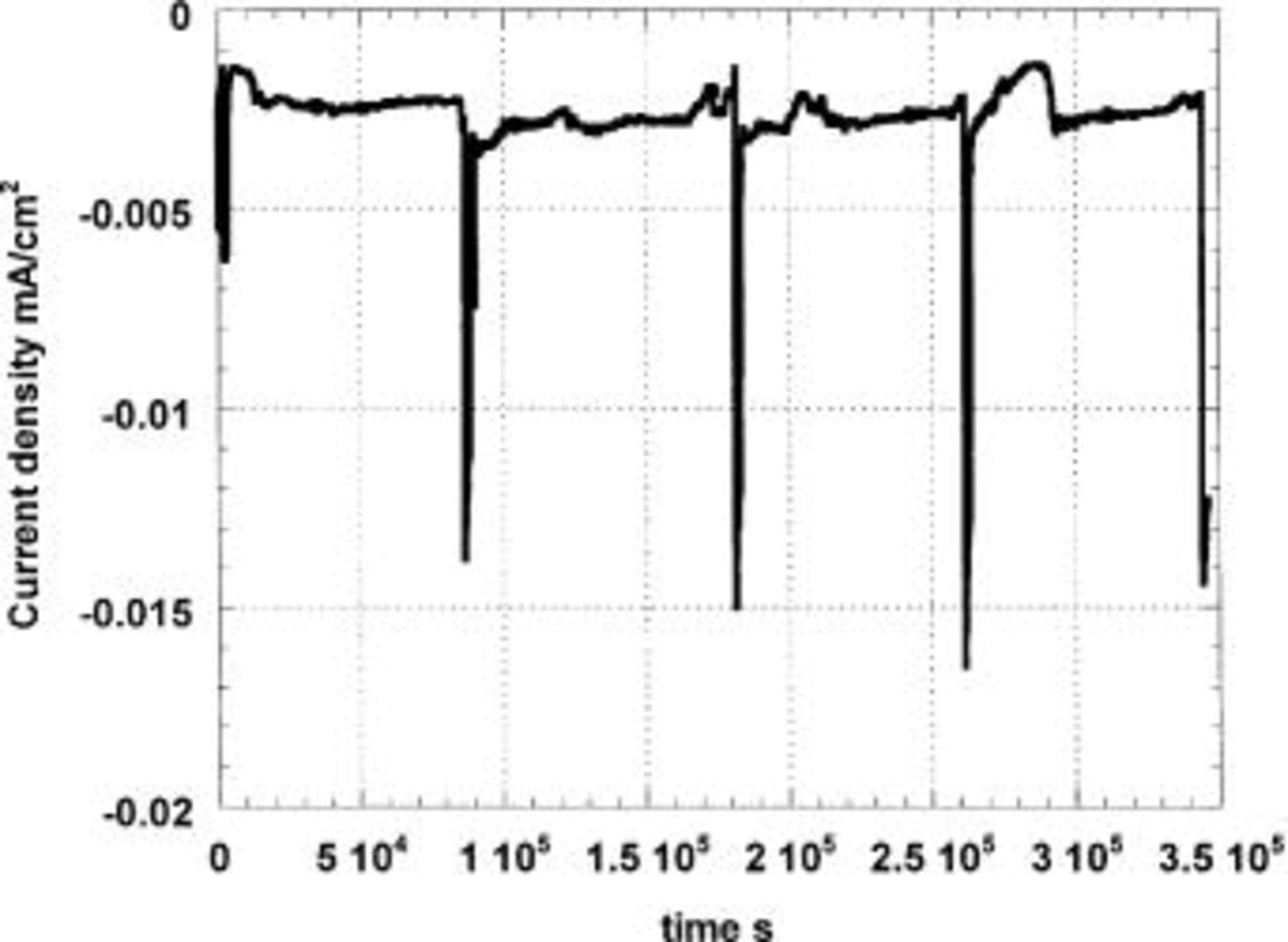
Figure 6. Cathodic polarization in naturally aerated solution at −800 mV (SCE) of Al-4% Cu previously immersed 5 min in a pH 11 solution. The specimen is polarized in 0.1 M  and transferred to 0.1 M
and transferred to 0.1 M  (pH 7) every 24 h for 30 min.
(pH 7) every 24 h for 30 min.
In Fig. 5 and 6, we can observe that the final limiting cathodic current densities of −7.68 μA/cm2 for Al-1Cu and −12.00 μA/cm2 for Al-4Cu in the borate solution are very low. Clearly the accumulation of hydrous alumina on the metal surface has strongly restricted the rate of oxygen transport. It was also observed that the cathodic limiting currents in the borate solution were consistently two times (for Al-1Cu) or three times (for Al-4Cu) lower on the large 30 cm2 specimens than on small 0.5 cm2 specimens because alumina deposition depends strongly on specimen orientation (the small samples always faced downward). On the smaller specimens without buffer, the anodic partial charge densities will also be two or three times higher as the net charge densities do not depend on the surface areas.
Open-circuit potential measurements.—
Figure 7 shows the dependence of open-circuit potential on pH. For precorroded surfaces, the Al-1% Cu alloy dropped below −800 mV at around pH 8.7-9.2, and the Al-4% Cu alloy around pH 9.2-9.3.
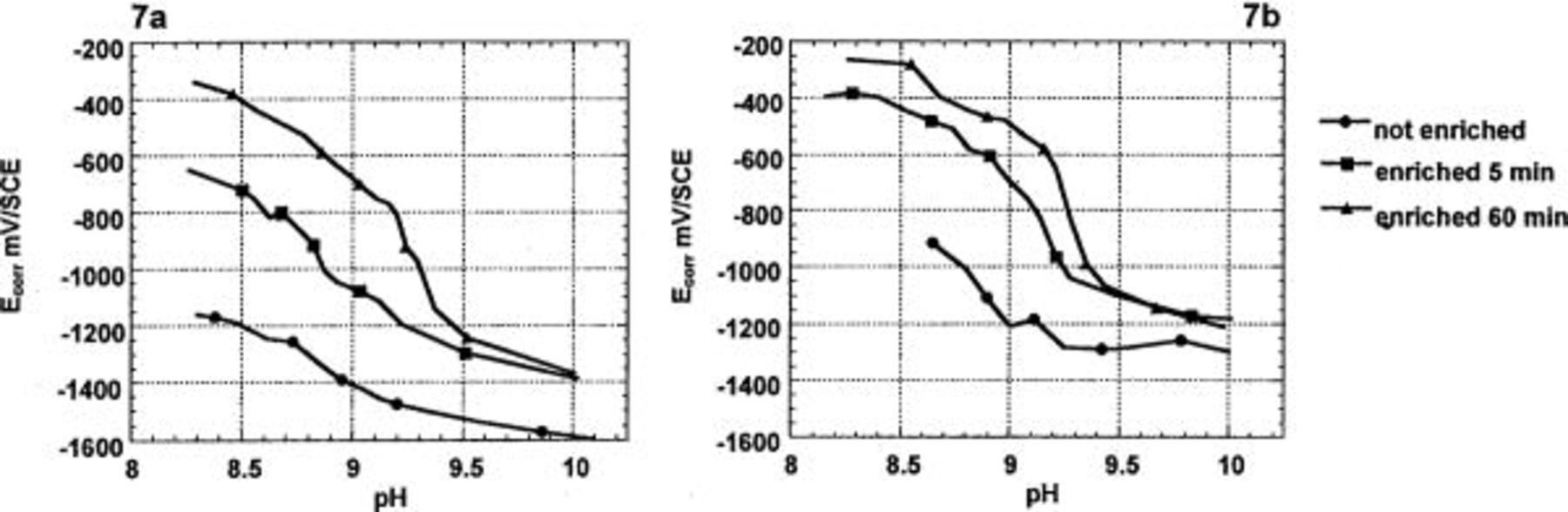
Figure 7. Evolution of the open-circuit potential of Al-1% Cu (a) and Al-4% Cu (b) as a function of the pH in aerated 0.1 M  with NaOH addition.
with NaOH addition.
Model
For the Al-1Cu alloy in unbuffered solution, we propose that cathodic corrosion of the AlCu solid solution between copper nanoparticles on the copper-enriched surface generates an overall net current that controls the surface pH very close to the value where the open-circuit potential in Fig. 7 meets the applied potential of −800 mV (since there is some cathodic current, the potential will always be slightly below the applicable open-circuit potential, but the change of the latter with pH is so large and so steep that this represents a small correction). This is a self-regulating system. The anodic and cathodic reactions on the cathodically polarized surface are strongly coupled and bound to proceed at mutually consistent rates. In a buffered solution there is no cathodic corrosion, and the net current is the cathodic current. The value of the apparent (net) limiting cathodic current density in unbuffered solution can be rationalized very roughly on this basis, as follows.
The limiting cathodic current density  for oxygen reduction on copper is 40 μA/cm2 for the present experimental conditions. Assuming that
for oxygen reduction on copper is 40 μA/cm2 for the present experimental conditions. Assuming that  diffuses three times faster than
diffuses three times faster than  this corresponds roughly to a surface concentration of
this corresponds roughly to a surface concentration of  two-thirds times the bulk concentration of oxygen (8 ppm or 0.25 mM), that is, 0.16 mM of
two-thirds times the bulk concentration of oxygen (8 ppm or 0.25 mM), that is, 0.16 mM of  or pH 10.2. We are interested in the ratio of
or pH 10.2. We are interested in the ratio of  with and without the buffer. This is determined by switching the solution back and forth over a short period during which the alumina deposit and thus the diffusion layer thickness should not change very much. This ratio should be simply the ratio of the
with and without the buffer. This is determined by switching the solution back and forth over a short period during which the alumina deposit and thus the diffusion layer thickness should not change very much. This ratio should be simply the ratio of the  concentration at the surface (0.16 mM, pH 10.2) to the ratio corresponding to the critical pH where the open-circuit potential equals −800 mV (0.005 mM, pH 8.7). In other words, we can rationalize a 30-fold decrease in the net cathodic-limiting current between the buffered and unbuffered solutions. This is in good agreement with the data for Al-1 Cu in Fig. 2.
concentration at the surface (0.16 mM, pH 10.2) to the ratio corresponding to the critical pH where the open-circuit potential equals −800 mV (0.005 mM, pH 8.7). In other words, we can rationalize a 30-fold decrease in the net cathodic-limiting current between the buffered and unbuffered solutions. This is in good agreement with the data for Al-1 Cu in Fig. 2.
Significance for corrosion behavior of aluminum alloys.—
For the solutions and test durations used in this work, the small net cathodic current density that can pass on an enriched AlCu solid-solution surface (no matter how enriched it is) severely restricts the corrosion current that could be used to drive a remote pit or crevice event. The cathodic current is wasted causing local uniform corrosion of the solid solution. However the presence of a modest amount of a weak acid can transform this situation. The addition of an amount of monobasic weak acid comparable to four times the oxygen molar concentration (more probably two or three times higher than that, i.e., 2 to 3 mM) will greatly enhance the apparent cathodic kinetics, reduce the alkaline corrosion of the matrix, and increase the rate of pit or crevice propagation. The amount of calcium bicarbonate buffer in natural waters is already close to this amount. This is a simple view. However there will be complications, for example
1. In the absence of alkaline dissolution of the matrix, it will take much longer to develop the full cathodic activity of the alloy.
2. The accumulation of deposited alumina may eventually dominate the interesting effects connected with cathodic corrosion (this accumulation happens anyway on particulate cathodic sites in alloys such as 2024, and one of the detrimental roles of copper is to make available the adjacent matrix as a cathode, albeit a weak one).
3. Much longer periods of corrosion may lead to dense copper-rich areas that can function as pure cathodes irrespective of the cathodic corrosion that is going on elsewhere (we already saw this happening on the 4% Cu alloy).
4. Copper may become electrically detached, corrode, and redeposit as dendritic particles that can function as pure cathodes.6
The model presented in this paper takes a simplified view of the surface pH evolution, and a full treatment would have to take into account the partitioning of the corrosion product between solid and dissolved forms, since the alkaline dissolution of aluminum will always decrease the pH to some extent.
Conclusions
1. Cathodic corrosion is a finely tuned, self-regulating process that limits the net cathodic current density that can be supplied by an AlCu solid solution to a remote corrosion site.
2. Buffering increases the net cathodic current density that can be supplied and thus the severity of localized corrosion.
3. Increasing the Cu content of the alloy from 1 to 4% can introduce enrichment sites that may function as pure cathodes.
4. Deposition of hydrous alumina is an essential complication in any model of corrosion in such systems.
5. The buffer capacity of natural waters should have effects on e.g., crevice corrosion propagation that can be interpreted using these concepts.
6. The mechanism of oxygen reduction on pure Cu deserves further attention, even if the observed slow transition from 4e to a 2e  reduction is an impurity effect.
reduction is an impurity effect.
Acknowledgment
This research was supported by a subcontract from Arizona State University under AFOSR MURI F49620-96-1-0475. Discussions with A. J. Davenport, K. Sieradzki, and N. Dimitrov are gratefully acknowledged.
University of Manchester Institute of Science and Technology assisted in meeting the publication costs of this article.