Article Text

Abstract
Objective Analyse the integrated safety profile of evobrutinib, a Bruton’s tyrosine kinase inhibitor (BTKi), using pooled data from multiple sclerosis (MS), rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) trials.
Methods Phase II, randomised, double-blind, placebo-controlled trial data were analysed (N=1083; MS: n=213, 48 weeks (W); RA: n=390, 12W; SLE: n=480, 52W). The analysis included all patients who received ≥1 dose of evobrutinib (25 mg or 75 mg once daily, or 50 mg or 75 mgtwice daily) or placebo. Descriptive statistics and exposure-adjusted incidence rates (EAIR) were used to report treatment-emergent adverse events (TEAEs).
Results Data from 1083 patients were pooled: evobrutinib, n=861; placebo, n=271 (sum >1083 due to MS trial design: n=49 received both placebo (W0–24) and evobrutinib 25 mg (W25–48)); median follow-up time (pt-years): evobrutinib, 0.501; placebo, 0.463. Across indications, the proportion of patients with TEAEs and the EAIR were similar for evobrutinib and placebo (66.2% (247.6 events/100 pt-years) vs 62.4% (261.4 events/100 pt-years)). By indication, the EAIR (events/100 pt-years) of TEAEs for evobrutinib versus placebo were: MS: 119.7 vs 148.3; RA: 331.8 vs 306.8; SLE: 343.0 vs 302.1. Two fatal events occurred (in SLE). The serious infections EAIR was 2.7 and 2.1 events/100 pt-years for evobrutinib and placebo. For previously reported BTKi-class effects, the EAIR of transient elevated alanine aminotransferase/aspartate aminotransferase TEAEs (events/100 pt-years) with evobrutinib versus placebo was 4.8 vs 2.8/3.5 vs 0.7, respectively. IgG levels were similar in evobrutinib/placebo-treated patients.
Conclusions This is the first BTKi-integrated safety analysis that includes patients with MS. Overall, evobrutinib treatment (all doses) was generally well tolerated across indications.
Trial registration numbers NCT02975349, NCT03233230, NCT02975336.
- IMMUNOLOGY
- MULTIPLE SCLEROSIS
- RHEUMATOLOGY
- SLE
Data availability statement
Data are available upon reasonable request. Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s Data Sharing Policy. All requests should be submitted in writing to Merck’s data sharing portal https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. When Merck has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavour to gain agreement to share data in response to requests.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Evobrutinib is an orally administered, central nervous system-penetrating, highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor, currently in phase III for multiple sclerosis (MS) and has also been investigated in patients with rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) up to phase II. In the three phase II trials (one for each indication), evobrutinib was well tolerated with no dose effect on the incidence of adverse events.
WHAT THIS STUDY ADDS
The results from this integrated phase II safety analysis demonstrate that evobrutinib treatment (all doses) was generally well tolerated across indications. This analysis was based on the extensive evobrutinib safety database of over 1000 patients enrolled in the phase II trials and represents the only integrated safety analysis of a BTK inhibitor that includes patients with MS.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
This integrated safety analysis adds confidence in the safety profile of evobrutinib—being generally well tolerated in patients with MS, RA and SLE—and supports the continued development of evobrutinib for MS in the ongoing phase III trial.
Introduction
Bruton’s tyrosine kinase (BTK), a member of the Tec family of non-receptor tyrosine kinases, is expressed in B cells, macrophages and microglia, and is involved in signal propagation and modulation of signal responsiveness in these cells.1–3 Overexpression of BTK has been shown to be associated with certain autoimmune diseases due to an increase in autoreactive B cells and autoantibodies.3 This is exemplified in the multiple sclerosis (MS) setting where elevated BTK levels have been detected in B cells of relapsing-remitting MS and secondary-progressive MS patients as well as in microglia within progressive MS lesions.4 5 Therefore, BTK represents a rational therapeutic target in autoimmune diseases and, as a result, several BTK inhibitors are being investigated in autoimmune conditions including MS, rheumatoid arthritis (RA) and/or systemic lupus erythematosus (SLE).6 7 BTK inhibitors impact B cell function via inhibition of BTK signalling rather than depleting the B cell population as per the effect of anti-CD20 therapies currently used for the treatment of MS, RA and SLE, which have been associated with hypogammaglobulinemia and increased risks of infections.3 8–11 Additionally, BTK inhibitors target innate immune cells involved in the pathogenesis of MS, including macrophages, microglia and astrocytes.7 Published evidence suggests that as central nervous system (CNS) resident cells, microglia and astrocytes may play a critical role in neurodegeneration and disease progression.12–15
Evobrutinib is an orally administered, CNS-penetrating, highly selective, covalent BTK inhibitor, with a low potential for off-target-related adverse effects.16–18 The covalent binding of evobrutinib to BTK results in continued target inhibition even after it has been cleared from the circulation (geometric mean half-life ~2 hours), but due to the continuous turnover of endogenous BTK protein, this inhibition is reversable following drug withdrawal.19 20 In preclinical studies, evobrutinib has been demonstrated to decrease the activation, proliferation and cytokine release of B cells, as well as inhibit proinflammatory macrophage differentiation.21 22 In the clinical setting, evobrutinib has been investigated in patients with MS, RA and SLE in three phase II trials.23–26 In the double-blind, randomised controlled trial in patients with relapsing MS, evobrutinib met its efficacy endpoints and no clinically relevant changes in immunoglobulin (Ig) or immune cell levels were observed.23 24 The RA and SLE trials did not meet their primary endpoints.25 26 In all three phase II trials, evobrutinib was well tolerated with no dose effect on the incidence of adverse events. The open-label extension (OLE) period from the MS trial is currently ongoing, but initial data have demonstrated that the safety of evobrutinib was maintained for over 2 years.27 28 The clinical development of evobrutinib in relapsing MS is ongoing, with two phase III trials that have recently completed enrollment (evolutionRMS 1 and 2; NCT04338022 and NCT04338061).29
We analysed the integrated safety profile of evobrutinib using pooled data from the three phase II trials in MS, RA and SLE, to further characterise the overall safety profile of evobrutinib in autoimmune indications. This is the first BTK inhibitor integrated safety analysis that includes patients with MS.
Methods
Trials included in the analysis
Data from three phase II, randomised, double-blind, placebo-controlled trials of evobrutinib in MS, RA or SLE were analysed. All three trials have been described previously and are summarised in table 1.23 25 26 The trials are registered with ClinicalTrials.gov and were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice Guidelines.
Overview of trials included in the integrated evobrutinib safety analyses
Endpoints
Safety endpoints for all trials included the assessment of the nature, severity and occurrence of treatment-emergent adverse events (TEAEs) as well as vital signs, electrocardiograms and clinical laboratory safety parameters. Ig and CD19+ B cell levels were assessed in each trial.
Statistical analyses
The analysis included all patients who received ≥1 dose of evobrutinib or placebo. TEAEs were summarised by treatment group, severity (using NCI-CTCAE V.4.03 criteria) and relationship to evobrutinib. Descriptive statistics and exposure-adjusted incidence rates (EAIR) were used to report TEAEs. EAIR in each treatment arm was defined as the ratio of the number of patients with an event to the sum of the duration of exposure to treatment of the patients up to the time of the first event or the end of observation (whichever occurred first) and was expressed as the rate per 100 patient-years (pt-years). Overall, results are reported (as appropriate) as evobrutinib (pooled doses) versus placebo across indications (integrated analysis) and by individual indication. The data cut-offs for each trial were: MS, 13 July 2018; RA, 25 October 2019; SLE, 21 August 2020.
Changes in IgG, A and M levels and CD19+ B cell levels were summarised across each treatment group by individual indication from baseline to Week 48 (MS), Week 12 (RA) and Week 52 (SLE) after randomisation.
In the MS trial, the placebo group switched to evobrutinib 25 mg once daily (QD) at 24 weeks. Therefore, data from Weeks 0–24 are included for the placebo analysis group (n=54) and data from Weeks 25–48 for the evobrutinib analysis group (n=49).
Results
Patient disposition and demographics
Overall, data from the three trials (MS: n=213; RA: n=390; SLE: n=480) were pooled and included 1083 patients. Of these, 861 patients were treated with evobrutinib and 271 patients with placebo (note: the sum of the groups is >1083 due to the MS trial design, whereby 49 patients in the MS trial are included in the placebo group for Weeks 0–24 and the evobrutinib group for Weeks 25–48; see Methods section for additional details). Treatment exposure was 145.1, 185.5, 168.3, 120.2 and 44.9 pt-years for placebo, evobrutinib 25 mg QD, 75 mg QD, 50 mg twice daily (BID) and 75 mg BID, respectively (median follow-up time (pt-years): evobrutinib, 0.501; placebo, 0.463).
The demographics and baseline characteristics were balanced between evobrutinib and placebo within the same indication (table 2). Per RA and SLE trial designs, concomitant immunosuppressants/immunomodulators (RA: methotrexate; SLE: azathioprine, 6-mercaptopurine, mycophenolate, methotrexate, sulfasalazine and leflunomide), non-steroidal anti-inflammatory drugs and corticosteroids were permitted. Given the different background therapies for these three disease areas, anticipated differences between indications were observed for immunosuppressants/immuno-modulators and corticosteroids at baseline (online supplemental eTable 1). As expected, a greater proportion of patients in the SLE trial used analgesics, immunosuppressants/immuno-modulators and corticosteroids at baseline compared with the MS and RA trials. Within each trial, the proportions of patients using analgesics, immunosuppressants/immuno-modulators and corticosteroids were similar for those patients treated with placebo and those treated with evobrutinib.
Supplemental material
Patient disposition and baseline characteristics
Overall TEAEs
Overall, the proportion of patients with TEAEs and the rate of events were similar for evobrutinib and placebo by indication and across trials (table 3 and figure 1). A total of 570/861 (66.2%) and 169/271 (62.4%) patients treated with evobrutinib or placebo had a TEAE, respectively. The EAIR of TEAEs was 247.6 events/100 pt-years for evobrutinib versus 261.4 events/100 pt-years for placebo. The majority of these TEAEs were mild or moderate. In the MS and RA trials, there were no fatal TEAEs in either treatment group. Two TEAEs in the evobrutinib treatment group from the SLE trial were fatal. One of these events was considered to be treatment related by the investigator (additional details in table 3). By indication, the EAIR of TEAEs for evobrutinib versus placebo were: MS: 119.7 vs 148.3; RA: 331.8 vs 306.8; SLE: 343.0 vs 302.1 events/100 pt-years. The majority of these TEAEs occurred in patients with SLE.
Overall TEAEs

EAIRs of TEAEs, serious TEAEs and infections among evobrutinib-treated and placebo-treated patients in MS, RA, SLE and across indications. EAIR, exposure-adjusted incidence rates; MS, multiple sclerosis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; TEAEs, treatment-emergent adverse events.
The most common TEAEs reported by evobrutinib-treated patients were urinary tract infections (9.5% vs 8.5% placebo), headache (9.2% vs 10.3% placebo), nasopharyngitis (7.3% vs 5.5% placebo), diarrhoea (6.2% vs 4.8% placebo) and alanine aminotransferase (ALT) increase (5.0% vs 3.0% placebo).
Serious TEAEs were reported by 48 patients (5.6%; EAIR 9.5 events/100 pt-years) treated with evobrutinib and 14 patients (5.2%; EAIR 9.8 events/100 pt-years) treated with placebo (table 3 and figure 1). The most frequent serious TEAEs in the evobrutinib treatment group were related to infections and infestations, primarily occurring in patients with SLE (table 4 and online supplemental eTable 2).
EAIRs of BTK inhibitor or concomitant drug class-associated TEAEs by preferred term
Serious infections
Across trials, there was no imbalance in the EAIR of serious infections between evobrutinib doses (25 mg QD: 4.4; 75 mg QD: 2.4; 50 mg BID: 1.7; 75 mg BID: 0.0 events/100 pt-years) and no notable differences compared with placebo (2.1 events/100 pt-years). In the MS trial, no serious infections in any of the evobrutinib treatment arms were observed. By indication, there were no serious infections reported with the highest evobrutinib dose in the MS and RA trials, while the EAIR for the highest evobrutinib dose in the SLE trial was similar to placebo (50 mg BID: 2.1 vs 2.0 events/100 pt-years).
Liver-related TEAEs
A higher EAIR of ALT and aspartate aminotransferase (AST) elevations was observed with evobrutinib compared with placebo across trials (table 4). These events occurred at a higher EAIR in the MS and RA trials compared with the SLE trial (online supplemental eTable 2). However, these elevations in ALT and AST levels were typically asymptomatic and reversible on withdrawal of evobrutinib.
Other potential BTK inhibitor or concomitant drug class-associated TEAEs
Cardiovascular disorders were similar between evobrutinib and placebo (1.9 vs 2.1 events/100 pt-years) across trials with a slightly higher frequency occurring in the SLE trial compared with the MS and RA trials (table 4 and online supplemental eTable 2). An increased EAIR of bleeding-associated or bruising-associated events was not observed with evobrutinib across trials or within each indication compared with placebo. Across trials, increase in amylase levels occurred at a similar EAIR with evobrutinib and placebo, which was also reflected in the individual trials. The EAIR of increase in lipase levels was higher in the evobrutinib treatment arm versus placebo (4.0 vs 2.8 events/100 pt-years), due to a higher EAIR observed in the SLE trial (3.4 vs 2.0 events/100 pt-years). Neoplasms occurred at a lower EAIR with evobrutinib compared with placebo across trials and within the MS and SLE trials (no events were observed in the RA trial).
The EAIR of other TEAEs known to be associated with concomitant medications typically used by patients with RA and SLE, such as hypersensitivity, herpes zoster infection, osteonecrosis and diarrhoea, were similar between placebo and evobrutinib across trials (table 4 and online supplemental eTable 2). The EAIR of hypertension was higher in the evobrutinib treatment arm versus placebo (3.7 vs 0.7 events/100 pt-years), due to a higher EAIR observed in the SLE trial (4.4 vs 0.0 events/100 pt-years).
Ig levels
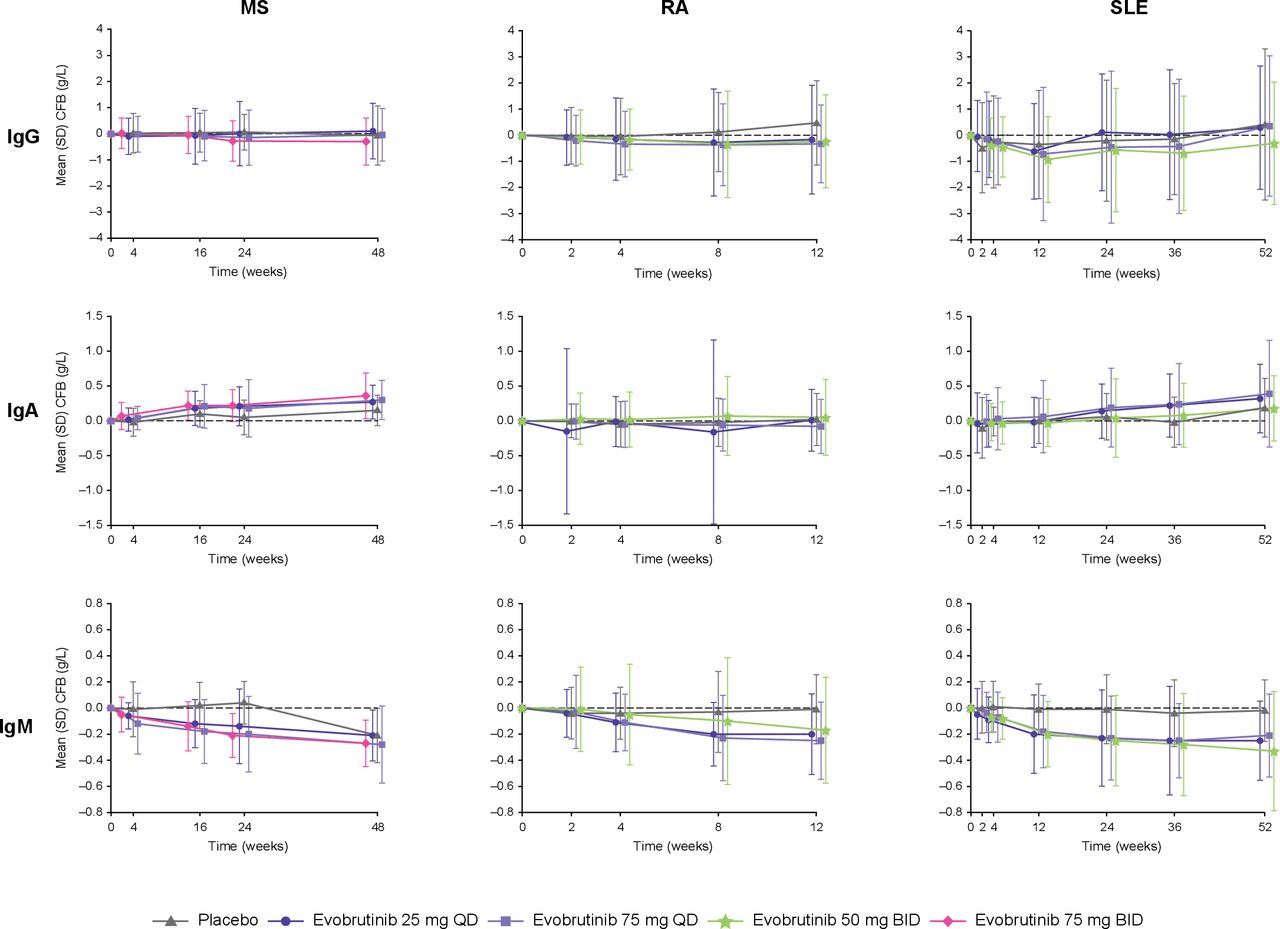
For all trials, IgG and subtype levels were maintained, and no relevant changes were observed at the end of each trial (MS: Week 48; RA: Week 12; SLE: Week 52) relative to baseline for evobrutinib and placebo (figure 2). In the MS and SLE trials, IgA levels increased in both the placebo and evobrutinib treatment arms; however, in the RA trial, IgA levels were maintained. Across all three trials, IgM levels decreased in the evobrutinib treatment arms but remained comparable with baseline levels in the placebo treatment arm except in the MS trial; after Week 24, IgM levels decreased to similar levels as the evobrutinib treatment arms due to patients switching from placebo to evobrutinib 25 mg QD. Overall, IgA and IgM levels remained within normal ranges for both treatment groups across the three trials.

Ig levels change from baseline over time. Normal ranges (g/L): IgG: 7–16; IgA: 0.7–4.0; IgM: 0.4–2.3. BID, twice daily; CFB, change from baseline; Ig, immunoglobulin; MS, multiple sclerosis; QD, once daily; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Immune cell levels
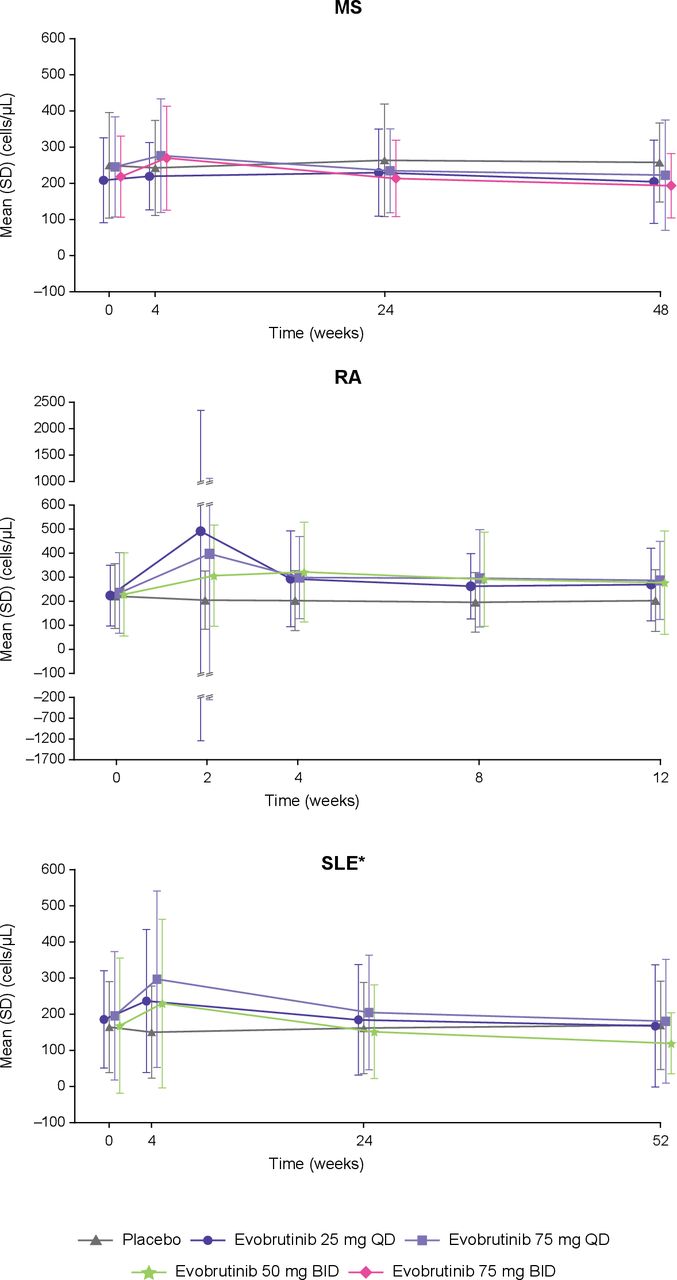
Total CD19+ B cells decreased in the MS and SLE trials and increased in the RA trial in the evobrutinib treatment groups compared with baseline; however, overall, these levels remained within the normal range (107–698 cells/µL; figure 3). For the SLE trial, an increase in B cells was initially observed with evobrutinib at week 4 (percent change from baseline at Week 4: 40–59%) before decreasing.

{kind=link}
{kind=link}
{kind=link}
CD19+ B cells levels over time. Normal range: 107–698 cells/µL. *B cell levels for the SLE trial are total B cell levels; the other trials are CD19+ B cell levels. BID, twice daily; MS, multiple sclerosis; QD, once daily; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Discussion
This is the first integrated analysis of evobrutinib safety data derived from phase II trials across MS, RA and SLE indications. Data from over 1000 patients treated with either evobrutinib or placebo contributed to this analysis. The majority of TEAEs were mild or moderate. An enhanced risk of serious infections (despite background immunosuppressant therapy) was not observed with evobrutinib treatment. A higher rate of elevations in ALT and AST were observed with evobrutinib compared with placebo. These ALT and AST elevations were typically asymptomatic and reversible following drug discontinuation. Other drug class-associated TEAEs, such as bleeding/bruising events or neoplasms, were not observed to be differential between evobrutinib and placebo with the exception of hypertension and increased lipase. Overall, the evobrutinib safety profile was comparable with that of placebo across indications, with no apparent relationship to evobrutinib dose.
Current treatment options for MS, RA and SLE are associated with a positive–benefit risk ratio in terms of adverse outcomes. However, targeted therapies that have a safety profile conducive to long-term use as necessitated by the chronic nature of these diseases may be beneficial for patients. In the RA and SLE settings, long-term use of corticosteroids and immunosuppressants/immunomodulators is associated with infections, cardiovascular events, malignancies, renal disorders and damage to multiple organ systems.30–34 In MS, several, currently available B cell-targeting treatments are highly effective at reducing inflammation but are associated with some tolerability and safety concerns, notably an increased risk of infections and malignancies.35–39 In addition to the above, treatments used in MS, RA and SLE are also associated with liver-related adverse events. The results from our analysis demonstrate that treatment with evobrutinib for up to a year is not associated with an increased risk of adverse events common to current therapies used in the treatment of MS, RA and SLE, except for an association with elevated levels of ALT and AST, which was in line with the observations from the individual MS, RA and SLE trials.23 25 26 However, as was observed in the MS and SLE trials, these elevations were typically transient and resolved following withdrawal of evobrutinib.23 26 In addition, in the MS trial, these elevations were observed only in the first 24 weeks of the trial.23 Furthermore, results from the MS trial OLE have demonstrated that the evobrutinib safety profile in the double-blind period was maintained at 2 years with no new safety signals identified.27 28 These results are substantiated further by the latest data from over 2.5 years of OLE treatment, whereby 59/213 patients (27.7%) had a treatment-related TEAE; six serious TEAEs were deemed treatment related.40 Severe/opportunistic infections (Grade ≥3) were reported by 9/213 patients (4.2%) of which three were fatal and not considered treatment related (COVID-19 pneumonia (n=2) and E. coli sepsis (n=1)). At OLE Week 120, most patients had IgG (91%), IgA (88%) and IgM (82%) levels within normal ranges. Overall mean CD19+ B cell levels were 0.218×106 cells/mL (OLE baseline) and 0.122×106 cells/mL (OLE Week 96). ALT and AST elevations were observed only in patients previously receiving dimethyl fumarate or evobrutinib 25 mg QD in the double-blind period and occurred within 12 weeks of OLE initiation (ie, evobrutinib 75 mg QD). Amylase and lipase increases occurred in 6 (2.8%) and 24 (11.3%) patients, respectively, but without clinical signs and symptoms.40 The above indication of the acceptable tolerability of evobrutinib with no new safety signals is likely due to both the BTK specificity of evobrutinib with minimal off-target kinase interactions as well as its mode of action that modulates B cell function by inhibiting BTK signalling rather than depleting the B cell population by cell lysis.3 17 41
As a class, earlier forms of BTK inhibitors, namely, ibrutinib used in oncology, have been associated with an increased risk of bleeding, cardiovascular events, skin reactions, diarrhoea and infections.6 42 Evidence suggests that these events are likely due to the low selectivity of ibrutinib. Ibrutinib has been shown to inhibit 22/221 off-target kinases by ≥80% compared with evobrutinib that inhibits 2/221 off-target kinases to the same degree.41 Interaction with the off-target kinases Tec and members of the epidermal growth factor receptor family has been implicated in the adverse events associated with ibrutinib.6 The ibrutinib-associated adverse events were not identified as common events with evobrutinib suggesting that the improved selectivity of evobrutinib (a second-generation BTK inhibitor) has overcome these class-associated TEAEs.
Overall, mean Ig levels remained within normal ranges with evobrutinib treatment, and across trials, there was no imbalance in the rate of serious infections between evobrutinib doses. Unlike BTK inhibitors, use of anti-CD20 therapies results in a near-complete depletion of B cells, which carries a risk of hypogammaglobulinaemia. Hypogammaglobulinaemia is associated with an increased susceptibility to infections.8–11 While mild hypogammaglobulinaemia may be asymptomatic, more severe cases of hypogammaglobulinaemia usually result in recurrent infections. In MS, there are currently no data on Ig levels from other BTK inhibitors; however, treatment with fenebrutinib in RA and SLE reduced IgG levels from baseline by approximately 1 g/L and 2.25 g/L, respectively, but levels remained within the normal range.43 Similarly, IgG levels in the evobrutinib RA and SLE trials also remained within the normal range (RA: −0.24 g/L; SLE: 0.11 g/L). However, it should be noted that studies have observed that the incidence of hypogammaglobulinaemia increases with longer treatment durations, with infrequent events observed in the first years of treatment with anti-CD20s.44–46 Therefore, continued monitoring of the ongoing extension phases from BTK inhibitor trials will provide further insights into the longer term effects of BTK inhibitors on Ig levels and the associated risk of infections.
A similar integrated safety analysis from another BTK inhibitor, fenebrutinib, has been conducted in RA, SLE and chronic spontaneous urticaria patients.43 In terms of overall TEAEs, serious TEAEs, serious infections and aminotransferase elevations, similar proportions of patients experienced these events with fenebrutinib as was observed in the evobrutinib trials.
This analysis has some limitations. The different trial designs, trial durations and patient populations may have restricted the extent of the comparisons that could be made between trials. In addition, the short duration of the RA trial limited the number of TEAEs and malignancies reported by this patient group. Furthermore, the baseline concomitant medications were different for these trials, in particular, for MS versus the RA and SLE trials. The greater level of concomitant medications, including use of corticosteroids and immunosuppressants, required by RA and SLE patients may explain the higher EAIR of infections in these trials compared with the MS trial. Finally, the stopping rules for evobrutinib varied across the trials; however, the variation was relatively minimal and is not expected to have impacted the results considerably. However, this analysis benefits from having the largest BTK inhibitor patient data set across MS, RA and SLE indications to date with a long cumulative treatment exposure across indications. Furthermore, the patients with SLE were using a greater level of concomitant medications, including immunosuppressants, which may have subsequently contributed to the higher EAIR of infections.
Conclusion
These results represent the only integrated safety analysis of a BTK inhibitor that includes patients with MS. The analysis was based on the extensive evobrutinib safety database of over 1000 patients with a long cumulative treatment exposure from three phase II trials across autoimmune indications. Overall, this analysis adds to our understanding of the safety profile of evobrutinib, confirms evobrutinib treatment was well tolerated in MS, RA and SLE, and supports the continued development of evobrutinib for MS and the ongoing phase III programme.
Data availability statement
Data are available upon reasonable request. Any requests for data by qualified scientific and medical researchers for legitimate research purposes will be subject to Merck’s Data Sharing Policy. All requests should be submitted in writing to Merck’s data sharing portal https://www.merckgroup.com/en/research/our-approach-to-research-and-development/healthcare/clinical-trials/commitment-responsible-data-sharing.html. When Merck has a co-research, co-development, or co-marketing or co-promotion agreement, or when the product has been out-licensed, the responsibility for disclosure might be dependent on the agreement between parties. Under these circumstances, Merck will endeavour to gain agreement to share data in response to requests.
Ethics statements
Patient consent for publication
Ethics approval
This trial involves human participants and was approved by Lead IRB for each trial included in our integrated safety analysis: Study code: MS200527-0086. Lead IRB details: Parc de Salut MAR DREC, Dr. Aiguader, 88 08003, Barcelona, Spain, ceic-psmar@imim.es, www.parcdesalutmar.cat. Study code: MS200527-0060. Lead IRB details: Stanford Institutional Review Board, 3000 El Camino Real, Five Palo Alto Square, 4th Floor, Palo Alto, CA, USA 94306. Study code: MS200527-0018. Lead IRB details: Copernicus Group IRB, One Triangle Drive, Suite 100, Durham, NC, USA 27713. Participants gave informed consent to participate in the trial before taking part.
Acknowledgments
The authors would like to thank the patients, investigators, co-investigators and the trial teams at each of the participating centres; Emily Martin (EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA) and Daniela Sera and Karin Broeder (Merck Healthcare KGaA, Darmstadt, Germany) for providing assistance with the statistical analyses; and Francesca Hemingway of Bioscript Group Ltd., Macclesfield, UK for providing medical writing and editorial support (funded by Merck).
References
Footnotes
Correction notice This article has been corrected since it was first published. The open access licence has been updated to CC BY.
Contributors All authors confirm that they meet the International Committee of Medical Journal Editors uniform requirements for authorship. All authors contributed to drafting/critically revising the article, approval of the final manuscript and sharing in the final responsibility for the content of the manuscript and the decision to submit it for publication. XM acts as guarantor and accepts full responsibility for the finished work, had full access to the data in the trial, and made the final decision to submit the manuscript for publication.
Funding The trial was sponsored by Merck Healthcare KGaA (CrossRef Funder ID: 10.13039/100009945).
Competing interests XM has received speaking honoraria and/or travel expenses for participation in scientific meetings, and/or has been a steering committee member of clinical trials and/or participated in advisory boards of clinical trials in the past years with Actelion, Alexion, Biogen, Bristol-Myers Squibb/Celgene, EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA, Excemed, Genzyme, Hoffmann-La Roche, Immunic, Janssen Pharmaceuticals, Medday, Merck Healthcare KGaA, MSIF, Mylan, Nervgen, NMSS, Novartis, Sandoz, Sanofi-Genzyme, Teva Pharmaceutical and TG Therapeutics. DW has received consultant fees from Amgen, Eli Lilly, EMD Serono Research & Development Institute, Inc., Billerica, MA, USA (an affiliate of Merck KGaA), Merck, Celgene and Janssen. MCG has received personal compensation from AbbVie, Astellas, EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA, Galapagos, Genentech/Roche, Gilead, Incyte, Eli Lilly, Pfizer, Sanofi and Vertex. DT is an employee of Ares Trading SA, Eysins, Switzerland, an affiliate of Merck KGaA, and received stock or an ownership interest from Novartis. DP-R was an employee of EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA at the time of the study, and is currently an employee of and has received stock from Pfizer. CLB and HG are employees of Merck Healthcare KGaA, Darmstadt, Germany. AHK is an employee of and received stock or an ownership interest from EMD Serono Research & Development Institute, Inc., Billerica, MA, USA, an affiliate of Merck KGaA.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.