Article Text

Abstract
Introduction Lateral elbow tendinopathy (LET) is a highly prevalent disease among the middle-aged population, with no consensus on optimal management. Non-operative treatment is generally accepted as the first-line intervention. Ultrasound (US) therapy has been reported to be beneficial for various orthopaedic diseases, including tendinopathy. The purpose of this study is to investigate the efficacy of US for LET treatment.
Methods and analysis This protocol entails a three-arm, prospective, multicentre, randomised controlled trial. Seventy-two eligible participants with clinically confirmed LET will be assigned to either (1) US, (2) corticosteroid injections or (3) control group. All participants will receive exercise-based therapy as a fundamental intervention. The primary outcome is Patient-rated Tennis Elbow Evaluation. The secondary outcomes include Visual Analogue Scale for pain, shortened version of the Disabilities of the Arm, Shoulder and Hand for upper limb disability, pain free/maximum grip strength, Work Limitations Questionnaire-25 for functional limitations at work, EuroQol-5D for general health, Hospital Anxiety and Depression Scale for mental status, Global Rating of Change for treatment success and recurrence rate, and Mahomed Scale for the participant’s satisfaction. Adverse events will be recorded. Intention-to-treat analyses will be used.
Ethics and dissemination Ethics committees of all clinical centres have approved this study. The leading centre is Shanghai Sixth People’s Hospital, whose approval number is 2021–153. New versions with appropriate amendments will be submitted to the committee for further approval. Final results will be published in peer-reviewed journals and presented at local, national and international conferences.
Trial registration number ChiCTR2100050547.
- elbow & shoulder
- musculoskeletal disorders
- orthopaedic sports trauma
- rehabilitation medicine
- sports medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- elbow & shoulder
- musculoskeletal disorders
- orthopaedic sports trauma
- rehabilitation medicine
- sports medicine
Strengths and limitations of this study
Exercise-based therapy as a fundamental intervention for all participants.
The first randomised controlled trial (RCT) to compare the efficacy between ultrasound therapy and corticosteroid injections in lateral elbow tendinopathy treatment.
Multicentre RCT with blinded outcome assessor and statistician.
Use of several patient-reported outcome measures as well as objective parameters.
Participants and treating surgeons not blinded.
Introduction
First described by Runge,1 lateral elbow tendinopathy (LET), also widely known as tennis elbow, has an estimated prevalence of 1%–3% in the general population, and peaks at the fourth and fifth decades of life, with an equal gender distribution.2 LET causes a great burden on the social economy, with an annual sickness absence rate as high as 5% in working-aged adults.3 Though previously considered as ‘tendinitis’, histological analysis suggests a degenerative rather than an inflammatory process in LET, which is now commonly converted to be considered as a ‘tendinosis’.4 A LET diagnosis is usually straightforward, with clear clinical signs and symptoms. The patient most often presents with pain at or around the bony surface of the upper half of the lateral epicondyle and is likely to have a history of strenuous overuse relating to particular repetitive actions in the affected upper limb.5 6
Though LET usually is a self-limiting condition, complaints may last up to 2 years or longer,7 therefore, it has great clinical value to find a better and faster recovery process. General principles of LET treatment should be orientated towards pain relief, movement restoration, grip strength and endurance improvement, return to normal function and life quality, and control of further clinical deterioration.8 Treatments can be divided into operative and non-operative therapies. Invasive treatments commonly include open, arthroscopic and percutaneous release of the common extensor origin.9 Among these, ultrasonic percutaneous tenotomy, a recently developed method, appealing to many researchers for its good durability of pain relief and functional recovery,10 has satisfactory long-term (90 months) outcomes reported by Ang et al.11 However, surgery is usually considered for patients with persistent pain and disability after a course of well-performed conservative therapy, with a proportion as low as 3% in the whole LET population;2 therefore, non-operative treatment is suggested as first-line treatment.12 Generally, non-surgical methods include injections (like corticosteroid, platelet-rich plasma, autologous blood, sodium hyaluronate, and so on), physiotherapy, extracorporeal shock-wave therapy (ESWT), ultrasound (US), topical glyceryl trinitrate, or oral naproxen, and so on.13 14
So far, despite the wide range of treatments, there is no successful and universally accepted regimen. In a cross-sectional survey of UK practice in managing LET, 81% of experts recommended exercise-based therapy (EBT) as the first choice of intervention.15 EBT was also supported by high-quality clinical trials16–18 and systematic reviews,19 20 regarded as the most cost-effective treatment for LET.21 The survey also showed that, as the mainstream treatment for a long time, corticosteroid injection (CI) was still the most recommended intervention second to EBT,15 due to its quick pain relief and physical functional improvement, though the recurrence rate may be high and prognosis may be worsened in the long term.16–18 In addition, systematic reviews have shown that the effects of other conservative treatments like autologous blood or hyaluronate injection,22 platelet-rich plasma injection,23 ESWT24 and acupuncture25 remain controversial or provide little to no benefit.
US is widely used for imaging purposes and regarded as an adjunct to physiotherapy. US can reduce muscle spasms and pain, and facilitate tissue repair by increasing local blood flow and stimulating inflammatory mediators.26 US has been widely reported to be treatment beneficial in fracture non-unions,27 28 osteoarthritis,29 30 chronic muscle pain,31 32 soft tissue injury,33 and so on. As for tendinopathy, US is also a potential non-invasive treatment modality for frozen shoulder,34 35 rotator cuff,36 achilles37 38 and patellar39 tendinopathy. Some studies have reported the efficacy of US in LET treatment, but with low grade of study design and data,40 and most of them focused on the comparison between US and ESWT.41–45 Both Yalvaç B43 and Özmen T41 have shown significant improvements in pain, upper limb function, strength and quality of life from baseline after treatment with US. However, they did not have a control group, which would make it unclear whether the efficacy comes from US itself or the passing time, as LET is a self-limiting disease. Therefore, the role of US in LET treatment still needs to be further explored by high-quality studies. Additionally, to our best knowledge, no study has compared the efficacy between US and CI in LET treatment yet.
Therefore, the purpose of the current three-arm, prospective, randomised, multicentre trial is to investigate the efficacy of US in treatment for LET, that is, US versus CI versus control, with a fundamental intervention of EBT, on clinical and functional outcomes, including Patient-rated Tennis Elbow Evaluation (PRTEE). In view of recent literatures, CI should be discouraged in LET;22 46 however, it is still common in clinics due to the ability to satisfy patients’ need for quick pain relief.15 Thus, a change in the paradigm of LET treatment is necessary. This change will come about through proposed evidence-based treatment guidelines. There have been some ongoing clinical trials on LET treatment in recent years,47–49 and our prospective randomised controlled trial (RCT) proposes to complement and add to this relevant and much needed scientific effort.
Methods
Study design
The design of this study is a three-arm, prospective, multicentre RCT that will enrol participants with a diagnosis of chronic symptomatic LET from four municipal tertiary hospitals (Shanghai Sixth People’s Hospital, Shanghai East Hospital, Shanghai Tenth People’s Hospital, and Pudong New Area People′s Hospital of Shanghai). This manuscript is written according to the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines.50 The name of this trial is Ultrasound versus Corticosteroid Injections versus Control in Lateral Elbow Tendinopathy (UCICLET).
Participant and public involvement
This study was done without participant involvement. Participants were not invited to comment on the design, consulted to choose patient-relevant outcomes, invited to contribute to the writing or editing of this manuscript for readability or accuracy. The final results and related publications will be disseminated to the public via mass media. Participants as a whole will be acknowledged at the end of our publications and presentations.
Participant recruitment
Figure 1 shows the participant flow chart throughout this study. Participants will be recruited over a period of 5 months, from the intake clinics of four principals of each subcentre. Additionally, participants will also be recruited through other physicians and healthcare professionals. Those interested will contact the research assistant who will provide further information about the study objectives and procedures and will perform an initial eligibility screening interview by telephone.
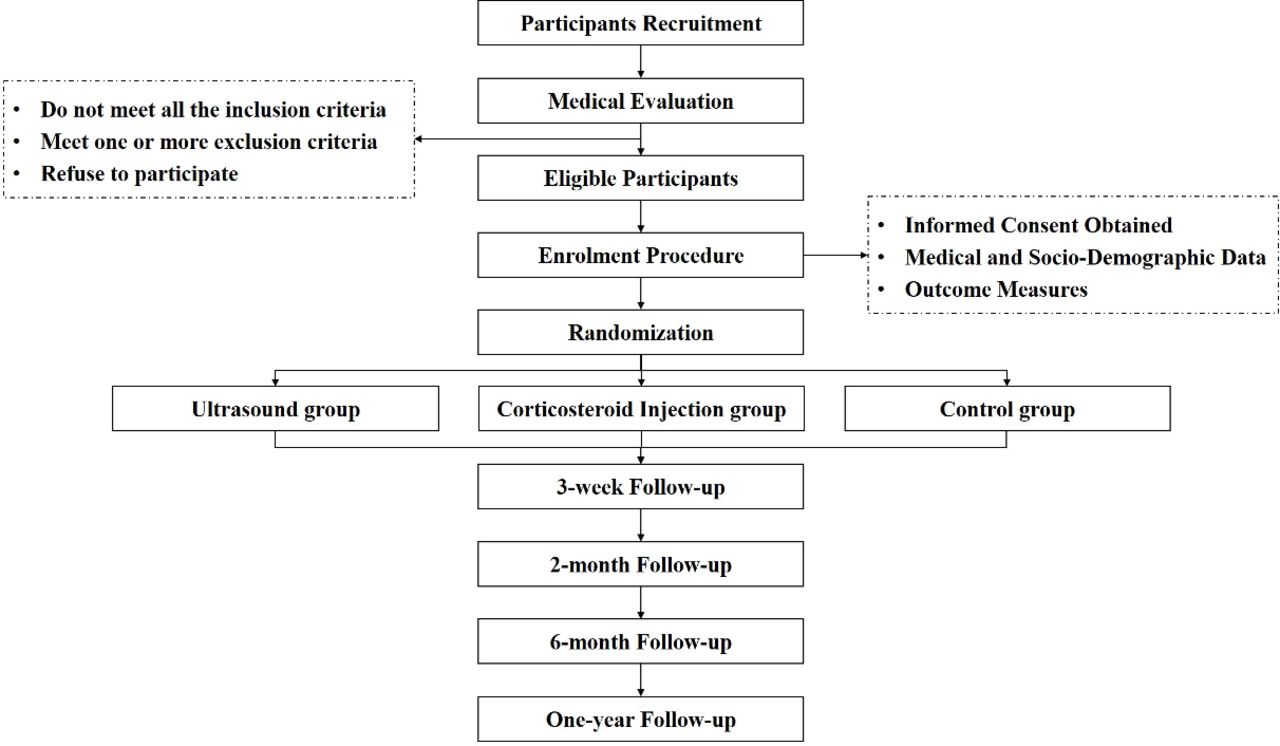
{kind=link}
Participant flow chart.
Medical evaluation and enrolment procedure
Participants potentially eligible will be invited to attend a medical examination to confirm the LET diagnosis and assess eligibility to participate in the research project.
Inclusion criteria
Age ≥18 years old;
Unilateral lateral elbow pain longer than 6 weeks duration;
Pain severity over the lateral humeral epicondyle greater than 30 mm on a 100 mm Visual Analogue Scale (VAS), provoked by at least two of the following: gripping, palpation, resisted wrist or middle finger extension, or stretching of forearm extensor muscles with reduced pain-free grip;16 49
Able to read and write in simplified Chinese (Mainland), understand and complete the questionnaire, and provide informed consent.
Exclusion criteria
Concomitant musculoskeletal pain conditions reported by participants to be their predominant complaint within the past 6 months;
History of symptoms suggesting radicular, neurological, inflammatory or systemic arthritic conditions;
Treatment by physiotherapy, electrophysical therapy, or injection within the past 6 months, or previous tennis elbow surgery;
Contraindications to US, including dermatological conditions, abnormal sensation in the affected arm, indwelling electrical pumps/pacemakers, epilepsy, pregnancy or breastfeeding, and so on;
Contraindications to CI, including hypertension, gastrointestinal ulcers, diabetes, mental illness, and so on.
Following the medical evaluation, a research assistant will meet with the eligible participants and obtain written informed consent. Demographic variables will be reported before treatment (baseline) of all participants regarding age, sex, body mass index, affected elbow, dominant arm, lifestyle (smoking and drinking) and previous medical history. Participants will also be asked relevant questions about the duration of symptoms and previous treatments (rehabilitation exercises, injections or others). Others like occupation, employment characteristics (full-time or part-time work, manual or non-manual labour), employment status (whether on sickness absence), professional activity characteristics (repetitive movements for >4 hours/day; wrist flexion or extension for >2 hours/day; use of computer keyboard/mouse or vibrating instruments (how many hours/day)), and sports activities (how many hours/week, activity type, team or individual sports)51 will also be collected.
Randomisation and blinding
Participants will be randomised into three intervention groups (either US or CI or control arm) in a ratio of 1:1:1, using a computer-generated randomised sequence with varying unknown block sizes (either three or six) for all study centres, without stratification. A research assistant with no involvement in the clinical care or outcome evaluations will prepare sequentially numbered, opaque, sealed envelopes according to the randomisation lists, with security in place to ensure allocation data cannot be accessed or influenced by any person. At the appropriate time, this assistant will open the envelope and assure coordination of the therapeutic interventions.
The outcome assessor and statistician will be blinded to group allocation and not involved in treatment procedures.
Intervention
At the beginning, all participants will receive standardised education and advice on adjusting activity patterns and managing pain, which will be distributed in the form of printed brochures and orally assessed on their understanding of the content. Participants will be told that absolute rest of the arm will not be advocated, and activities that do not cause elbow pain should be encouraged. The primary physical impairment in LET, which occurs in the muscle system, is best characterised as a deconditioning response of the forearm muscles to the pain. Therefore, all participants will receive the internationally best recommended fundamental intervention, EBT programme, for the forearm muscles.15 The EBT in this study will follow a standard protocol that has been adopted and used by several high-quality RCTs,16 18 52 53 mainly for addressing motor impairments, relieving pain and stimulating tendon remodelling. Thirty minutes per day, including basic tasks (pain-free (1) gripping and (2) extension exercise) and appendage tasks ((3) flexion, (4) supination and pronation, and (5) radial and ulnar deviation exercise). Various kinds of resistance and loads can be used, like free weights, rubber bands, manual resistance, isokinetic dynamometry or isometric contractions. (6) It is essential that all exercises performed for the upper limb be done with sound alignment of the spine, trunk and proximal arm.
Pain-free gripping exercise with exercise putty, which allows practice of various gripping actions.
Forearm extensor muscle exercise using a free-standing dumbbell. Note that the forearm is fully stabilised by the bench and upper body in sound postural alignment. Duration per repetition lasts about 6–10 s.
Dumbbell weight exercise for the forearm flexor muscle with 6–10 s per repetition. The postural is the same as 2).
Exercises for forearm supinator and pronator muscles using an imbalanced adjustable dumbbell weight with 6–10 s per repetition, from end range of supination to pronation with the participant maintaining full active control of the weight. The elbow bent to 90° with the arm stabilising beside the trunk. Progressions in load imposed on the muscles can be achieved by increasing the weight or the distance between weight and hand.
Radial and ulnar deviation exercises are performed with similar equipment and guidelines in 4.
Education on recognition and correction of the poor posture from the pelvis to neck. Once the spine and trunk are aligned more optimally, the upper limb position should be addressed.
Participants in the US group will receive continuous mode US (Shanghai, China) at a frequency of 1 MHz and intensity of 1.0 W/cm2 for 10 min, 5 days per week for 3 weeks on the maximum pain region of the lateral elbow.
Participants allocated to the CI group will receive a single local infiltration of 1 mL triamcinolone acetonide (10 mg/ mL) and 1 mL lidocaine 1%. Local CI was administered to the most painful area on pressure around the lateral epicondyle. Participants will be advised to wait for 20 min following injection and inform their doctor if there is any suggestion of infection or other adverse events. All adverse reactions will be managed by a committee chaired by the chief investigator. Rest from all strenuous activity for 1–2 weeks following injection will be strongly recommended, followed by a gradual return to normal activities. Participants will be instructed to avoid an aggressive return to activities even if substantial relief is obtained to minimise the potential recurrence of their symptoms.
Participants randomised to the control group will neither receive US therapy nor CI. They will only receive the fundamental intervention, EBT programme.
We discourage additional treatments to that assigned (that is, not per protocol) during the intervention period, but we allow the use of simple analgesics as needed. Participants will report all not-per-protocol treatments, such as drugs, in a diary.
Data management
Data will be collected during the participants’ visits to the hospital at baseline, 3 weeks, 2 months and 6 months, and 1 year after random assignment (table 1). In order to maximise participant compliance in follow-up completion, reminder emails and a telephone call by the research assistant will be programmed. Registered participants will be withdrawn from the study if: (1) Participant withdraws his/her consent, and (2) Exclusion criteria is discovered after registration. The reason and date of discontinuation will be recorded. Consent to use the data already collected prior to a participant’s withdrawal will be included in the consent form.
Study evaluation procedures and timeline
Outcome measures
Primary outcome
The primary outcome measure will be the difference in PRTEE. The PRTEE, formerly known as the Patient-Rated Forearm Evaluation Questionnaire, is a well-validated composite scale measuring pain (five items, with 0=no pain and 10=worst imaginable) and physical function (six items for specific activities and four items for usual activities, with 0=no difficulty and 10=unable to do),54 ranging from 0 to 100 points, with higher scores representing worse possible pain and more loss of function. The pain (intraclass correlation coefficients, ICC=0.89), physical function (ICC=0.83) and the total (ICC=0.89) scores all demonstrate excellent reliability.55 A variation of 11/100 points or 37% of baseline scores are reported for clinical significance defined as ‘much better’ or ‘completely recovered’.56 We use a validated Hong Kong Chinese version57 of the PRTEE translated into simplified Chinese (Mainland) because the culture and language are the same.
Secondary outcome
Secondary outcome measures will be the differences in VAS58 for pain, shortened version of the Disabilities of the Arm, Shoulder and Hand (Quick-DASH)59 for upper limb disability, pain-free/maximum grip strength, Work Limitations Questionnaire-25 (WLQ-25)60 for functional limitations at work, EuroQol-5D (EQ-5D)61 for life quality and health status, the Hospital Anxiety and Depression Scale (HADS)62 for anxiety and depression status, Global Rating of Change (GROC) for treatment success and recurrence rate, and Mahomed Scale63 for participants’ satisfaction.
Pain
VAS will be used for pain evaluation, which consists of a 100 mm horizontal numbered line anchored at one end (0) with the words ‘no pain’ and at the other end (100) with the words ‘worst pain imaginable’. The score is determined by the distance between the left end of the line and the participant’s mark in mm.58 VAS is considered to be the most sensitive of all pain scoring scales and has been specifically validated in the LET population with high reliability (r=0.89) and a moderate correlation with pain-free grip strength (r=0.47).64 Participants will be asked to score their pain on this line during rest (at time of measure), provocation and maximum grip strength. The provocation test is conducted on the outpatient clinic by resisted wrist dorsiflexion during full elbow extension. Clinically relevant improvement will be defined when a 50% decrease in VAS is observed before and after the treatment.65 The consumption of rescue medication taken by each patient will be also recorded at each follow-up visit.
Upper limb disability
The well-validated simplified Chinese (Mainland) version of Quick-DASH66 will be used for elbow function evaluation, consisting of eleven questions scored on a 5-point scale similar to DASH.59 Total and individual module scores will be calculated out of 100, with a higher score indicating a worse status. A minimal clinically important difference of 15.91 points has been reported.67
Grip strength
Pain-free/maximum grip strength will be measured using a dynamometer (CAMRY, City of Industry, California, USA). The participants will be asked to take a shoulder-width stance and allow their arms to hang loose, holding their arm adducted along the body and the elbow in full extension. The pain-free grip strength will be measured, followed by the maximum grip strength, and the affected side will be measured first and then the unaffected side. The measurement readings will be not revealed to the subjects until the completion of the test. The pain-free grip strength will be measured up to the point when the subject slowly squeezes the dynamometer until the occurrence of pain. The maximum grip strength will be measured at the maximum grip level. The mean of three consecutive trials, separated by a 20 s pause, will be calculated. Results will be presented as a ratio of values of the symptomatic side/asymptomatic side × 100.68
Functional limitations at work
In order to gather the information that is complementary to the pain and disability scales, functional limitations at work will be measured with WLQ-25. It contains 25 items arranged under four subscales addressing four dimensions of job demands: time demands, physical demands, mental/interpersonal demands and output demands.60 A five-level ordinal response scale ranging from 0 (all of the time) to 4 (none of the time) with an additional sixth option (does not apply to my job) is used. The total scores range from 0 to 100 points, and a 13-point (out of 100) improvement for the summed score is established for clinically important differences.69
Life quality and health status
The EQ-5D is a widely validated generic health-related quality of life (HRQol) measure known for its simplicity.61 It contains a five-dimension descriptive system (mobility, self-care, usual activities, pain/discomfort and anxiety/depression) and a VAS, ranging from 0 to 1, in which one represents perfect health. All the dimensions are grouped into three levels (no problem, some problem and extreme problem). We used a validated Chinese version70 of the EQ-5D, which has been recommended by China Guidelines for Pharmacoeconomic Evaluations 2011 for a measure for HRQol and health utility.71
Anxiety and depression status
HADS will be used to identify and quantify two of the most common psychological disorders, anxiety and depression.62 There is evidence of increased levels of anxiety and depression in people with LET.72 HADS is a 14-item scale independent of somatic symptoms, which consists of two 7-item subscales measuring depression and anxiety, respectively. A 4-point scale (from 0 representing the absence of symptoms to 3 representing the maximum symptomatology) is used. The total scores for each subscale range from 0 to 21 points, with higher scores indicating higher levels of disorder. HADS has two cut-offs for categorisation: 0–7, ‘non-case’; 8–10, ‘possible or doubtful case’; 11–21, ‘probable or definite case’.73
Treatment success and recurrence rate
Participants’ treatment impressions of change regarding their condition will be recorded on a 6-point Likert Scale (from ‘completely recovered’, ‘much improved’, ‘somewhat improved’, ‘same’, ‘worse’ to ‘much worse’). Success rates will be calculated by dichotomising responses. Participants who report their overall condition as ‘completely recovered’ or ‘much improved’ since the beginning of the study will be counted as successes, while other responses will be counted as failures.16 18 Recurrence will primarily be defined as occurring when a participant rates a success at 3 weeks and a failure at 2 months or 6 months or 1 year on the GROC.16 18
Additional treatments will also be recorded after the failure of management in this study (that is, not per protocol), if any, including subsequent interventions and even surgery.
Participants’ satisfaction
Similarly, participants’ level of satisfaction on the evolution of their condition will be determined on a validated 4-point Likert Scale ranging from ‘very satisfied’, ‘somewhat satisfied’, ‘somewhat dissatisfied’ to ‘very dissatisfied’.74
Adverse events
All adverse events, defined as any negative or unwanted reactions to intervention, will be recorded through the symptoms reported by the participants, and observations by the researcher at every visit. US treatment may cause mild local swelling, spot-like bleeding, ecchymosis, enhanced local pain response, and local hyperesthesia or decrease. CI-related adverse events are divided into acute and long-term ones. Acute events include dizziness, skin flushing, local bleeding, and some may even develop rarer physical reactions, such as arrhythmias. Therefore, all participants must take at least 20 min in the outpatient room to observe and even manage any acute adverse reactions following the injection. Long-term events may cause skin pigmentation, local calcification and infection.
Sample size calculation
Sample size and power calculation are based on the primary outcome of the PRTEE Score. All sample size calculations assume two-sided analysis with a power of 90% (1-β=0.90) at a significant level of α=0.05. A SD of 5.1 points on the PRTEE Score will be used based on the previous trial.75 To detect a minimum clinically significant difference of 11.0 points56 (superiority margin) between the US and control groups (assuming a true difference of 15.6 points),43 75 a total of 22 participants in each group is required. Allowing for an up to 10% dropout rate, we aim to enrol at least 24 participants in each group to complete the study.
Analysis plan
Baseline characteristics will be summarised for the three treatment groups using appropriate descriptive statistics. Both primary and secondary analyses will be conducted blinded to treatment allocation and analysed on the intention-to-treat76 approach with all randomised participants retaining their original randomised group. Multiple imputation by chained equations will be used to address missing data caused by loss to follow-up and non-responses if these missing data are judged to be random.
The primary comparisons for the PRTEE Scores will be made using linear regression. In secondary analyses, a repeated measures mixed model77 will also be used to examine the associations between treatments and repeated outcome measures, with terms of treatment, time, trial centre and corresponding baseline values as covariates (age, gender, body mass index, and so on). Linear regression will be used for numerical outcomes and logistic/ordinal regression for any categorical outcomes.
Quality assurance/monitoring/management
To standardise staff training and study procedures like participant recruitment, outcome measurement, data import, security, management and analysis, a Manual of Operations and Procedures and a case report form will be developed as per protocol, which also include the monitoring plans to assure participant protection and data integrity, thus facilitating consistency in protocol implementation and data collection. The investigators, physicians, research assistants, outcome assessors and statisticians are different people and should receive Good Clinical Practice training. A trained project manager will visit each centre for monitoring to ensure data quality and compliance with the trial protocol.
All data obtained will be kept strictly confidential and stored electronically in a database with secured access. Encryption will be used for data transfer, with removal of any information that could identify individuals. De-identification of data will only be permitted at the final of this trial for analysis.
Study duration
Recruitment will begin in November 2021, and 1-year follow-up for all participants is anticipated to be completed by March 2023. See table 1 for time points and recruitment progress.
Ethics and dissemination
The trial has been approved by all four medical ethics committees: Ethics Committee of Shanghai Sixth People’s Hospital (the leading clinical centre, approval No. 2021–153), Ethics Committee of Shanghai East Hospital (EC.D(BG).016.03.1-2021-096), Ethics Committee of Shanghai Tenth People’s Hospital (SHSY-IEC-4.1/21-193/01) and Ethics Committee of Pudong New Area People’s Hospital (IRBY2021-005). The potential risks of this clinical trial are considered to be minimal and are addressed in the protocol and consent forms. A written consent (online supplemental file 1) will be obtained by clinical practitioners from each participant. The trial was registered on the www.chictr.org website (registration number ChiCTR2100050547). Data will be published in peer-reviewed journals and presented at conferences, both nationally and internationally.
Supplemental material
Limitation
This study will have one limitation. Participants and treating surgeons are inevitably not blinded, which may produce bias. However, we will strictly control the outcome assessors and statisticians to be blinded to group allocation and not involved in treatment procedures to reduce the bias.
Ethics statements
Patient consent for publication
Acknowledgments
The authors thank the Base for Interdisciplinary Innovative Talent Training, Shanghai Jiao Tong University and Youth Science and Technology Innovation Studio of Shanghai Jiao Tong University School of Medicine for their support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
ZS, SC and WL are joint first authors.
ZS, SC and WL contributed equally.
Contributors ZS and SC are the primary investigators. ZS, SC, WL, YZ, CF participated in the development of the study design. ZS, SC, WL, GS, JL, JW, WW, YZ and CF participated in the study conduct. ZS, SC and WL drafted the manuscript under CF’s supervision. CF contributed to applying for and gaining funding. All authors contributed to the content and critical revision and approved the final draft of the manuscript.
Funding This study will be supported by General Project of National Natural Science Foundation of China (8217090787); Shanghai Engineering Technology Research Center and Professional Technology Service Platform project of 2020 ‘Science and Technology Innovation Action Plan’ of Shanghai (20DZ2254100); Municipal Hospital Clinical Skills and Innovation Capacity of Three-year Action Plan Program of Shanghai Shenkang Hospital Development Center (SHDC2020CR2039B, SHDC2020CR6019-002); Biomedical Technology Support Special Project of Shanghai ‘Science and Technology Innovation Action Plan’ (20S31900300, 21S31902300); Clinical Research Center (CRC) of Shanghai University of Medicine and Health Sciences (20MC2020001).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.