Article Text

Abstract
Objective To investigate how genetics influence the risk of smoking-related systemic lupus erythematosus (SLE) manifestations.
Methods Patients with SLE (ndiscovery cohort=776, nreplication cohort=836) were genotyped using the 200K Immunochip single nucleotide polymorphisms (SNP) Array (Illumina) and a custom array. Sixty SNPs with SLE association (p<5.0×10−8) were analysed. Signal transducer and activator of transcription 4 (STAT4) activation was assessed in in vitro stimulated peripheral blood mononuclear cells from healthy controls (n=45).
Results In the discovery cohort, smoking was associated with myocardial infarction (MI) (OR 1.96 (95% CI 1.09 to 3.55)), with a greater effect in patients carrying any rs11889341 STAT4 risk allele (OR 2.72 (95% CI 1.24 to 6.00)) or two risk alleles (OR 8.27 (95% CI 1.48 to 46.27)).
Smokers carrying the risk allele also displayed an increased risk of nephritis (OR 1.47 (95% CI 1.06 to 2.03)). In the replication cohort, the high risk of MI in smokers carrying the risk allele and the association between the STAT4 risk allele and nephritis in smokers were confirmed (OR 6.19 (95% CI 1.29 to 29.79) and 1.84 (95% CI 1.05 to 3.29), respectively).
The interaction between smoking and the STAT4 risk allele resulted in further increase in the risk of MI (OR 2.14 (95% CI 1.01 to 4.62)) and nephritis (OR 1.53 (95% CI 1.08 to 2.17)), with 54% (MI) and 34% (nephritis) of the risk attributable to the interaction. Levels of interleukin-12-induced phosphorylation of STAT4 in CD8+ T cells were higher in smokers than in non-smokers (mean geometric fluorescence intensity 1063 vs 565, p=0.0063).
Lastly, the IL12A rs564799 risk allele displayed association with MI in both cohorts (OR 1.53 (95% CI 1.01 to 2.31) and 2.15 (95% CI 1.08 to 4.26), respectively).
Conclusions Smoking in the presence of the STAT4 risk gene variant appears to increase the risk of MI and nephritis in SLE. Our results also highlight the role of the IL12−STAT4 pathway in SLE-cardiovascular morbidity.
- lupus erythematosus
- systemic
- smoking
- cardiovascular diseases
- polymorphism
- genetic
- lupus nephritis
Data availability statement
Data are available on reasonable request. Deidentified patient data and statistical code are available on request with project plan.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Neither traditional nor systemic lupus erythematosus (SLE)-related risk factors can fully account for the excess cardiovascular disease risk seen in patients with SLE, but interactions between traditional and SLE-specific risk factors have been scarcely investigated.
What does this study add?
Our results show that the signal transducer and activator of transcription 4 (STAT4) risk allele rs11889341 enhances the effect of smoking on the risk of myocardial infarction and nephritis and that smoking is associated with increased interleukin (IL)-12-induced phosphorylation of STAT4 in CD8+ T cells.
We further demonstrate that the IL12A SLE risk variant rs564799 is associated with an increased risk of myocardial infarction, which further highlights the importance of the IL12-STAT4 pathway in the aetiology of cardiovascular morbidity in SLE.
How might this impact on clinical practice or future developments?
Our results suggest that genetic profiling of patients with SLE may be useful for predicting comorbidities of the disease, impact of environmental factors and for targeted smoking cessation interventions.
Introduction
Systemic lupus erythematosus (SLE) is a disease characterised by loss of tolerance to self-antigens, formation of immune complexes and an activated type I interferon (IFN) system.1 A widely accepted view of the aetiology of SLE is that environmental factors trigger the disease in genetically susceptible individuals. The genetic background is complex, with more than 100 single nucleotide polymorphisms (SNPs) associated with risk for SLE.2 Exposure to certain environmental factors, including ultraviolet radiation and viral infections, is associated with SLE development and flare-ups of the disease.3 4 Several studies have evaluated smoking as a risk factor for SLE, with the largest meta-analysis to date showing a modest risk increase.5 While the results are not confirmed in prospective studies, both Cozier and Barbhaiya et al observed a trend of increased risk in smokers.6 7 The most extensive prospective study involving 286 cases with SLE demonstrated an association between smoking and development of SLE with increased anti-dsDNA, but no risk of overall SLE.7
Although death from active SLE has decreased since the 1950s,8 the mortality rate still exceeds that of the general population, with cardiovascular morbidity remaining considerably high and a strong risk factor for premature mortality.9–11 Both traditional and SLE-related risk factors, such as hypertension, nephritis and high disease activity have been identified as risk factors, but cannot fully account for the excess cardiovascular disease (CVD) risk seen in patients with SLE.12 13 To fully explain the aetiology of SLE or its comorbidities such as CVD, gene–gene or gene–environment interactions may be essential to consider. In rheumatoid arthritis, there is compelling evidence of a strong interaction between the HLA-DRB1*04 shared epitope and smoking on the development of anticitrullinated protein autoantibodies14 and a high prevalence of cardiovascular events (CVE).15
In SLE, a few studies have investigated the interaction between genetic risk factors and smoking on the development of the disease.16 17 Recently, Cui et al demonstrated that an additive interaction between smoking and the cumulative genetic risk of SLE increases the risk of the disease.18 However, gene–smoking interactions on the development of specific manifestations or co-morbidities of SLE have been scarcely studied. This study, therefore, aims to investigate the effect of smoking on the development of specific manifestations of SLE, including CVE, end-stage renal disease (ESRD) and nephritis, and examine how the effect is modulated by the presence of genetic variants associated with an increased risk of SLE development.
Methods
Patients of the discovery and replication cohort
The discovery cohort included 774 patients with SLE from Sweden. The replication cohort included 836 patients from Norway and Denmark. All subjects fulfilled ≥4 American College of Rheumatology (ACR)−82 and ACR-97 classification criteria for SLE and were of European descent. Clinical characteristics of the cohorts are described in table 1 and online supplemental table 1, respectively. Clinical data, including smoking status (ever-smoker, including current or a history of smoking, versus never-smoker), the ACR-82 classification criteria, antiphospholipid syndrome diagnosis, ESRD, renal biopsy data and CVE, defined as myocardial infarction (MI), ischaemic cerebrovascular disease (ICVD) or venous thromboembolism, was collected from medical records. For definitions, see online supplemental file. In the replication cohort, data regarding ESRD were not available, and data on smoking were available from 503 patients.
Supplemental material
Supplemental material
Prevalence of clinical manifestations in smokers (n=371) and non-smokers (n=387) in the discovery cohort
Genotyping and selection of SNPs
Genotyping of the discovery cohort was performed using the Illumina 200K Immunochip SNP array, for details, see online supplemental file. SNPs previously associated with SLE at genome-wide significance in the European population2 were selected. For SNPs not included on the Immunochip, the SNP-proxy with the highest linkage disequilibrium (LD) (r2 ≥0.96) was selected. All SNPs were filtered for independent signals, removing the variant with the lowest SLE-OR for SNPs in LD (r2 >0.2). In total, 4 HLA and 56 non-HLA SNPs were investigated for associations with MI (online supplemental table 2). Individuals in the replication cohort were genotyped for three single nucleotide variants using a custom assay on the MassARRAY system (see online supplemental file).
Interleukin-12-induced phosphorylation of STAT4
Interleukin 12 (IL-12)-induced phosphorylation of signal transducer and activator of transcription 4 (pSTAT4) was previously determined in 72 healthy blood donors from Uppsala Bioresource using flow cytometry.19 Smoking data were available from 45 of these donors, of which 20 were past or current smokers and 25 were non-smokers.
Statistical analysis
To investigate associations between smoking and clinical manifestations, logistic regression models were used. As smoking was associated with longer disease duration and higher age at follow-up (table 1), these variables were included as covariates. In analysis of associations between genetic variants and MI, SNPs were first analysed separately. Next, all variants demonstrating a positive association with MI were included in a forwards conditional multiple regression model. All analyses were adjusted for age and disease duration. Results considered statistically significant (unadjusted p<0.05) were reanalysed in the replication cohort using the same statistical model and covariates. Meta-analyses were performed on the two datasets and multiplicative and additive interactions between the STAT4 risk allele and smoking were studied in a combined dataset through addition of a STAT4*smoking interaction term in the logistic models and by calculating the attributable proportion due to interaction, respectively.20 21 Differences in levels of pSTAT4 were assessed by Student’s t-test and by a linear regression model allowing adjustment for age and the STAT4 risk allele. R22 was used for all analyses except the meta-analyses which were performed in PLINK.23
Results
Smoking is modestly associated with MI
Initially, we assessed the association between smoking and clinical manifestations (table 1). We found no evidence of any associations between smoking and the ACR criteria, except the haematological criterion, which was less prevalent in smokers (table 1). Elevated levels of red and white blood cells in smokers is a well-known phenomenon.24 Smoking was not associated with a history of DVT or ICVD, however, a significant association between smoking and MI was observed (OR 1.96 (95% CI 1.09 to 3.55), p=0.025) (table 1).
Increased risk of MI in SLE-smokers with the STAT4 risk allele
Next, we asked whether there are sub-groups of patients in which smoking plays a more prominent role in MI development. We initially examined 60 SNPs with established association with SLE (p<5.0×10−8) for association with MI (online supplemental table 2). We found that the Neutrophil Cytosolic Factor 2 (NCF2), Interleukin-12A (IL12A) and STAT4 risk alleles displayed independent, positive association with MI (table 2). In addition, patients carrying two alleles of both the STAT4 and the IL12A risk variants (n=37, 4.9% of the patients) displayed a substantially higher prevalence of MI compared with those with any other allele combination (27% vs 7%) (OR 5.88 (95% CI 2.44 to 14.17), p=7.9×10−5) (figure 1A).
Associations between SLE risk SNPs and myocardial infarction in the discovery and replication cohort

Prevalence of myocardial infarction (MI) in patients with different numbers of STAT4 and IL12A risk alleles. The prevalence of MI for patients in the discovery (A) and the replication (B) cohorts, stratified by number of risk alleles of STAT4 (rs11889341) and IL12A (rs564799). Patients carrying two risk alleles of both STAT4 and IL12a (ndiscovery cohort=37 (4.9%), nreplication cohort=28 (3.4%)) were compared with the remaining patients using logistic regression, adjusting for age at follow-up and disease duration. IL12, interleukin-12; STAT4, Signal Transducer and Activator of Transcription 4.
Next, we stratified patients by smoking status to determine whether each of the three SNPs displayed stronger association with MI in smokers. No significant associations were found for the NCF2 or IL12A risk alleles (OR 1.58 (95% CI 0.89 to 2.78), p=0.12 and OR 1.36 (95% CI 0.89 to 2.08), p=0.15, respectively). However, the STAT4 risk allele demonstrated a stronger association in smokers (OR 2.45 (95% CI 1.46 to 4.19), p=0.00086) (figure 2A). Next, we assessed the association between smoking and MI in patients carrying the STAT4 risk allele and observed an almost 3−fold increase in risk for the smokers compared with the non-smokers (OR 2.72 (95% CI 1.24 to 6.00), p=0.013). In patients carrying two risk alleles, the risk was more than eightfold higher for smokers (OR 8.27 (95% CI 1.48 to 46.27), p=0.016). In contrast, we could not demonstrate a significant association between smoking and MI in patients without the risk allele (OR 1.20 (95% CI 0.49 to 2.96) p=0.55).

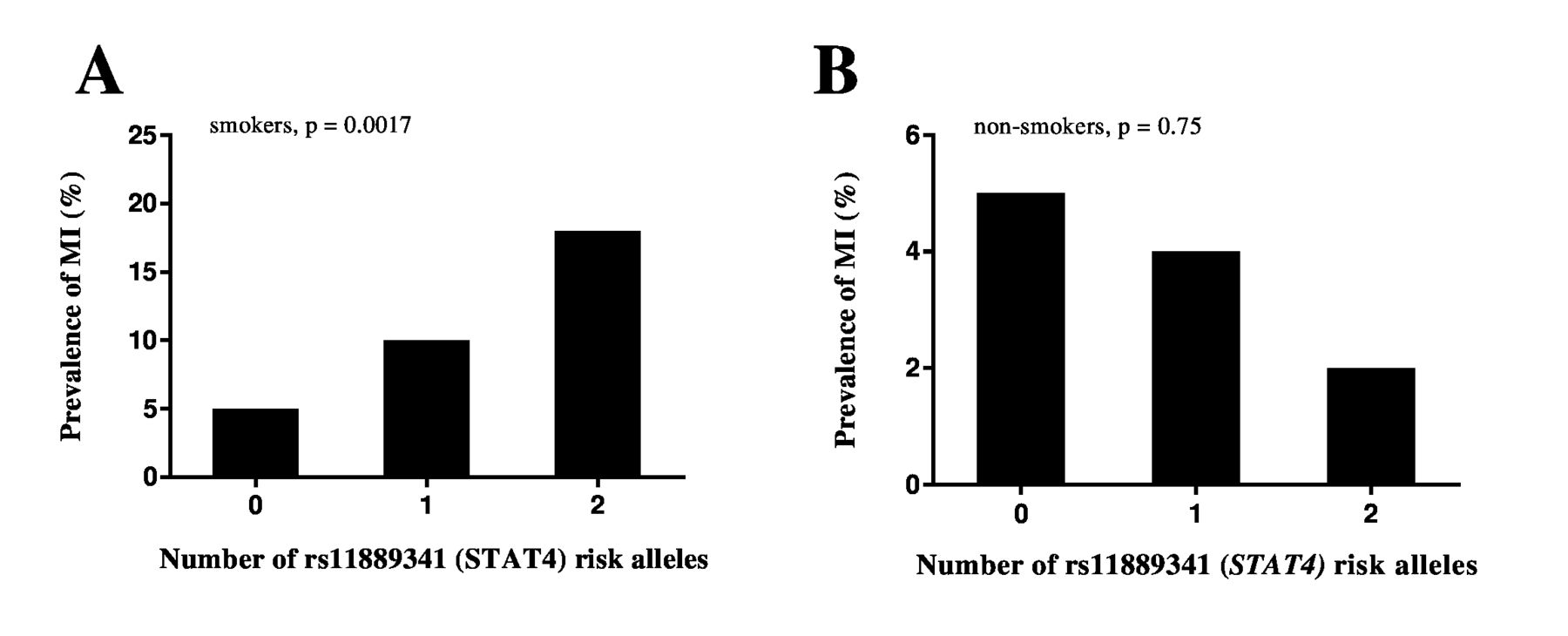
Prevalence of myocardial infarction (MI) and end-stage renal disease (ESRD) in the discovery cohort. Prevalence of MI (A) and ESRD (B) in the discovery cohort in all patients (n=776), smokers only (n=371) and non-smokers only (n=405), stratified by the number of STAT4 (rs11889341) risk alleles. Patients with 0, 1 or 2 risk alleles in each group were compared using logistic regression, adjusting for age at follow-up and disease duration. STAT4, Signal Transducer and Activator of Transcription 4.
As patients with nephritis have previously been shown to have a higher prevalence of both MI and the STAT4 risk allele.25–27 we hypothesised that the results would be similar if using nephritis, rather than MI, as the outcome variable. Without stratifying for smoking, the association between the STAT4 risk allele and nephritis reached suggestive significance (OR 1.23 (95% CI 0.98 to 1.54), p=0.072). The effect was more pronounced in the smokers only (OR 1.47 (95% CI 1.06 to 2.03), p=0.020). In addition, we found moderate evidence that patients with nephritis carrying the STAT4 risk allele were at a greater risk of developing ESRD (OR 1.85 (95% CI 0.96 to 3.59), p=0.068), and this risk was enhanced in smokers (OR 2.52 (95% CI 1.04 to 6.10). p=0.040) (figure 2B). Of note, despite the non-smoking group including more patients with nephritis (n=140 vs n=129), no evidence of an association between the STAT4 risk allele and nephritis or ESRD could be demonstrated in this group (OR 1.07 (95% CI 0.77 to 1.46), p=0.70 and OR 1.10 (95% CI 0.38 to 3.16), p=0.86, respectively).
To validate our significant findings, we performed the same analyses in an independent cohort of patients with SLE (online supplemental table 1). Analysis of the genetic variants demonstrated that the IL12A risk allele was the only gene variant significantly associated with MI when not accounting for smoking (table 2). Patients with two risk alleles of both STAT4 and IL12A (n=28, 3.2% of the patients) were found to have a significantly higher prevalence of MI than patients without this combination of risk alleles (OR 7.21 (95% CI 1.36 to 38.27, p=0.020) (figure 1B). Similar to in the discovery cohort, we found a significant association between the STAT4 risk allele and MI in smokers (OR 2.11 (95% CI 1.04 to 4.26), p=0.038). In addition, smoking was associated with MI in patients carrying the STAT4 risk allele (OR 6.19 (95% CI 1.29 to 29.79), p=0.023). No evidence of these associations could be observed in the non-smoking group (OR 0.58 (95% CI 0.09 to 3.90), p=0.58 and OR 1.32 (95% CI 0.23 to 7.34), p=0.75, respectively). In patients carrying two risk alleles (n=51), the logistic regression could not be performed due to a ‘perfect separation’ between groups, with 9% of smokers having had a MI compared with 0% of never-smokers. As in the discovery cohort, we found an association between the STAT4 risk allele and nephritis in smokers (OR 1.84 (95% CI 1.05 to 3.29), p=0.035), whereas the effect size was non-significant in non-smokers (OR 0.78 (95% CI 0.41 to 1.45), p=0.44).
Meta-analysis of the two cohorts showed the risk of MI to more than double with each additional STAT4 risk allele in smokers (OR 2.28, p=0.00010). In contrast, no association could be detected in the never-smoker group (OR 0.80, p=0.52). Similarly, in meta-analysis of patients with 2 STAT4 risk alleles (n=282, 22%), smoking was found to be a strong risk factor for MI (OR 8.27, p=0.016). For nephritis, each STAT4 risk allele increased the risk by ~50% in smokers (OR 1.52, p=0.00051), whereas no increase in risk was found in the non-smoker group (OR 0.82, p=0.33).
Interaction between STAT4 risk allele and smoking results in a higher risk of MI and nephritis
We subsequently performed interaction analyses on all patients and found a significant multiplicative interaction between the STAT4 risk allele and smoking on the development of MI (OR 2.14 (95% CI 1.01 to 4.62), p=0.049) as well as nephritis (OR 1.53 (95% CI 1.08 to 2.17), p=0.020) (online supplemental table 3). Next, we examined additive interaction and observed an attributable proportion due to interaction of 0.54 (95% CI 0.24 to 0.83, p=0.00019) and 0.34 (95% CI 0.080 to 0.61, p=0.0051) for MI and nephritis, respectively.
To determine whether the effect of the STAT4 risk allele on nephritis in smokers could explain the association with MI, we performed stratification of the combined dataset and investigated the association between the STAT4 risk allele and MI in smokers and non-smokers without nephritis. In the smokers, the association between the STAT4 risk allele and MI remained significant (n=428, OR 2.43 (95% CI 1.40 to 4.27), p=0.0017) (figure 3). Similarly, the association between the STAT4 risk allele and nephritis in smokers remained significant after excluding patients with MI from the analysis (n=623, OR 1.54 (95% CI 1.20 to 1.98), p=0.00076).

The prevalence of myocardial infarction (MI) in patients without nephritis. To investigate whether the association between the rs11889341 (STAT4) allele and MI was dependent on the association between the STAT4−nephritis association, all patients with nephritis were excluded and the prevalence of MI was subsequently plotted for smoking (A) and non-smoking (B) patients with 0, 1 or 2 of the STAT4 risk allele. Patients with 0, 1 or 2 risk alleles in each group were compared using logistic regression, adjusting for age at follow-up and disease duration. STAT4, signal transducer and activator of transcription 4.
Lastly, as both SLE risk alleles in STAT4 and smoking have shown association with the development of aPL in previous studies,28 29 we assessed the association between the STAT4 risk allele and aPL in smokers, however, it was not significant (OR 1.56 (95% CI 0.71 to 3.72, p=0.26). Next, we performed a multiple regression model in the smoking group including the STAT4 risk allele, any aPL, nephritis, age at follow-up, and disease duration as covariates. We found the association between the STAT4 risk allele and MI to remain significant (OR 3.26 (95% CI 1.15 to 9.20), p=0.026), whereas neither aPL nor nephritis displayed significant association with MI (p=0.87 and p=0.50, respectively), (online supplemental table 4).
Levels of activated STAT4 are increased in smokers
The STAT4 risk allele is associated with higher levels of pSTAT4 in CD8+ T cells from SLE patients on stimulation with IL-12.19 Furthermore, higher levels of STAT4 expression have demonstrated association with increased cardiovascular damage in patients with SLE.30 To investigate whether smoking also increases the levels of pSTAT4, we analysed IL-12 stimulated CD8+ T cells from healthy blood donors who were smokers (n=20) or never-smokers (n=25). We found the levels of pSTAT4 to be higher in smokers (p=0.0063), with a mean value of 1063 compared with 565 in non-smokers (figure 4). When adjusting for age and the STAT4 risk allele, which is in LD (r2=1.00) with a STAT4 risk variant previously shown to influence levels of pSTAT4 in these individuals,19 31 the association between smoking and levels of pSTAT4 remained significant (β=396, p=0.023).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Levels of pSTAT4 in CD8+ T cells after stimulation with IL-12. The levels of IL-12−induced pSTAT4 were compared between healthy blood donors who were smokers (current or past) (n=20) and never-smokers (n=25) by Student’s t-test. IL-12, interleukin-12; pSTAT4, phosphorylated Signal Transducer and Activator of Transcription 4.
Discussion
In the present study, we demonstrate that smoking substantially increases the risk of MI in a subset of patients with SLE carrying a variant of the STAT4 SLE-risk gene. In both the discovery and replication cohorts, the effect size increased with an increasing number of STAT4 risk alleles, with smoking giving rise to a more than 8-fold risk of MI in homozygous individuals. We believe that our results add important knowledge in the understanding of how SLE-risk alleles can modulate the effect of traditional risk factors.
The prevalence of MI is higher in SLE patients with nephritis than patients without renal manifestations26 32 and SLE-risk alleles in STAT4 have previously been linked to both nephritis and severe renal insufficiency.25 27 We, therefore, speculated that the smoking-STAT4 risk allele interaction did not directly affect MI, but rather, was a consequence of an interaction between the STAT4 risk allele and smoking on the development of nephritis. Indeed, we found that this gene-environment combination also results in a higher risk of nephritis, as well as ESRD. Interestingly, however, the STAT4 risk allele/smoking effect on MI did not decrease when adjusting the model for nephritis or when completely removing the patients with nephritis from the analysis. Similarly, the effect of the STAT4 risk allele/smoking on nephritis remained significant after excluding all patients with MI from the analysis, indicating that the associations were independent.
Based on these results, we hypothesised that the increased risk in individuals who smoke and carry the risk allele is connected with the levels of activated STAT4 in these individuals. Hagberg et al have shown that the rs7574865 STAT4 risk allele—which is in perfect LD (r2=1.00) with the SNP used in this study31—is associated with increased levels of pSTAT4 in activated CD8 +T cells of SLE patients.19 Therefore, we assessed whether smoking elevates pSTAT4 in this cell type and found that the levels were almost twofold higher in smokers. This observation is in line with previous findings by Di Stefano et al, who demonstrated higher levels of pSTAT4 in bronchial T cells from healthy smokers compared with non-smokers.33 When STAT4 is activated and phosphorylated, it homodimerises and translocates to the nucleus where it induces expression of hundreds of genes, resulting in production of IFN-γ, T-helper type 1 and 17 differentiation and activation of monocytes.34 Increased STAT4 mRNA expression is associated with increased cardiovascular damage in patients with SLE,30 and several studies on animal models indicate a link between STAT4 and the development of atherosclerosis.35 The mechanism of how smoking leads to increased levels of activated STAT4 is unclear, however, we speculate that epigenetics may constitute the bridge between smoking, genetics and SLE. It is well known that smoking affects both overall DNA methylation and specific gene promotors.36 37 Epigenetic regulation is further believed to play an important role both in cardiovascular biology and in SLE development.36 38 Whether smoking is associated with epigenetic changes in SLE-specific genes, and if such changes are associated with specific manifestations of SLE, deserves further studies.
The analyses of individual SLE risk alleles identified the SLE-risk SNP IL12A to be associated with an increased risk of MI, and that patients in the discovery and replication cohort carrying two alleles of both the IL12A and STAT4 risk SNPs had a more than fivefold and eightfold risk of MI, respectively. The IL12A SNP is located within the fourth intron of the IL12A gene, which encodes the p35 subunit of the IL-12 protein. On binding to its receptor, IL-12 induces phosphorylation of STAT4.34 We believe that the association of the IL12A risk allele, in addition to the STAT4 risk allele, with MI points to the importance of this pathway in the development of the comorbidity. Previous work has demonstrated that JAK-inhibitors efficiently block the increase in pSTAT4 levels, and ameliorate murine lupus as well as its associated vascular dysfunction.19 39 Due to the potential therapeutic strategy of JAK-inhibitors for patients with SLE displaying an altered activity in this particular pathway, we believe that further studies of the effect of this pathway on development of CVE are warranted.
This study’s strength is the large, well-characterised discovery cohort, that results were validated in a second large cohort, and the analysis of healthy control cells, which confirmed that pSTAT4 levels are higher in smokers. In addition, the quality control of genetic data was rigorous, and the patients’ long mean follow-up time of 17 years allowed for many outcome variables such as MI to be recorded. There are, however, some limitations. First, our study is based on retrospective data and we lacked data on the year of smoking cessations. As patients who were past smokers at the last follow-up may have been active smokers at the time of their CVE, we could not analyse previous and current smoking separately. Second, we did not have data on number of pack-years, which may have generated more precise results. Third, the study includes only Scandinavian patients with SLE, and whether the associations are generalisable to patients of other ethnicities needs further investigation.
Conclusion
We demonstrate that smokers carrying the STAT4 risk allele are at an increased risk of MI and nephritis and that the IL12-STAT4 pathway may be important for the development of MI. Our results stress the importance of smoking cessation in SLE and particularly among those carrying this risk allele.
Data availability statement
Data are available on reasonable request. Deidentified patient data and statistical code are available on request with project plan.
Ethics statements
Ethics approval
The study protocol was approved by the Regional Ethical Review Board, Uppsala (DNR 2020−05065) and the local ethics committees.
Acknowledgments
We thank the patients and healthy volunteers for participating, clinicians and researches contributing to the replication cohort and Maija-Leena Eloranta, Cane Yaka, Rezvan Kiani and Karolina Tandre.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors SR, DL and LR designed the study. SR, NH, JKS, AA, PP, CS, AJ, IG, A-CS, AMT, AV, AAB, ES, LR and DL collected the data for the discovery cohort. KL, AMT, AV, ØM and SJ collected the data for the replication cohort. SR, DL and LR analysed the data. SR, DL and LR wrote the manuscript. All authors revised the manuscript critically for important intellectual content and approved the final version of the manuscript.
Funding This work was supported by grants from the Swedish Research Council for Medicine and Health (D0283001), the Swedish Rheumatism Association, King Gustav V’s 80-year Foundation, the Swedish Society of Medicine and the Ingegerd Johansson donation, Åke Wiberg’s foundation, Gustafsson’s foundation, Selander’s foundation, Norsk Revmatikerforbunds Forskningsfond, Pahles legat og Stortuens legat and the Swedish Society for Medical Research (S20-0127).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.