
Abstract
Many cells are capable of maintaining viability in a non-dividing state with minimal metabolism under unfavorable conditions. These are germ cells, adult stem cells, and microorganisms. Unfortunately, a resting state, or dormancy, is possible for tuberculosis bacilli in a latent form of the disease and cancer cells, which may later form secondary tumors (metastases) in different parts of the body. These cells are resistant to therapy that can destroy intensely dividing cells and to the host immune system. A cascade of reactions that allows cells to enter and exit dormancy is triggered by regulatory factors from the microenvironment in niches that harbor the cells. A ratio of forbidding and permitting signals dictates whether the cells become dormant or start proliferation. The only difference between the cell dormancy regulation in normal and pathological conditions is that pathogens, mycobacteria, and cancer cells can influence their own fate by changing their microenvironment. Certain mechanisms of these processes are considered in the review.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Oncology diseases are still among the leading causes of deaths. This is explained by the fact that the disease may recur months, years, or even decades after diagnosis and removal of the primary tumor. The development of secondary, metastatic disease prevents effective treatment and causes many deaths [1]. The mechanism of metastasis is still poorly understood.
Like oncology diseases, tuberculosis caused by Mycobacterium tuberculosis (Mtb) killed more people than any other infection before the pandemic of coronavirus disease. According to the World Health Organization, more than 10 million new cases of tuberculosis infection were diagnosed and more than 1.5 million deaths were due to tuberculosis in 2020 [2]. Tuberculosis is still a major healthcare problem, mostly because infection is latent in a substantial portion of patients and may reactivate several months or several decades after diagnosis [3].
Antibiotic treatment, which is most commonly used in tuberculosis, is similar to anticancer chemotherapy and radiotherapy in that only rapidly proliferating cancer cells and mycobacteria are eliminated. However, undividing or dormant (from the Latin dormire, “to sleep”) cells, which are in a resting state, may survive and cause disease recurrence.
DORMANCY OF Mycobacterium tuberculosis
Entering lung alveoli with inhaled air, Mtb bacilli inhabit macrophages, where they are replicated and evade the host defense mechanisms by various means [4, 5].
Culturable Mtb cells were isolated from patient sputum samples and counted over 14 days of drug therapy. The Mtb count rapidly decreased in the first two days of therapy, and then the elimination rate decreased [6]. The observation indicates that a substantial portion of mycobacteria might be preserved in a less drug-sensitive (dormant) physiological state [7].
Macrophages are known to internalize mycobacteria to produce phagosomes, which have higher levels of acidity, reactive oxygen species, and reactive nitrogen species and lack nutrients. Bacterial cells may be destroyed on exposure to low pH, which arises in phagosomes as a result of proton pumb function, and reactive oxygen and reactive nitrogen species, which are produced by phagocytic NADPH oxidase and NO synthase. Macrophage activation processes in response to bacterial infection are controlled by the host immune system, for instance, the cytokine interferon γ. Microbial metabolism is decelerated at the same time [8, 9]. It was believed that the host immune response determines the formation of dormant Mtb forms, resulting in granulomas. The granuloma is a separate structure and initially consists of innate immunity cells, such as infected macrophages and neutrophils. Then antigen-specific T cells are recruited and infected macrophages activated. The formation of granulomas facilitates dissemination of infected macrophages [10]. As a result, Mtb spreads from dying macrophages to newly recruited ones. A central core is a hallmark of a granuloma and contains infected macrophages surrounded by foamy macrophages, phagocytes of a monocytic origin, and T cells. The core was thought to provide a niche where dormant bacilli persist during latent infection [11]. Granuloma formation was considered as the host’s attempt to localize infection. The resulting infection foci with unfavorable conditions were thought to facilitate the induction and maintenance of the dormant state of bacilli for long periods of time (years or even decades) [12]. It is known now that bacteria play an active role in granuloma formation [11, 13]. Experiments where Danio embryos were infected with Mycobacterium marinum showed that the bacterial 6-kDa virulence protein ESAT-6 (early secretory antigenic target 6) stimulates the epithelium to produce matrix metalloproteinase 9 (MMP9), which alters the structure of the extracellular matrix (ECM), and activates macrophage recruitment to the infection site [14].
In addition, bacilli actively recruit mesenchymal stem cells (MSCs) into the granuloma interior, where MSCs act to suppress T-cell immunity by producing nitric oxide, tumor necrosis factor α (TNF-α), and the chemokine RANTES (regulated on activation, normal T cell expressed and secreted), which is encoded by human CCL5 [4]. Macrophage infection was additionally found to arrest the mycobacterial cell cycle at the G0/G1 transition. The mechanism probably allows mycobacteria to avoid being eliminated by cytotoxic T cells [15]. Products of the mycobacterial gene for the σ factor SigH, which regulates transcription intiation, most likely play a substantial role in modulating the host immune response by affecting chemotaxis- and apoptosis-related processes. The product of this gene was shown to partly decrease expression of β-chemokines, which are the main chemoattractants that attract cells of the immune system to the infection site. This ensures Mtb protection. It is known additionally that the induction of proinflammatory factors during infection is accompanied by the activation of prostaglandin synthesis driven by cyclooxygenase 2, which decreases the intracellular concentration of proapoptogenic arachidonic acid and distorts p53-dependent apoptosis. Stimulation of cyclooxygenase expression by SigH prevents apoptosis in infected cells and promotes the persistence of infection [16].
It was thought earlier that nondividing bacilli survive in the lung within MSC-containing granuloma regions, which are poorly accessible to therapeutics. More recent studies showed that MSCs are also infected by Mtb [17, 18]. Mycobacteria can long remain viable within human bone marrow (BM) stem cells in vitro. In addition, mycobacteria are found in BM stem cells of mice with dormant tuberculosis infection. Finally, viable bacilli were observed in BM stem cells of patients after successful long-term drug therapy. Genes for ATP-dependent ABC transporters are intensely expressed in MSCs to ensure their drug resistance [19]. A study of the effect of Mtb infection on human MSC metabolism showed that mycobacteria induce expression of quiescence markers, the FOXO3a transcription factor (a member of the Forkhead box O3 family), NOTCH1 (a member of the transmembrane protein family), and SOX9 (sex determining region Y-box transcription factor 9), which are characteristic of stem cells. At the same time, they inhibit expression of cell cycle progression markers. In contrast, expression of the proliferation markers S-phase kinase-associated protein 2 (SKP2) and cyclin A1 (CCNA1) is upregulated in infected macrophages. Thus, Mtb occurs in a dormant state in infected MSCs during latent infection [20].
A specific microenvironment of infected human MSCs was found in vitro to facilitate more intense synthesis of mycobacterial Rv1734, which triggers dormancy, and the α-crystallin homolog HspX, which is associated with the cell wall and is responsible for its gradual thickening upon a transition to the nonreplicative dormant state. An accumulation of lipid bodies was additionally observed in MSCs. Expression of the virulence protein ESAT-6 is downregulated. It is of interest that the above changes occur in early infection, as soon as bacilli find their way into MSCs, but never take place in infected macrophages. Early experimental attempts to induce Mtb dormancy in human macrophages and dendritic cells in vitro did not meet with success [21–23].
Normally, MSCs are capable of self-renewal, produce chemokines, cytokines, and growth factors to maintain homeostasis, to ensure wound healing, and to inhibit inflammatory responses [24, 25]. It is known that human MSCs exert direct and indirect antimicrobial activities against Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumonia by secreting the antimicrobial peptide LL-37 [26]. MSCs act to suppress the immune response mediated by T cells, dendritic cells, and macrophages and thus create the microenvironment for drug-resistant Mtb [27]. Mycobacterial infection of MSCs activates the MSC production of exosomes, which are nanovesicles of 30–150 nm in diameter and are produced by many cells. Nanovesicles are encircled by a lipid bilayer and contain a variety of biomolecules, including regulatory proteins, glycans, lipids, RNA, DNA, and metabolites [28, 29]. Exosomes can be captured by other cells, and their contents are internalized and affect the phenotype of the recipient cell; i.e., exosomes act as important mediators of cell-to-cell communication. MSC exosomes are captured by macrophages, thus triggering the inflammatory response and upregulating expression of TNF-α, RANTES, and inducible nitric oxide synthase (iNOS) [30].
Human immunodeficiency virus type 1 (HIV-1) is also capable of persisting in a latent state for decades and reactivating afterwards. It is therefore of interest to study the mechanism of HIV-1 reactivation induced by Mtb infection [31]. Earlier studies showed that the mechanism involves a standard set of regulator factors, such as inflammation, major histocompatibility complex class II (MCH-II) processing, the Toll-like receptor signaling pathway, expression of the CXCR4/CCR5 chemokine receptors, and activation of the regulators of transcription from the HIV LTR. In combined infection, exosomes secreted by Mtb-containing macrophages were observed to reactivate HIV-1 by inducing oxidative stress. A proteome analysis of the exosomes revealed the host signaling molecules that are capable of reactivating HIV-1 by altering redox metabolism with subsequent development of inflammation and the immune response [31].
The following features are now commonly accepted for the dormant Mtb phenotype.
(1) Replication is impossible. The state is defined as “viable but nonculturable” or “nongrowing but metabolically active” [32]. The state is due to expression of many mycobacterial genes. For example, relA, which codes for (p)ppGpp synthase, controls many synthetic processes, including ATP and GTP syntheses, DNA replication, and protein synthesis [16].
(2) Dormancy genes are expressed. There are 4173 genes in the Mtb genome, and more than one-fourth of them are responsible for entering and exiting the dormant state [3, 33]. A proteome analysis was carried out with dormant Mtb cells and cells that are exiting dormancy [34]. The functional aspects of dormancy-associated Mtb genes were reviewed in detail [35]. For example, the DosR (dormancy survival regulator) regulon consists of 48 genes. Their expression is activated in the granuloma in conditions of oxygen deficiency and NO stress and facilitates the transition of intensely replicating bacteria into the dormant state to withstand stress and to ensure long-term survival in the host and reactivation in favorable conditions [35]. Cells occurring in the dormant state for a long period of time (8 months) are characterized by an accumulation of enzymes responsible for cell protection from oxidative stress (superoxide dismutases and catalases-peroxidases), DNA-binding proteins (НupB/Rv2986 and IniB/Rv0341), and protein aggregation-preventing chaperones [36].
(3) Metabolism changes. All metabolic changes are generally associated with inhibition of cell processes and, eventually, an arrest of the cell cycle and division. However, increased synthesis is observed for the enzymes that are atypical to the usual cell state. For example, the glyoxylate pathway is activated to allow Mtb to utilize lipids as the only carbon source, energy metabolism changes to a lower intracellular ATP content and a higher NADН/NAD ratio, and changes occur in lipid and nitrogen metabolism [37].
(4) Cell morphology changes. These changes usually accompany functional reorganization of nonreplicating bacilli and involve their size, shape, and ultrastructure. Sporulation of certain Gram-positive bacteria provides the most brilliant example of such changes and is thought to be a truly dormant state [38]. Although Mtb is known to be incapable of sporulation, altered shapes, including an ovoid shape, were described for dormant bacilli [39]. Forms that have a changed staining pattern and contain lipid bodies were detected for Mtb. Lack of nutrients and oxygen was similarly found to induce a gradual transition from rod-like to ovoid shapes in nonpathogenic M. smegmatis [40]. Scanning electron microscopy showed that dormant Mtb cells change in appearance and substantially decrease in length, while their initial morphology is gradually restored after a restart of aeration [41].
(5) Drug tolerance is acquired. Many molecular and cell mechanisms of tolerance are still poorly understood. It is known that ABC transporters are expressed in MSCs harboring dormant Mtb forms, thus preventing drug accumulation in the cell [19]. Dormant cells do not express catalase-peroxidase, which is necessary for activation of the antituberculosis antibiotic Isoniazid [42]. Cell-wall thickening, which is observed in dormant Mtb cells within MSCs, is possibly accompanied by structural changes that also provide for antibiotic resistance [23].
(6) The state is reversible. A modeling of Mtb dormancy in vitro showed that many factors play a role in the process, including a decrease in oxygen content, an increase in nitric oxide and carbon oxide contents, nitrogen starvation, deficient or excessive metal ions, potassium deficit, lipids as the only carbon source, and antibacterial drugs [5]. The physiology and general transcriptomic profiles of Mtb were studied during reactivation from a hypoxia-induced nonreplicative state, and the results showed a lag phase, in which physiology and metabolism are restored prior to the start of cell division. Reaeration led to repression of dormancy-associated regulons, including DosR, MprA, SigH, SigE, and ClgR and inhibition of lipid utilization metabolic pathways. Regulons and metabolic pathways that are upregulated at the exit from the nonreplicative state were identified and were found to include metal ion transport, DNA recombination and repair, and syntheses of major components of the cell wall [43]. Factors that impair host immunity are known to promote reactivation of dormant Mtb forms. The set includes HIV infection, organ transplantation with immunosuppressive therapy, silicosis, tight contacts with patients with active tuberculosis, use of blockers of proinflammatory TNF-α, chronic kidney failure with hemodialysis, corticosteroid administration, tobacco smoking, and metabolic disorders (diabetes mellitus and rheumatoid arthritis) [44].
CANCER CELL DORMANCY
A concept of tumor dormancy, or quiescence, was proposed as a model where an increase in cell number in the tumor as a result of proliferation is balanced with a decrease in cell number as a result of cell death [45, 46].
Another type of tumor quiescence is dormancy of individual disseminated tumor cells. Their dormancy is induced by factors that trigger growth arrest programs and include signals from the microenvironment and the extracellular matrix of metastatic niches, hypoxia, and various stress factors [47]. Cell dormancy is characterized by minimal proliferation, minimal death, and reversibility. A quiescent state (dormant status) is observed in stem cells of various plant and animal tissues and cancer stem cells [48]. A direct relationship was established between cancer recurrence in the form of metastasis and dormancy of cancer cells [49].
The history of cancer cell dormancy studies was briefly described in a recent review, starting from the 1st century AD [50]. In 2019, the dormant cell transcriptome was described for human myeloma [51]. A time scale was constructed to demonstrate the development of the concepts of dormancy of individual cancer cells and dormancy of the whole tumor. The idea of cancer recurrence formed as early as the 1st century, when Celsus, a Greco-Roman philosopher, was the first to observe that after a tumor was removed and even a scar has formed, the disease may recur and cause death [50]. In 1934, Rupert Willis noted that metastases found in patients without recurrence at the tumor excision site indicate that “neoplastic cells must have lain dormant in the tissues in which they were arrested”; the term “dormancy,” or quiescence, was coined at that time [50].
Convincing evidence that dormant cancer cells can cause the disease to recur after a long quiescent period is provided by the cases where tumors were detected in patients who underwent transplantation of donor organs. Inadvertent transfer of cancer cells through transplants from seemingly healthy donors was first described in a case where a patient had kidney transplantation and was diagnosed with renal squamous cell cancer 8 months after surgery. It was found out later that the donor had lung cancer. In another study, a prostate tumor was found in a heart donor upon postmortem examination, and the recipient was diagnosed with multiple metastasis in the vertebral column, sacral region, and ribs 10 months after heart transplantation. Histochemistry with antibodies against prostate antigens showed that biopsy material from a rib (but not the prostate) of the recipient contained cancer cells characteristic of metastases that prostate cancer cells produce [46, 52, 53].
COMPARISON OF EMBRYONIC DIAPAUSE AND CANCER CELL DORMANCY
Quiescence of cancer and Mtb cells is not a unique phenomenon and seems to employ the universal dormancy mechanisms that work in embryonic cells and adult stem cells of plants and animals [47, 54]. According to current concepts described in a recent review, environmental signals (light, temperature, and nitrate content) control dormancy of embryonic cells in plant seeds by changing the hormone levels [55]. Regulatory gene networks responsible for dormancy of plant stem cells were considered in our review [56]. A quiescent period may occur in the normal embryonic development in animals. The period is known as embryonic diapause in mammals and the Dauer larva period in the nematode Caenorhabditis elegans, and its activation helps to preserve the offspring in adverse environmental conditions [57–59]. Diapause mechanisms can be activated in C. elegans, Fundulus fish, and even mice [60, 61]. For example, the majority of cells proved arrested at G1/G0 in samples isolated from Austrofundulus limnaeus fish embryos in stress due to oxygen starvation [60]. Gene expression profiling showed activation of approximately 100 genes in mouse embryos during diapause. Some of these genes are upregulated in dormant cancer cells as well [58]. For example, a specific regulation of cell cycle inhibitors and chromatin remodeling factors was observed in embryonic cells during diapause and dormant cancer cells. A transition of blastocysts (preimplantation mammalian embryos) to a quiescent state was shown to require sustained expression of pluripotency genes and a decrease in c-Myc protooncogene level, the finding relating dormant cancer cells with dormant blastocysts. The main role of DNA-binding factors of the Myc family is presumably not to maintain pluripotency in itself (in particular, in mouse embryonic stem cells), but rather to suppress early differentiation stages [59]. Expression of the main pluripotency markers Oct4 (octamer-binding transcription factor 4) and Nanog (from Keltic mythological “Tir nan Og”, Land of Eternal Youth) gradually decreases during differentiation. The pluripotency factors are seemingly coexpressed with early differentiation markers at the initial stages of differentiation. The quiescent state of dormant cells with intense expression of NANOG, OCT4, and SOX2 (SRY (sex determining region Y)-box transcription factor 2) coincides with lower c-Myc expression and a general decrease in expression of cell-cycle genes targeted by c-Myc [62, 63]. The immediate microenvironment that is in contact with blastocysts upon their implantation can also provoke hormonal reactivation of diapause cells. For example, expression of the heparin-binding EGF-like growth factor (HB-EGF) was decreased in diapause embryos. An increase in HB-EGF expression led to the establishment of a uterus–embryo connection, thus facilitating embryo implantation and growth [58]. It is of interest that embryonic diapause is an evolutionarily conserved phenomenon and can be induced in diapause-free mammals, such as sheep [61].
In vivo experiments with three-dimensional cultures showed that some cancer cells that survived chemotherapy display a reversible transcription program that is observed in embryo cells during embryonic diapause and is associated with suppression of the Myc-related factors. These cells are inactive in terms of biosynthesis and proliferation. Oppositely, induced Myc activation increased the efficacy of chemotherapy [64].
Inhibition of mTOR (mammalian target of rapamycin) and Myc activities and activation of autophagy factors determine the diapause-like state of breast cancer and colorectal cancer cells. However, a comprehensive transcriptome analysis showed that the quiescent state of cancer cells differs from embryonic diapause and rather resembles the quiescence of embryonic stem cells [65–67].
COMPARISON OF EMBRYONIC AND CANCER CELLS
Many cells form far away from the sites of their future location and function during embryo development and have to migrate over great distances. To start moving, embryonic epithelial cells undergo the so-called epithelial-to-mesenchymal transition (EMT), which destroys intercellular contacts and ensures cell mobility. Cells that have arrived to their destination have to undergo a reverse mesenchymal-to-epithelial transition (MET) to proliferate and differentiate into particular tissue cells [68, 69]. Movements of neural crest cells were studied most comprehensively [70]. Best evidence for EMT occurring in cancer cells is provided by the facts that individual cells break away from the primary tumor and that circulating and disseminated cancer cells display signs of EMT and high epithelial-mesenchymal plasticity [71, 72].
However, EMT is not an all-or-nothing transition. Partial EMT was observed in Drosophila during gastrulation and in amniotes (amphibians and fish) during early migration of groups of neural crest cells, when cells display both epithelial and mesenchymal features. The EMT program has already been triggered in these cells: the cells gradually decrease transcription of the gene for the cell adhesion protein E-cadherin, lose their apical–basal polarity, but still preserve their intercellular contacts and migrate in groups. Cancer cells with a transition phenotype were found to be the most aggressive [73, 74]. The hybrid phenotype allows cancer cells to separate from the primary tumor and to rapidly colonize new regions. Although EMT in embryo development and EMT in pathology have many common features, their differences are also evident. An EMT classification was developed. Type 1 EMT is characteristic of embryo development; type 2 EMT is observed in wound healing, tissue regeneration, and fibrosis of organs; and type 3 EMT occurs in carcinogenesis. Embryonic EMT includes a transition from epithelial to mesenchymal properties, but not induces an inflammatory response in embryos, while such a response is typical of type 2 and type 3 EMTs. Cancer cells undergo type 3 EMT during tumor development. Type 3 EMT ensures not only invasiveness and mobility, but also penetration of separated cells into lymphatic and blood vessels and back into tissues. These intravasation and extravasation phenomena are not observed in fibrosis or embryonic cell migration.
It is of interest that an epithelial phenotype is usually expressed by metastases, suggesting an important role in tumor development for epithelial-mesenchymal plasticity. In other words, there must be an EMT reversion, which was first hypothesized in [75] and was observed in a study of embryo development [76].
Like in embryos, cancer cell EMT coincides with a decrease in expression of EMT-associated transcription factors. MET includes not only a reversion to the epithelial phenotype, but also an increase in proliferation, which is important for the growth of both embryos and secondary tumors [68, 69, 77].
CIRCULATING CANCER CELLS
Hypoxia and nutrient deficiency arise in growing tumors. Subpopulations of aggressive invasive cells are presumably formed and selected under the influence of these factors [78–80]. These cells rapidly change their metabolism and start utilizing alternative energy sources, such as remains of necrotic cells or their lipids, which are internalized via macropinocytosis [81]. Chronic stress leads to a transformation of benign tumors with noninvasive cells to malignant tumors [69]. Genetic [82, 83] and epigenetic changes arise as a result in some cancer cells to allow their migration and invasion into the circulatory and lymphatic systems [79]. The cells undergo EMT, which is characterized by loss of intercellular contacts and apical–basal polarity and acquisition of mobility and invasiveness. The changes are accompanied by dramatic alterations in cell behavior, which are due to upregulation of EMT markers, including transcription factors of various families, such as SNAIL, TWIST, and ZEB [68, 69, 77].
Cancer cell exfoliation and dissemination into lymph nodes and the liver may occur in early development of the primary tumor, before the tumor is exerted or even detected [74].
Whichever the mechanism cells separate from the primary tumor, penetrate into the basement membrane, migrate to blood vessels, enter circulation, and circulate until reaching their destination, where they may produce secondary tumors, or metastases [79, 85]. Although all cells that underwent EMT have higher invasiveness and higher migration activity, only their minor specific subpopulation, known as circulating tumor cells (CTCs), survives in the blood and lymph. CTCs are resistant to chemotherapy and radiotherapy. Their resistance is due to the accumulation of somatic mutations, which abolish their sensitivity to signals of anoikis, which is a form of programmed cell death that is characteristic of normal cells when they detach from their support [86]. Anoikis occurs in cells detached from the extracellular matrix (ECM) and neighbor cells. The factor PHD2, which is also known as hypoxia-inducible factor (HIF) prolyl hydroxylase 2 HIF-PH2, is an oxygen-sensitive molecule and normally inhibits HIF. HIF hydroxylation and degradation are impossible in hypoxic conditions within the tumor, and HIF rapidly accumulates to trigger the adaptive response, where cells become capable of remodeling their metabolism, changing pH of their environment, and activate expression of the vascular endothelial growth factor (VEGF) [79]. HIF upregulates TWIST expression and triggers EMT and SNAIL1 expression, leading to loss of epithelial markers, such as E-cadherin and the epithelial cell adhesion molecule (EpCAM), and acquisition of mesenchymal markers, such as vimentin and N-cadherin [69]. Then the cells penetrate through the basement membrane, enter circulation, and become CTCs.
It should be noted that a similar situation occurs when neural crest cells exfoliate from the neuroepithelium in vertebrate embryos. Cells similarly undergo EMT and lose the cell adhesion proteins, thus acquiring the capability of migration [70, 87, 88].
To exit vessels and enter new niches for colonization and metastasis formation, CTCs lose mesenchymal features and undergo a reverse transition, MET. Two models were advanced to explain how colonization occurs. An EMT/MET model suggests that single CTCs change their phenotype and restore their epithelial properties at a colonization site. According to a collective migration model, circulating cells move in large clusters, varying in the degree of EMT. A cluster may harbor not only epithelial cells, but also fully mesenchymal cells and cells with a hybrid phenotype. Both of the models may be true in principle [73, 79].
CTCs remain in circulation no longer than 1–2 days, and the majority of them die. Cells of the immune system were shown to play a certain role in CTC migration, the set including leukocytes, neutrophils, and macrophages attracted by growth factors and cytokines. Platelets greatly contribute to CTC survival in the blood. Their prometastatic effect is exerted at the physical and molecular levels. Physically, platelets quickly cover CTCs and protect them from mechanical damage in circulation. Tumor cells utilize their surface integrins to bind the platelet adhesion proteins (fibronectin and von Willebrand factor), thus causing platelet aggregation. In addition, platelets increase CTC adhesion to the vascular endothelium through their surface molecules selectins. Protection from the immune system is also ensured by platelets. Namely, platelets secrete the transforming growth factor β (TGF-β), which inactivates natural killer (NK) cells by downregulating expression of the NKG2-D antigen receptor. To further weaken the effect of the immune system, MHC molecules are transferred from the surfaces of granulated platelets to CTCs, thus providing them with platelet identity and disorienting NK cells [78].
CLUSTERS OF CIRCULATING TUMOR CELLS
Apart from single CTCs, CTC clusters are found in the patient blood [78]. CTC clusters are rarer and contain 2–50 cells each. Larger clusters, which are known as tumor microemboli, were observed in patients with lung and breast tumors [89, 90].
A shift in metabolism was observed to occur in CTC clusters and to confer stem cell features on CTCs; CTC clusters grown in vitro form spheroids [79]. This is due to higher secretion of plakoglobin and the transmembrane glycoprotein CD44, which protects cancer stem cells (CSCs). Plakoglobin gene silencing decreased the number of metastases in mice with breast tumors by a factor of 30–40. Overexpression of keratin-14 increased the likelihood of metastasis formation by these cells [91]. In addition, binding sites for the transcription factors OCT4, NANOG, SOX2, and SIN3A, which are characteristic of stem cells, are hypermethylated in CTC clusters, as is typical for embryonic stem cells [92].
Large clusters can pass through microchannels of 50–300 µm in diameter [93]. It was found that a cluster unfolds with disruption of intercellular connections to form a cell chain, which squeezes through capillaries and forms a cluster again at a destination site. Morphological changes occur in the cells in the process. As cells are constricted, their nuclei are deformed and lose their rounded shape to become ellipsoid. These properties seem to ensure a high metastatic potential of CTC clusters [94].
Thus, CTCs are a highly heterogeneous cell subpopulation rather than a single general population. Single CTCs, CTC clusters, and CTCs with stem cell properties [94] are members of the same family of aggressive cells that derive from the primary tumor [95].
Clusters of CTCs may get stuck in small capillaries to produce small thrombi. Fibronectin is consequently activated and interacts with CTC integrins [96]. Resident cells also interact with CTC integrins to stimulate expression of S100, which regulates protein phosphorylation, cell growth, differentiation, and inflammation. Expression of this gene plays an important role in cytoskeletal dynamics and premetastatic niche preparation and triggers CTC settlement in metastatic niches [78, 79, 97].
DISSEMINATED TUMOR CELLS
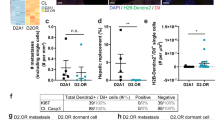
CTCs can leave the blood stream at any place, but can survive and subsequently start proliferating only in a particular organ, which depends on the origin and histological subtype of the primary tumor [98, 99]. It is known that only one out of 40 disseminated tumor cells (DTCs) forms a micrometastasis and that only one out of 100 micrometastases progresses to a macroscopic tumor [48, 50, 100]. Signs of metastatic lesions can develop several years or decades after primary tumor excision. Such clinical observations indicate that DTCs do not always start dividing at once, but can persist in a dormant state. Dormant DTCs survive chemotherapy courses by utilizing the conserved mechanisms of adaptation and survival that work in both embryonic and adult tissues [48, 54, 101].
The ability of dormant cells to slow down their own metabolism is of particular interest. The conditions in which DTCs occur were described as hypoxia, higher acidity, and low glucose [102]. Glycolysis is used to produce energy in rapidly proliferating cancer cells even when the available oxygen is sufficient for mitochondrial function. The phenomenon is known as the Warburg effect [103]. Acidification of the environment blocks glycolysis, and cell metabolism switches to fatty acid oxidation and slows down. The changes allow the cell to stop growing and to survive in adverse conditions.
CANCER STEM CELLS
The concept of cancer stem cells (CSCs) is based on a series of teratocarcinoma studies, which made it possible to assume that malignant neoplasms are a mixture of malignant stem cells, which have a pronounced potential to proliferate and a limited potential to differentiate, and the differentiated, possibly benign, progeny of these malignant cells [104].
Stem cells are cells that have a potential for self-renewal and differentiation into various cell types [105]. The capability of self-renewal is due to expression of telomerase, which prevents telomere shortening during cell division and provides for infinite proliferation [106]. It was believed that embryonic stem cells can differentiate in any adult cells, while adult stem cells can give origin to and fully replace only differentiated cells of the cognate tissue, thus maintaining the tissue and organ structure throughout life [107]. This model of unidirectional development from a pluripotent state to complete differentiation was put to doubt. A hypothesis was advanced that, in certain conditions, single stem cells can acquire a phenotype other than that of cognate tissue cells and that adult cells can be dedifferentiated to stem cells and give origin to cells of various tissues. Takahashi and Yamanaka [108] confirmed the hypothesis by demonstrating that adult differentiated cells are possible to reprogram to induced pluripotent stem cells, which are capable of differentiating to any endodermal, ectodermal, or mesodermal cell lineage. To allow the reprogramming, differentiated cells are engineered to overexpress the genes for stem cell-associated factors known as the Yamanaka factors: c-MYC, Kruppel-like factor 4 (KLF4), Sox2, and Oct-3/4 [109, 110]. The findings confirmed the dedifferentiation and transdifferentiation phenomena, which underlie the modern CSC theory. CSCs were first isolated from acute myeloid leukemia patients in 1997 [111]. It is thought that CSCs originate either from MSCs found in adult tissues or from differentiated cells that are reprogrammed to pluripotent cells via dedifferentiation. Normal stem cells and CSCs are similar in properties, including the capability for self-renewal, unlimited growth, invasiveness, and blocked differentiation [112]. The properties allow CSCs to initiate and sustain tumor growth. The factors SOX2, OCT4, and NANOG are components of the main regulatory chain that ensures the maintenance of self-renewal potential and pluripotency in stem cells [110]. CSCs have high plasticity and can change their phenotype and functions in response to the changes that radiotherapy and chemotherapy induce in the tumor microenvironment.
It should be noted that overlapping signaling pathways regulate the maintenance of stem-cell properties and cell senescence [113]. Key signaling molecules that regulate cell senescence, the cyclin-dependent kinase inhibitors p16 (also known as Ink4a or Arf) and p21(Cip1), and the tumor suppressor p53 (also known as Trp53) play an important role in maintaining cell stemness as well [114]. For example, the senescence-triggering factors p53, p16, and active centromere component Suv39h1 prevent conversion of normal differentiated cells to induced pluripotent cells. Aging cells were recently found to secrete the cyclin-dependent kinase inhibitor p21, which helps to stop proliferation of aging cells and provides a signal to trigger immune surveillance [115].
Aging may directly facilitate CSC plasticity by inducing expression of genes characteristic of stem cells in nonstem tumor cells [116]. CSCs undergo EMT in many cancers, leading to metastasis.
It is often thought that CSCs and DTCs are the same cells [50]. DTCs most likely combine both stem and nonstem tumor cell populations [117]. In fact, single-cell studies showed that DTCs are highly heterogeneous and include groups of aging, quiescent, and intensely proliferating cells [118].
The mechanisms that regulate cell behavior in CSCs are highly similar to those of stem cells, including hematopoietic, muscle, nervous, and hair follicle stem cells. CSCs utilize similar programs of growth arrest, pluripotency, and epigenetic plasticity [48]. Activation of the mTOR pathways was observed to increase the CSC pool in DTCs within a bone marrow metastatic niche (MN) by promoting the release of the growth arrest specific 6 (GAS6) factor from osteoblasts [119]. An increase in p38-MAPK (mitogen-activated protein kinase)-to-ERK (extracellular signal-regulated kinase)–MAPK ratio was shown to induce dormancy in DTCs of various tumors [120] and prostate CSCs [121].
The Notch and Wingless (Wnt) signaling pathways, which control the CSC pool and the balance between quiescence and proliferation of dormant DTCs, can activate the growth of metastases originating from various solid tumors [46, 48, 100]. The pathways apparently activate the cell cycle through the c-Myc protooncogene, while their blockage induces CSC quiescence and tumor dormancy. In addition, c-Myc increases Bmi-1 (polycomb repressor complex 1 component Bmi-1) expression, which regulates the self-renewal capacity of breast CSCs and correlates with disease recurrence [122]. Another example of similarities between dormant DTCs and CSCs is provided by the interleukin 6 (IL-6)‒leukemia inhibitory factor (LIF)–LIF receptor (LIFR) cascade, which is necessary for maintaining dormancy and stemness in breast DTCs within bone marrow (BM). Finally, the mechanical properties of the ECM and the EMT process play a substantial role in the acquisition of stem-cell properties by tumor cells and their metastatic growth [123]. For example, ZEB1, which is a key EMT regulator and is responsible for the cell response to microenvironmental stimuli, such as local inflammation, and TGF-β activate the transcriptional program that helps DTCs to exit dormancy, confers stem-cell properties on DTCs, and ensures their capacity for proliferation [124].
PREMETASTATIC NICHES
While tumor cells are major cells in the primary tumor mass, DTCs enter a hostile aggressive environment. The idea underlies the well-known seed and soil hypothesis, which was advanced in 1889 and states that DTCs will grow only in a favorable environment, like seeds will grow only on fertile soil [125]. This classical concept emphasizes that the properties of the environment (soil) are as important for DTC survival as the properties of DTCs (seeds) are. The concept of a niche (from French niche or German Nische, a nest) was first coined in 1978 when describing immortality of hematopoietic stem cells (HSCs) [126]. It was stated that the stem cell should be considered in association with surrounding cells, which determine its behavior, and that this niche plays an active role in preventing the stem cell from maturation and proliferation. The concept defines the cell niche as a dynamic ecosystem where certain cell types interact with each other to perform certain functions. The function is to maintain the stem-cell properties in the case of the HSC niche. In fact, it was demonstrated that the malignant phenotype of tumor cells can be reversible and depend on their microenvironment [127].
Even before DTC spreading, factors determined by the primary tumor recruit immune cells to the sites where they change the conditions to produce a premetastatic niche (PMN) prepared for being colonized by DTCs [128, 129]. Namely, an immunosuppressive microenvironment is prepared. The microenvironment consists of myeloid suppressor cells, regulatory T cells (Treg), tumor-associated macrophages (TAMs), and tumor-associated neutrophils and does not hinder DTC colonization [130–133]. Systemically spreading exosomes can act as one of the factors [134]. Cells that express VEGF receptor 1 (VEGFR1) and originate from BM were found to settle in the lung before tumor cells arrive. The cells interact with the stroma and form sites for prospective metastatic cells [135]. Exosomes produced by the primary tumor act as key mediators of the process. For example, melanoma-derived exosomes, which contained the c-Met (tyrosine protein kinase) oncogene, were captured by BM cells and ensured their infiltration in the lung, where they contributed to the PMN formation by increasing the permeability of capillaries and facilitating metastasis [134]. In addition to recruiting immune cells to the lung and increasing the capillary permeability, exosomes of breast cancer and melanoma cells, but not pulmonary epithelial cells, were observed to modulate resident pulmonary fibroblasts by inducing S100 expression and thus stimulating cell proliferation and migration [136, 137].
Although the seed and soil (organotropism) concept was advanced more than 120 years ago to describe the propensity of particular tumors to produce metastases in certain organs [125], the mechanism of the phenomenon is still unclear. It is known that the PMN location in the lung, liver, and brain is partly determined by the exosomes of a tumor origin that contain certain integrins, namely, α6β1, α6β4, αvβ5, and αvβ3, which are associated with the ECM molecules laminin and fibronectin, and by certain cells of the target organs [136]. In addition, adhesion molecules and other components exposed on the exosome surface can determine organotropism of DTCs from various tumors. Studies of the exosome role showed that niches are not uniform and that unique properties are characteristic of pulmonary, bone, and liver PMNs. The contents of exosomes originating from various cells and their contribution to PMN formation and organ-specific metastasis were reviewed in [138].
Liver metastasis occurs more often than primary tumors and is characteristic of many tumors, especially gastrointestinal, breast, lung, and pancreatic cancers. Exosomes that are produced by pancreatic cancer cells with integrin αvβ5 expression and carry the macrophage migration inhibitory factor (MIF) were shown to induce TGF-β secretion from Kupffer cells and thus to lead to fibronectin synthesis in stellate cells of the liver [139]. In the lung, small nuclear RNAs contained in exosomes activate expression of MMP9, which remodels the ECM at the prospective niche site, and fibronectin to facilitate neutrophil recruitment [136, 137].
While immune cells are affected by exosomes in the case of lung and liver metastasis, stromal cells, osteoblasts, and osteoclasts act as exosome-modulated targets in bones.
Brain metastasis and the role of exosomes in the formation of PMNs in the brain are poorly understood. It is known that exosomes that contain integrin αvβ3, but not any other integrin, are captured by endothelial cells of the brain [136].
A unique method of co-culturing tumor cells with cells of the target organ was recently used to study the spatiotemporal aspects of breast cancer cell migration and invasion in pulmonary tissue. Changes that tumor cell exosomes induced in fibroblasts of the target organ facilitate the PMN formation, and, in turn, PMNs attract DTCs upon organotropic metastasis [140].
Exosomes of the primary tumor are not the only factor that facilitates dissemination of its cells. MSC exosomes were found to promote breast cancer cell migration upon activation of the Wnt signaling pathway [141].
METASTATIC NICHES
Once colonized by DTCs, a PMN becomes a metastatic niche (MN) and retains the spatial architecture and functional status of the PMN, including growing vessels, their permeability, and immunosuppressive conditions. This specific microenvironment ensures the survival of DTCs with or without stem-cell properties in secondary organs (BM, lymph nodes, the lung, the liver, and the brain) [46, 101].
Changes that occur in niches harboring DTCs and CSCs dictate whether the cells will survive, trigger long-term quiescence programs, or start proliferating.
Bone marrow is possible to consider as a “sacred space” where DTCs and CSCs originating from breast and prostate tumors hide for long periods of time. It is of interest that Notch2 induces tumor cell proliferation in the primary tumor in breast cancer [46, 142] and exerts an opposite effect in BM MNs, causing quiescence and long-term survival of disseminated breast CSCs [143]. The Wnt signaling pathway in its canonical variant regulates cell proliferation and stem-cell properties, but is associated with dormancy of prostate cancer cells in BM niches that utilize the noncanonical ROR2/Siah E3–SIAH2 pathway, which inhibits the canonical Wnt/β-catenin pathway. These opposite effects are possible to explain only by influences from microenvironmental factors in the MN. For instance, several repressor signals were identified, including TGF-β2, bone morphogenetic protein 7 (BMP7), GAS6, LIF, Wnt5α, and chemokine (C-X-C motif) ligand 12 (CXCL12). TGF-β2 is intensely expressed in BM and causes quiescence in CSCs [144] and DTCs, leading to a lower ERK/p38 kinase ratio. The BMP7 transcription factor is secreted by bone stromal cells and induces dormancy in stem-like prostate cancer cells by stimulating expression of the NRDG1 (N-myc downstream regulated 1) metastasis suppressor gene and p38 signaling [121].
GAS6 is secreted by osteoblasts and promotes dormancy of prostate DTCs by activating the MER tyrosine kinase receptor and mTOR [119]. This activation correlates with the appearance of a stem-cell phenotype in DTCs.
LIF is also secreted by the BM stroma and facilitates quiescence of breast CSCs in bones, while loss of LIFR determines the exit from dormancy and progression of metastasis by repressing the genes responsible for maintaining stemness. It is thought that osteoblasts induce DTC dormancy in BM [145], while osteoclasts play a role in exit from dormancy and osteolytic metastasis into bones [146]. These cells are known to physically interact with osteogenic cells, and the osteogenic niche, including osteoblasts, consequently serves as a calcium reservoir for micrometastasis. Breast and prostate cancer cells contain calcium-sensitive receptors, and calcium ions activate the regulatory cascades that inhibit apoptosis and stimulate proliferation in these cells. In addition, calcium ions induce secretion of the parathyroid hormone-related protein (PTHRP), which facilitates further resorption of bone tissue and the release of calcium. Calcium ions can additionally serve as a chemoattractant for breast cancer cells and determine their localization in bones [147]. Age-related phenotypic changes and senescence of osteoblasts increase metastasis [148].
It is probably no accident that the normal HSC niche provides a site for DTCs to stop in BM [119]. The role of Notch2 was established. To induce dormancy of breast DTCs in BM, Notch2 mimics the internal cell mechanisms that are responsible for HSC quiescence [143]. As numerous studies demonstrated, the specific factors BMP7, TGF-β2, BMP4, and GAS6, which ensure quiescence of normal stem cells, induce DTC dormancy in pulmonary and BM niches [121, 149, 150]. Strict control of HSC dormancy in BM suggests similar dormancy mechanisms for HSCs and DTCs [150–152]. Moreover, dormant breast DTCs express the same stem-cell genes that HSCs do. However, these DTCs additionally have certain embryonic stem cell features, such as higher-level expression of the pluripotency-associated genes NR2F1, SOX9, SOX2, OCT4 (POU5F1), and NANOG, which provides DTCs with higher plasticity and renders their complete differentiation less likely [54].
Brain metastases are most often observed at sites of contacts between gray and white matters and at the boundaries between territories of neighbor blood vessels. The blood velocity in these regions is low enough to allow CTCs to penetrate the blood–brain barrier and to exit the blood stream. However, tumor cells inevitably die if moving away from vessel walls. Metastases of malignant tumors are found, in fact, along the outer walls of cerebral blood vessels. Neuronal stem cells similarly occur in the same perivascular space, and their maintenance and differentiation in normal neurogenesis are sustained by growth factors, such as VEGF. The same factors ensure the growth of cancer cells. It is of interest to note that cancer cells are sensitive to normal neuronal stem cell signaling and, in turn, release BMP-2 and thus force neuronal stem cells to differentiate into astrocytes for their own purposes because, in contrast to astrocytes, neuronal stem cells are capable of preventing tumor growth [154, 155].
As is known, niches of normal adult stem cells are protected from the immune system [156], and this is favorable for dormant DTCs. Lower antigenicity is characteristic of both normal stem cells and dormant DTCs. The fact agrees with the assumption that evasion from immune surveillance is their common feature. Expression of FBXW7 ubiquitin ligase is possibly another common feature. FBXW7 inhibition distorts quiescence in lymphoma stem cells and lung adenocarcinoma cells and facilitates resuscitation and, consequently, chemotherapeutic elimination of dormant breast DTCs [48, 157, 158].
The lung provides another shelter to dormant DTCs and CSCs of many tumors. Like in BM, BMP4 (a member of the TGF-β family) is secreted by resident cells and prevents self-renewal of breast cancer cells. DTCs are capable of interacting with the ECM through their integrin receptors when colonizing the lung, and this capability also facilitates their dormancy and survival [46, 159].
Perivascular niche. Because blood and lymphatic vessels are used by DTCs to leave the primary tumor, the vascular endothelium is the first barrier that DTCs encounter in new environments [78]. It is not surprising that breast, lung, and melanoma DTCs were tightly associated with the vascular basement membrane in various experimental models. Their microenvironment is known as the perivascular niche (PVN) and is involved in normal tissue differentiation and development. PVN contains oxygen, nutrients, and paracrine factors to support DTC and CSC proliferation [46, 160]. Stem cells of various organs are known to reside in PVNs at the ends of capillaries, where their growth is regulated and maintained by factors produced in the vascular endothelium. The maintenance of the stem cell state agrees with the induction and maintenance of DTC dormancy in PVNs. BM PVNs induce long-term dormancy of DTCs and protects them from chemotherapy by ensuring their interactions through integrins with the von Willebrand factor and the integrin ligand VCAM1 [161]. Distortion of these contacts with the use of integrin-binding antibodies led to DTC resuscitation and chemotherapeutic elimination.
It is of interest that different loci are occupied in perivascular regions by dormant and proliferating breast cancer cells. Proliferating cells are closer to the bone surface in macrometastases, while dormant cells are closer to perisinusoidal venules. The venules express the adhesion molecules E-selectins, which are characteristic of vascular cells in inflammation, and their expression facilitates tumor cell penetration into BM. Stromal cell-derived factor 1 (SDF-1), which is characteristic of stromal cells, is also expressed to ensure cell fixation in the niche by interacting with C-X-C chemokine receptor type 4 (CXCR4) [162].
MSCs of various human and animal organs and tissues, including the umbilical cord, and their roles in DTC dormancy and reactivation was reviewed in detail in [163].
FACTORS AFFECTING DORMANCY MAINTENANCE AND EXIT FROM DORMANCY IN DTCs
Role of the Microenvironment
The concept of reversibility of the malignant phenotype of tumor cells suggests that tumor cells are capable of remodeling their epigenetic programs in a certain microenvironment, such as the embryonic one [164]. An extract of axolotl embryos was observed to suppress the growth of breast tumor cells and to induce their dormancy, including activation of expression of the cell-cycle inhibitor p27 and inhibition of phosphorylation of the tumor suppressor retinoblastoma protein (RB) and key signaling pathways of cell proliferation [165]. The findings indicate that homeostasis of the microenvironment in secondary target organs apparently maintains tumor cell dormancy in a manner similar to that utilized in niches of quiescent adult stem cells.
While conditions created in PMNs facilitate DTC fixation and survival in the dormant state, any change in MN triggers the cell cycle in dormant DTCs and results in metastatic growth [48, 54, 100]. Various MN components were tested for the ability to convert dormant DTCs from quiescence (like in stem cells) to the state of self-renewal and proliferation. The conversion was due to EMT induction in DTCs and their acquisition of stem-cell properties in many cases [109, 166].
The extracellular matrix (ECM), which is defined as the noncellular component of a dormant DTC niche, is a quickly changing and physiologically active structure that surrounds cells and plays an important role in cell–cell interactions. ECM is highly structured in embryo development and tissue homeostasis, but becomes unstructured in metastasis. ECM provides a background in which physical and chemical factors create the conditions for quiescence or proliferation for dormant cells. The ERK/p38MAPK kinase activity ratio upon binding with ECM was shown to act as a molecular switch of dormancy. The higher the ratio, the lower is the proportion of dormant cells. Thus, ECM determines dormancy of DTCs by affecting their survival and proliferation [47, 48, 167–170].
In the state of long-term dormancy, cells are tightly associated with the rigid matrix via integrin α5β1 adhesion and Rho-associated kinase (ROCK)-generated tension. Moreover, the possibility to exit dormancy is most likely due to fibronectin 1 degradation in ECM by matrix metalloproteases and a weakening of the above association [118].
In addition, the urokinase plasminogen activator surface receptor (uPAR), which is a membrane glycoprotein and ensures cell interactions with ECM, was shown to bind with integrins of squamous cell head-and-neck cancer cells, leading to inactivation of their mitogenic cascades and thus inducing dormancy [168, 171].
Osteopontin and tenascin C are components of PMN ECM and also regulate the CSC survival, self-renewal, and resuscitation by regulating expression of the Wnt, Nanog, and Oct4 (POU5F1) transcription factors [46].
It is important that, apart from the effects of cellular MN structures on the DTC state, resident DTCs can themselves create favorable conditions for their growth or quiescence. For example, breast DTCs were shown to activate stromal cells located in their immediate vicinity to release periostin and tenascin C. In turn, these activate the Wnt, Nanog, and Oct4 stem-cell proliferative pathways in dormant DTCs, leading to metastatic growth [100, 172, 173]. Interstingly, periostin secreted by newly growing blood capillaries in PMNs acts together with TGF-β1 to induce proliferation of dormant breast tumor cells, while thrombospodin 1 secreted by preexisting normal vessels leads to dormancy of these cells [174].
Roles of Autophagy and Apoptosis
It is known that autophagy may be induced by distorted adhesion of DTCs to ECM. Autophagy is an evolutionarily conserved mechanism that is triggered to maintain energy balance in cells exposed to metabolic stress and consists in degradation of damaged proteins, organelles, and part of the cytosol. In fact, autophagy was shown to regulate the CSC survival [175]. Autophagy is tightly associated with stress signals and metabolic changes. Autophagy is characteristic of dormant cancer cells to a greater extent than of their proliferating counterparts and allows them to survive stress in a quiescent state [157]. Inhibition of autophagy leads to exit from the dormant state [120, 158, 176]. Types and fine mechanisms of autophagy were reviewed in detail [177].
Proteins of the Bcl-2 mitochondrial apoptosis inhibitor family are known to regulate apoptosis. Bcl-2 overexpression is characteristic of breast and prostate CSCs. In the latter case, Bcl-2 overexpression is due to upregulation of Notch and the Hedgehog morphogenetic factor, which are responsible for CSC self-renewal and differentiation [178]. The canonical Wnt/β-catenin signaling pathway provides another example of the relationships between apoptosis and CSC maintenance. Inhibition of the pathway leads to cell exit from the dormant state [179, 180].
Role of Neoangiogenesis
Processes related to the formation of new vessels are also involved in remodeling the microenvironment of dormant DTCs. Dormant cells reside in PMN due to interactions with its endothelial cells through thrombospondin 1, and new growing vessels stimulate dormant cell reactivation and tumor growth by producing periostin and TGF-β1 [174]. The Coco membrane factor, which acts as an inhibitor of the BMP pathway (members of the TGF-β family), facilitates reactivation of breast cancer cells in the lung [149]. The mechanism is apparently possible to consider as a universal organ-independent mechanism of dormant cell reactivation [50].
Several factors are known to regulate angiogenesis, including the fibroblast growth factor (FGF), PDGF, VEGF, and-IL-8 [181].
It is known also what changes occur in the tumor cell that undergoes the so-called angiogenic switch [182]: thrombospondin (angiogenesis inhibitor) expression decreases, while upregulation is observed for genes that have not been related to tumor dormancy as of yet, including ESM1 (endothelial cell specific molecule 1), TIMP3 (tissue inhibitor of metalloproteinase 5'-ectonucleotidase), EGFR (epidermal growth factor receptor), IGF1R (insulin-like growth factor 1 receptor), PI3K (phosphatidylinositol 3-kinase, a signaling pathway component), EphA5 (ephrin A5 receptor), and H2BK (histone Н2BK) [46, 183]. A balance between the angiogenic switch and dormancy is finely regulated by microenvironmental factors, including proangiogenic VEGF, PDGF, antiangiogenic thrombospondin 1, angiostatin, and endostatin [184].
Role of the Inflammatory Response and Fibrosis
The processes that occur in chronic inflammation and, in particular, promote the activation of metastatic growth were reviewed in detail [185]. Chronic inflammation leads to immunosuppression in the microenvironment of dormant tumor cells as a result of recruitment of M2 macrophages, Treg cells, myeloid suppressor cells, and other cells and cytokines. This is accompanied by oncogene activation, stimulation of cell proliferation, and metastasis. The process is facilitated by various epigenetic alterations, including DNA methylation, histone modification, chromatin remodeling, and synthesis of noncoding RNAs. For example, the inflammatory response induced by renal tumor cells was observed to promote metastatic growth in the lung. Epigenetic chromatin remodeling led to transcriptional activation of inflammation-associated genes in the process. Inflammation is often accompanied by recruitment of tumor-associated fibroblasts, which are responsible for the accumulation of collagen and various ECM components that facilitate cell proliferation and angiogenesis. The fibroblasts additionally produce a variety of cytokines and chemokines, including osteopontin, CXCL1, CXCL2, CXCL12, CXCL13, IL-6, IL-1β, and CCL-5, which change the behavior of surrounding epithelial cells and promote tumor cell proliferation. Induction of the inflammatory reaction in the lungs with bacterial polysaccharides or tobacco smoke led to EMT induction, ZEB1 expression, and reactivation in dormant DTCs [186]. A study performed to understand the relationship between damage to normal tissues, inflammation, and tumor growth showed that neutrophils trigger the Notch-dependent signaling pathway of cell proliferation to ensure regeneration of damaged tissue. Neutrophils are therefore also responsible for creating favorable conditions that allow DTCs to acquire stem-cell properties and to subsequently start metastatic growth [187].
Distortions of wound healing result in fibrosis. The formation of fibrosis-like foci enriched in type 1 collagen and fibronectin creates the environment that is permissive for reactivation of dormant DTCs. Collagen and fibronectin were shown to trigger the integrin 1β (Intβ1) signaling pathway by activating focal adhesion kinase (FAK). Then ERK kinase is activated and, in turn, activates myosin light-chain kinase (MLCK). As a result, F-actin stress fibrils form, and tumor cells pass from the quiescent state to proliferation. Inhibition of MLCK activation or Intβ1 expression prevented DTC reactivation both in vitro and in vivo [53, 188].
The hypoxia-induced multifunctional factor LOXL2 (lysyl oxidase-like 2) was observed to cause posttranslational crosslinking of fibroblast-produced type 1 collagen in pulmonary fibrosis-like foci. This increased the mechanical rigidity of ECM and created favorable conditions for tumor cell colonization and subsequent metastatic growth. Inhibition of LOXL2 prevented colonization and metastasis [189]. It is of interest that, apart from playing a role in the formation of fibrosis foci outside the cell, intracellular LOXL2 induces EMT, promotes invasiveness, and confers stem-cell properties on cells, leading to their transition from dormancy to proliferation and metastatic growth [190]. When the inflammatory response in the lung was modeled in mice via inhalation of tobacco smoke or nasal administration of a polysaccharide solution, neutrophil extracellular traps (NETs) were observed to form in the extracellular space from chromatin regions associated with proteolytic enzymes that neutralize foreign substances. Chromatin-associated proteases remodeled laminin, which accumulates in inflammation, in such a manner that laminin acted to activate integrin α3β1 on the tumor cell membrane, and the integrin triggered the FAK/ERK/MLCK/YAP in the cell. The YAP (yes-associated protein) transcription factor activated the genes that are responsible for proliferation of dormant breast cancer cells and metastasis in the lung [191].
Cancer and tuberculosis are highly similar immunologically. However, tuberculosis studies focus predominantly on the means to prevent the disease, while cancer studies are mostly aimed at eliminating the existing disease by activating the patient’s immune system [192]. Administration of the common anti-tuberculosis BCG (bacillus Calmette–Guérin) vaccine is one of the most effective methods to treat bladder cancer at early stages. Apart from inducing the immune response, BCG stimulates NET formation. However, in vitro experiments and studies with a mouse model of bladder cancer showed that incubation of cancer cells with NET preparations decreased their mobility, arrested the cell cycle, and induced apoptosis, thus exerting a dose-dependent cytotoxic effect [193]. To explain the discrepancy, it is possible to assume that the NET mechanisms of action differ depending on the NET concentration and cell type. The NET concentration is possibly low in niches of dormant cancer cells, and the activating effect of NETs is mediated by ECM modifications. A cytotoxic effect is exerted when NETs are used to treat primary tumor cells in vitro.
It is of interest to note that surgery-induced systemic inflammation stimulates proliferation of dormant immunogenic DTCs in various organs, while administration of nonsteroidal anti-inflammatory drugs prior to surgery prevents DTC reactivation in the lung [194].
Role of Immune Control
Two mechanisms are possible for tumor cell dormancy. One is a proliferation arrest, when the cell cannot divide. The other is a balance between T-cell cytotoxicity and angiogenesis in tumor cell niches and subsequent tumor cell reactivation [167]. When activity of the immune system prevails, tumor cells are eliminated. When active angiogenesis prevails, tumor cells proliferate. Therefore, tumor cells remain dormant when the two processes are balanced. Analyses of organ transplantation results shed the first light on the role that the immune system plays in metastasis. It is known that metastasis usually becomes detectable 20–35 months after the removal of the primary tumor [195]. The period between transplantation and possible detection of metastasis in the recipient is shorter, 3–36 months, depending on the tumor type and the organ transplanted. The observations suggest immune control over latent neoplasms and active growth of tumor cells in conditions of drug-induced immunosuppression, which is essential for preventing transplant rejection and abolishes the immune control [196]. However, organ transplantation is a traumatic intervention and causes the inflammatory response, which may also activate dormant tumor cells.
Apart from promoting PMN colonization with CTCs, cells of the immune system affect the DTC fate to determine whether DTCs will persist in the dormant state or will be eliminated upon attempted reactivation [79, 197]. Treg cells were shown to promote DTC dormancy. Treg; cells release adenosine, which protects quiescent cells from oxidative stress [198]. In addition, dormant cells are protected from T cells, while proliferating cells are sensitive to their cytotoxic effect [197].
Studies with a mouse model showed that NK cells selectively increase in number in the immediate surrounding of dormant breast cancer cells in the liver [199]. Adjuvant immunotherapy based on IL-15 increased the NK cell number, thus facilitating the maintenance of cancer cell dormancy through interferon-γ signals, preventing the development of metastases in the liver, and increasing the lifespan. Exit from dormancy and metastatic growth were due to a substantial reduction of the NK cell area and a competitive accumulation of activated stellate cells. The chemokine CXCL12 secreted by stellate cells was found to block NK cell activity by binding to their CXCR4. The findings demonstrate that the proportion of NK and stellate cells acts as a major switch of tumor cell dormancy in the liver [199].
It is of interest that coexistence of growing tumors and T cells was described rather long ago [200]. Intracellular and external factors that render T cells incapable of eliminating tumors cells are known now [201]. The respective state is known as T-cell exhaustion and is characterized by gradual loss of function up to a complete disappearance of T cells. A similar phenomenon is observed when antigenic load remains high for a long period of time in chronic virus infections and oncology diseases. RNA sequencing in single cells from tumor, peritumoral, and blood samples of 316 patients with 21 tumor types made it possible to conclude that transcriptional programs and the state of T cells depend to a substantial extent not only on the tumor type, but also on the tumor microenvironment, including TGF-β, TNF-α, interferons, interleukins, and T-helper and Treg cells [202].
CONCLUSIONS
Dormancy is a reversible nonproliferative quiescent state and is characteristic of many animal, plant, and microbial cells exposed to unfavorable conditions. In particular, dormancy is known for adult stem cells; embryonic cells; DTCs, which produce distant metastases after long periods of time; and Mtb in latent infection. The main regulatory pathways that establish dormancy are universal according to published data. For example, hypoxia triggers expression of the transcription factors that stop cell proliferation, and nutrient deficiency decreases cell metabolic activity or switches metabolism from glucose utilization to nonstandard pathways, such as utilization of lipids as the only carbon source. These prohibiting signals are characteristic of normal stem cell niches. PMNs form in response to signals from the primary tumor before being colonized with DTCs and occur close to or within normal stem cell niches in the lung, the liver and BM. Cells remain undetectable by the immune system and are protected from the cytotoxic effect of chemotherapeutics in these niches. The activity of mycobacteria as true intracellular parasites is regulated by the signals that act in infected cells. Namely, Mtb cells become dormant only in MSCs. Dormant mycobacteria are similarly protected from immune cells and drugs. It is important that macrophages, which are a classical Mtb host, cannot ensure Mtb cell dormancy. This fact determines the efficacy of drug therapy in acute disease.
Normal stem cells exit dormancy and start proliferating in response to permissive signals, which may be elicited by any changes in the body, for example, traumatic impairment of cell integrity. Growth factors are released in the process, inflammation develops, and new blood vessels grow. Assuming that CSCs constitute a fraction of DTCs, their reactivation will naturally take place in response to microenvironmental changes induced by similar factors. This provides additional evidence for the universal character of cell dormancy-regulating mechanisms. There are still certain specifics. Both Mtb as an intracellular parasite and tumor cells as pathogens occurring in the body are capable of remodeling their microenvironment to trigger parallel processes of self-reactivation. For example, mycobacteria induce infected MSCs to produce exosomes, which are captured by macrophages and trigger the inflammatory response and disease development. Tumor cells induce stromal cells of their niche to release the matrix factors that activate the regulatory pathways ensuring tumor cell proliferation.
REFERENCES
Dillekås H., Rogers M.S., Straume O. 2019. Are 90% of deaths from cancer caused by metastases? Cancer Med. 8, 5574–5576.
Global tuberculosis report 2021. Geneva: World Health Organization. 2021. Licence: CC BY-NC-SA 3.0 IGO.
Peterson E.J.R., Abidi A.A., Arrieta-Ortiz M.L., Aguilar B., Yurkovich J.T., Kaur A., Pan M., Srinivas V., Shmulevich I., Baliga N.S. 2020. Intricate genetic programs controlling dormancy in Mycobacterium tuberculosis. Cell Rep. 31, 107577.
Khan A., Hunter R.L., Jagannath C. 2016. Emerging role of mesenchymal stem cells during tuberculosis: the fifth element in cell mediated immunity. 101, 45–52.
Batyrshina Y.R., Schwartz Y.S. 2019. Modeling of Mycobacterium tuberculosis dormancy in bacterial cultures. Tuberculosis (Edinb.). 117, 7–17.
Jindani A., Aber V.R., Edwards E.A., Mitchison D.A. 1980. The early bactericidal activity of drugs in patients with pulmonary tuberculosis. Am. Rev. Respir. Dis. 121, 939–949.
Wayne LG. 1994. Dormancy of Mycobacterium tuberculosis and latency of disease. Eur. J. Clin. Microbiol. Infect. Dis. 13, 908–914.
Schubert O.T., Ludwig C., Kogadeeva M., Zimmermann M., Rosenberger G., Gengenbacher M., Gillet L.C., Collins B.C., Röst H.L., Kaufmann S.H., Sauer U., Aebersold R. 2015. Absolute proteome composition and dynamics during dormancy and resuscitation of Mycobacterium tuberculosis. Cell Host Microbe. 18, 96–108.
Gengenbacher M., Kaufmann S.H.E. 2012. Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532.
Davis J.M., Ramakrishnan L. 2009. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. 136, 37–49.
Kundu M., Basu J. 2021. Applications of transcriptomics and proteomics for understanding dormancy and resuscitation in Mycobacterium tuberculosis. Front. Microbiol. 12, 642487.
Mayito J., Andia I., Belay M., Jolliffe D.A., Kateete D.P., Reece S.T., Martineau A.R. 2019. Anatomic and cellular niches for Mycobacterium tuberculosis in latent tuberculosis infection. J. Infect. Dis. 219, 685–694.
Paige C., Bishai W.R. 2010. Penitentiary or penthouse condo: the Tuberculous granuloma from the microbe׳s point of view. Cell. Microbiol. 12, 301–309.
Volkman H.E., Pozos T.C., Zheng J., Davis J.M., Rawls J.F., Ramakrishnan L. 2010. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. 327, 466–469.
Cumming B.M., Rahman M.A., Lamprecht D.A., Rohde K.H., Saini V., Adamson J.H., Russell D.G., Steyn A.J.C. 2017. Mycobacterium tuberculosis arrests host cycle at the G1/S transition to establish long term infection. PLoS Pathog. 13, e1006389.
Dutta N.K., Mehra S., Martinez A.N., Alvarez X., Renner N.A., Morici L.A., Pahar B., Maclean A.G., Lackner A.A., Kaushal D. 2012. The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS One. 7, e28958.
Raghuvanshi S., Sharma P., Singh S., Van Kaer L., Das G. 2010. Mycobacterium tuberculosis evades host immunity by recruiting mesenchymal stem cells. Proc. Natl. Acad. Sci. U. S. A. 107, 21653–21658.
Das B., Kashino S.S., Pulu I., Kalita D., Swami V., Yeger H., Felsher D.W., Campos-Neto A. 2013. CD271+ bone marrow mesenchymal stem cells may provide a niche for dormant Mycobacterium tuberculosis. Sci. Transl. Med. 5, 170ra13.
Khan A., Mann L., Papanna R., Lyu M., Singh C.R., Olson S., Eissa N.T., Cirillo J., Das G., Hunter R.L., Jagannath C. 2017. Mesenchymal stem cells internalize Mycobacterium tuberculosis through scavenger receptors and restrict bacterial growth through autophagy. Sci. Rep. 7, 15010.
Fatima S., Kamble S.S., Dwivedi V.P., Bhattacharya D., Kumar S., Ranganathan A., Van Kaer L., Mohanty S., Das G. 2020. Mycobacterium tuberculosis programs mesenchymal stem cells to establish dormancy and persistence. J. Clin. Invest. 130, 655–661.
Garhyan J., Bhuyan S., Pulu I., Kalita D., Das B., Bhatnagar R. 2015. Preclinical and clinical evidence of Mycobacterium tuberculosis persistence in the hypoxic niche of bone marrow mesenchymal stem cells after therapy. Am. J. Pathol. 185, 1924–1934.
Tornack J., Reece S.T., Bauer W.M., Vogelzang A., Bandermann S., Zedler U., Stingl G., Kaufmann S.H., Melchers F. 2017. Human and mouse hematopoietic stem cells are a depot for dormant Mycobacterium tuberculosis. PLoS One. 12, e0169119.
Singh V.K., Mishra A., Bark S., Mani A., Subbian S., Hunter R.L., Jagannath C., Khan A. 2020. Human mesenchymal stem cell based intracellular dormancy model of Mycobacterium tuberculosis. Microbes Infect. 22, 423–431.
Kim S., Kim T.M. 2019. Generation of mesenchymal stem-like cells for producing extracellular vesicles. World J. Stem Cells. 11, 270–280.
Wang L.T., Ting C.H., Yen M.L., Liu K.J., Sytwu H.K., Wu K.K., Yen B.L. 2016. Human mesenchymal stem cells (MSCs) for treatment towards immune- and inflammation-mediated diseases: review of current clinical trials. J. Biomed. Sci. 23, 76.
Chow L., Johnson V., Impastato R., Coy J., Strumpf A., Dow S. 2020. Antibacterial activity of human mesenchymal stem cells mediated directly by constitutively secreted factors and indirectly by activation of innate immune effector cells. Stem Cells Transl. Med. 9, 235–249.
Khan A., Jagannath C. 2019. Interactions of Mycobacterium tuberculosis with human mesenchymal stem cells. In Tuberculosis Host–Pathogen Interactions. Cirillo J., Kong Y., Eds. Cham: Springer, 95–111.
Wortzel I., Dror S., Kenific C.M., Lyden D. 2019. Exosome-mediated metastasis: communication from a distance. Dev. Cell. 49, 347–360.
Mathieu M., Martin-Jaular L., Lavieu G., Théry C. 2019. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication Nat. Cell Biol. 21, 9–17.
Liu M., Wang Z., Ren S., Zhao H. 2021. Exosomes derived from Mycobacterium tuberculosis-infected MSCs induce a pro-inflammatory response of macrophages. Aging (Albany NY). 13, 11595–11609.
Tyagi P., Pal V. K., Agrawal R., Singh S., Srinivasan S., Singh A. 2020. Mycobacterium tuberculosis reactivates HIV-1 via exosome-mediated resetting of cellular redox potential and bioenergetics. mBio. 11, e03293-19.
Li L., Mendis N., Trigui H., Oliver J.D., Faucher S.P. 2014. The importance of the viable but non-culturable state in human bacterial pathogens. Front. Microbiol. 5, 258.
Salina E.G., Mollenkopf H.J., Kaufmann S.H.E., Kaprelyants A.S. 2009. M. tuberculosis gene 749 expression during transition to the “non-culturable” state. Acta Nat. 1, 73–77.
Gopinath V., Raghunandanan S., Gomez R.L., Jose L., Surendran A., Ramachandran R., Pushparajan A.R., Mundayoor S., Jaleel A., Kumar R.A. 2015. Profiling the proteome of Mycobacterium tuberculosis during dormancy and reactivation. Mol. Cell. Proteomics. 14, 2160–2176.
Joshi H., Kandari D., Bhatnagar R. 2021. Insights into the molecular determinants involved in Mycobacterium tuberculosis persistence and their therapeutic implications. Virulence. 12, 2721–2749.
Trutneva K.A., Shleeva M.O., Demina G.R., Vostroknutova G.N., Kaprelyans A.S. 2020. One-year old dormant, “non-culturable” Mycobacterium tuberculosis preserves significantly diverse protein profile. Front. Cell Infect. Microbiol. 10, 26.
Chang D.P.S., Guan X.L. 2021. Metabolic versatility of Mycobacterium tuberculosis during infection and dormancy. Metabolites. 11, 88.
Rittershaus E.S.C., Baek S.H., Sassetti C.M. 2013. The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe. 13, 643–651.
Shleeva M.O., Kudykina Y.K., Vostroknutova G.N., Suzina N.E., Mulyukin A.L., Kaprelyants A.S. 2011. Dormant ovoid cells of Mycobacterium tuberculosis are formed in response to gradual external acidification. Tuberculosis. 91, 146–154.
Anuchin A.M., Mulyukin A.L., Suzina N.E., Duda V.I., El-Registan G.I., Kaprelyants A.S. 2009. Dormant forms of Mycobacterium smegmatis with distinct morphology. Microbiology. 155, 1071–1079.
Raghunandanan S., Jose L., Gopinath V., Kumar R.A. 2019. Comparative label-free lipidomic analysis of Mycobacterium tuberculosis during dormancy and reactivation. Sci. Rep. 9, 3660.
Karakousis P.C., Williams E.P., Bishai W.R. 2008. Altered expression of isoniazid-regulated genes in drug-treated dormant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 61, 323–331.
Du P., Sohaskey C.D., Shi L. 2016. Transcriptional and physiological changes during Mycobacterium tuberculosis reactivation from non-replicating persistence. Front. Microbiol. 7, 1346.
Ai J.-W., Ruan Q.-L., Liu Q.-H., Zhang W.-H. 2016. Updates on the risk factors for latent tuberculosis reactivation and their managements. Emerg. Microbes Infect. 5, e10.
Endo H., Inoue M. 2019. Dormancy in cancer. Cancer Sci. 110, 474–480.
Sistigu A., Musella M., Galassi C., Vitale I., De Maria R. 2020. Tuning cancer fate: tumor microenvironment’s role in cancer stem cell quiescence and reawakening. Front. Immunol. 11, 2166.
Aguirre-Ghiso J.A. 2007. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer. 7, 834–846.
Risson E., Nobre A.R., Maguer-Satta V., Aguirre-Ghiso J.A. 2020. The current paradigm and challenges ahead for the dormancy of disseminated tumor cells. Nat. Cancer. 1, 672–680.
Boire A., Coffelt S.B., Quezada S.A., Vander Heiden M.G., Weeraratna A.T. 2019. Tumour dormancy and reawakening: opportunities and challenges. Trends Cancer. 5, 762–765.
Phan T.G., Croucher P.I. 2020. The dormant cancer cell life cycle. Nat. Rev. Cancer. 20, 398–411.
Khoo W.H., Ledergor G., Weiner A., Roden D.L., Terry R.L., McDonald M.M., Chai R.C., De Veirman K., Owen K.L., Opperman K.S., Vandyke K., Clark J.R., Seckinger A., Kovacic N., Nguyen A., Mohanty S.T., Pettitt J.A., Xiao Y., Corr A.P., Seeliger C., Novotny M., Lasken R.S., Nguyen T.V., Oyajobi B.O., Aftab D., Swarbrick A., Parker B., Hewett D.R., Hose D., Vanderkerken K., Zannettino A.C.W., Amit I., Phan T.G., Croucher P.I. 2019. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood. 134, 30–43.
Loh E., Couch F.J., Hendricksen C., Farid L., Kelly P.F., Acker M.A., Tomaszewski J.E., Malkowicz S.B., Weber B.L. 1997. Development of donor-derived prostate cancer in a recipient following orthotopic heart transplantation. JAMA. 277, 133–137.
Sauer S., Reed D.R., Ihnat M., Hurst R.E., Warshawsky D., Barkan D. 2021. Innovative approaches in the battle against cancer recurrence: novel strategies to combat dormant disseminated tumor cells. Front. Oncol. 11, 659963.
Aguirre-Ghiso J.A., Sosa M.S. 2018. Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis. Ann. Rev. Cancer Biol. 2, 377–393.
Sano N., Marion-Poll A. 2021. ABA metabolism and homeostasis in seed dormancy and germination. Int. J. Mol. Sci. 22, 5069.
Voronina A.S., Pshennikova E.S. 2020. Plant stem cells. Mol. Biol. (Moscow). 54, 163–177.
Fukuyama M., Rougvie A.E., Rothman J.H. 2006. C. elegans DAF-18/PTEN mediates nutrient-dependent arrest of cell cycle and growth in the germline. Curr. Biol. 16, 773–779.
Hamatani T., Daikoku T., Wang H., Matsumoto H., Carter M.G., Ko M.S., Dey S.K. 2004. Global gene expression analysis identifies molecular pathways distinguishing blastocyst dormancy and activation. Proc. Natl. Acad. Sci. U. S. A. 101, 10326–10331.
Scognamiglio R., Cabezas-Wallscheid N., Thier M.C., Altamura S., Reyes A., Prendergast Á.M., Baumgärtner D., Carnevalli L.S., Atzberger A., Haas S., von Paleske L., Boroviak T., Wörsdörfer P., Essers M.A., Kloz U., Eisenman R.N., Edenhofer F., Bertone P., Huber W., van der Hoeven F., Smith A., Trumpp A. 2016. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell. 164, 668–680.
Meller C.L., Meller R., Simon R.P., Culpepper K.M., Podrabsky J.E. 2012. Cell cycle arrest associated with anoxia-induced quiescence, anoxic preconditioning, and embryonic diapause in embryos of the annual killifish Austrofundulus limnaeus. J. Comp. Physiol. B. 182, 909–920.
Ptak G.E., Tacconi E., Czernik M., Toschi P., Modlinski J.A., Loi P. 2012. Embryonic diapause is conserved across mammals. PLoS One. 7, e33027.
Yoshida G.J. 2018. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 37, 173.
Adam A.P., George A., Schewe D., Bragado P., Iglesias B.V., Ranganathan A.C., Kourtidis A., Conklin D.S., Aguirre-Ghiso J.A. 2009. Computational identification of a p38SAPK regulated transcription factor network required for tumor cell quiescence. Cancer Res. 69, 5664–5672.
Dhimolea E., de Matos Simoes R., Kansara D., Al’Khafaji A., Bouyssou J, Weng X., Sharma S., Raja J., Awate P., Shirasaki R., Tang H., Glassner B.J. Liu Z., Gao D., Bryan J., Bender S., Roth J., Scheffer M., Jeselsohn R., Gray N.S., Georgakoudi I., Vazquez F., Tsherniak A., Chen Y., Welm A., Duy C., Melnick A., Bartholdy B., Brown M., Culhane A.C., Mitsiades C.S. 2021. An embryonic diapause-like adaptation with suppressed myc activity enables tumor treatment persistence. Cancer Cell. 39, 240–256.
Rehman S.K., Haynes J., Collignon E., Brown K.R., Wang Y., Nixon A.M.L., Bruce J.P., Wintersinger J.A., Singh Mer A., Lo E.B.L., Leung C., Lima-Fernandes E., Pedley N.M., Soares F., McGibbon S., He H.H., Pollet A., Pugh T.J., Haibe-Kains B., Morris Q., Ramalho-Santos M., Goyal S., Moffat J., O’Brien C.A. 2021. Colorectal cancer cells enter a diapause-like dtp state to survive chemotherapy. Cell. 184, 226–242.
Santos-de-Frutos K., Djouder N. 2021. When dormancy fuels tumour relapse. Commun. Biol. 4, 747.
Orford K.W., Scadden D.T. 2008. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 9, 115–128.
Nieto M.A. 2013. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 342, 1234850.
Lamouille S., Xu J., Derynck R. 2014. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 15, 178–196.
Taneyhill L.A., Schiffmacher A.T. 2017. Should I stay or should I go? Cadherin function and regulation in the neural crest. Genesis. 55, e23028.
Yu M., Bardia A., Wittner B.S., Stott S.L., Smas M.E., Ting D.T., Isakoff S.J., Ciciliano J.C., Wells M.N., Shah A.M., Concannon K.F., Donaldson M.C., Sequist L.V., Brachtel E., Sgroi D., Baselga J., Ramaswamy S., Toner M., Haber D.A., Maheswaran S. 2013. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 339, 580–584.
Raimondi C., Gradilone A., Naso G., Vincenzi B., Petracca A., Nicolazzo C., Palazzo A., Saltarelli R., Spremberg F., Cortesi E., Gazzaniga P. 2011. Epithelial-mesenchymal transition and stemness features in circulating tumor cells from breast cancer patients. Breast Cancer Res. Treat. 130, 449–455.
Jie X.X., Zhang X.Y., Xu C.J. 2017. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: mechanisms and clinical applications. Oncotarget. 8, 81558–81571.
Ryser M.D., Mallo D., Hall A., Hardman T., King L.M., Tatishchev S., Sorribes I.C., Maley C.C., Marks J.R., Hwang E.S., Shibata D. 2020. Minimal barriers to invasion during human colorectal tumor growth. Nat. Commun. 11, 1280.
Brabletz T., Jung A., Reu S., Porzner M., Hlubek F., Kunz-Schughart L.A., Knuechel R., Kirchner T. 2001. Variable b-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. U. S. A. 98, 10356–10361.
Hay E.D. 1991. Collagen and other matrix proteins in embryogenesis. In Cell Biology of the Extracellular Matrix. Hay E.D., Ed. New York: Plenum.
Ksiazkiewicz M., Markiewicz A., Zaczek A.J. 2012. Epithelial-mesenchymal transition, a hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 79, 195–208.
Dasgupta A., Lim A.R., Ghajar C.M. 2017. Circulating and disseminated tumor cells: harbingers or initiators of metastasis? Mol. Oncol. 11, 40–61.
Tinganelli W., Durante M. 2020. Tumor hypoxia and circulating tumor cells. Int. J. Mol. Sci. 21, 9592.
Barrak N.H., Khajah A., Luqmani Y.A. 2020. Hypoxic environment may enhance migration/penetration of endocrine resistant MCF7- derived breast cancer cells through monolayers of other non-invasive cancer cells in vitro. Sci. Rep. 10, 1127.
Micalizzi D.S., Haber D.A., Maheswaran S. 2017. Cancer metastasis through the prism of epithelial-to-mesenchymal transition in circulating tumor cells. Mol. Oncol. 11, 770–780.
Fitzgerald D.M., Hastings P.J., Rosenberg S.M. 2017. Stress-induced mutagenesis: Implications in cancer and drug resistance. Annu. Rev. Cancer Biol. 1, 119–140.
Sun H., Lu Z., Singh A., Zhou Y., Zheng E., Zhou M., Wang J., Wu X., Hu Z., Gu Z., Campbell J.L., Zheng L., Shen B. 2021. Error-prone, stress-induced 3' flap-based Okazaki fragment maturation supports cell survival. Science. 374, 1252–1258.
Vera-Ramirez L., Hunter K.W. 2017. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Res. 6, 2134.
Lambert A.W., Pattabiraman D.R., Weinberg R.A. 2017. Emerging biological principles of metastases. Cell. 168, 670–691.
Paoli P., Giannoni E., Chiarugi P. 2013. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta. 1833, 3481–3498.
Meulemans D., Bronner-Fraser M. 2004. Gene-regulatory interactions in neural crest evolution and development. Dev. Cell. 7, 291–299.
Pshennikova E.S., Voronina A.S. 2019. Neural crest—an unusual population of embryonic cells. Mol. Biol. 53, 227–236.
Aceto N., Toner M., Maheswaran S., Haber D.A. 2015. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer. 1, 44–52.
Al-Mehdi A.B., Tozawa K., Fisher A.B., Shientag L., Lee A., Muschel R.J. 2000. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat. Med. 6, 100–102.
Goto W., Kashiwagi S., Asano Y., Takada K., Takahashi K., Hatano T., Takashima T., Tomita S., Motomura H., Ohsawa M., Hirakawa K., Ohira M. 2017. Circulating tumor cell clusters-associated gene plakoglobin is a significant prognostic predictor in patients with breast cancer. Biomark. Res. 5, 19.
Gkountela S., Castro-Giner F., Szczerba B.M., Vetter M., Landin J., Scherrer R., Krol I., Scheidmann M.C., Beisel C., Stirnimann C.U., Kurzeder C., Heinzelmann-Schwarz V., Rochlitz C., Weber W.P., Aceto N. 2019. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell. 176, 98–112.
Au S.H., Storey B.D., Moore J.C., Tang Q., Chen Y.L., Javaid S., Sarioglu A.F., Sullivan R., Madden M.W., O’Keefe R., Haber D.A., Maheswaran S., Langenau D.M., Stott S.L., Toner M. 2016. Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. U. S. A. 113, 4947–4952.
Yang M.H., Imrali A., Heeschen C. 2015. Circulating cancer stem cells: the importance to select. Chin. J. Cancer Res. 27, 437–449.
Aceto N., Bardia A., Miyamoto D.T., Donaldson M.C., Wittner B.S., Spencer J.A., Yu M., Pely A., Engstrom A., Zhu H., Brannigan B.W., Kapur R., Stott S.L., Shioda T., Ramaswamy S., Ting D.T., Lin C.P., Toner M., Haber D.A., Maheswaran S. 2014. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 158, 1110–1122.
Hamidi H., Ivaska J. 2018. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer. 18, 533–548.
Hoshino A., Costa-Silva B., Shen T.L., Rodrigues G., Hashimoto A., Tesic Mark M., Molina H., Kohsaka S., Di Giannatale A., Ceder S., Singh S., Williams C., Soplop N., Uryu K., Pharmer L., King T., Bojmar L., Davies A.E., Ararso Y., Zhang T., Zhang H., Hernandez J., Weiss J.M., Dumont-Cole V.D., Kramer K., Wexler L.H., Narendran A., Schwartz G.K., Healey J.H., Sandstrom P., Labori K.J., Kure E.H., Grandgenett P.M., Hollingsworth M.A., de Sousa M., Kaur S., Jain M., Mallya K., Batra S.K., Jarnagin W.R., Brady M.S., Fodstad O., Muller V., Pantel K., Minn A.J., Bissell M.J., Garcia B.A., Kang Y., Rajasekhar V.K., Ghajar C.M, Matei I., Peinado H., Bromberg J., Lyden D. 2015. Tumour exosome integrins determine organotropic metastasis. Nature. 527, 329–335.
Nguyen D.X., Bos P.D., Massague J. 2009. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 9, 274–284.
Gao Y., Bado I., Wang H., Zhang W., Rosen J.M., Zhang X.H. 2019. Metastasis organotropism: redefining the congenial soil. Dev. Cell. 49, 375–391.
Hen O., Barkan D. 2019. Dormant disseminated tumor cells and cancer stem/progenitor-like cells, similarities and opportunities. Semin. Oncol. 60, 157–165.
Ghajar C.M. 2015. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer. 15, 238–247.
Guitart A.V., Hammoud M., Dello Sbarba P., Ivanovic Z., Praloran V. 2010. Slowcycling/quiescence balance of hematopoietic stem cells is related to physiological gradient of oxygen. Exp. Hematol. 38, 847–851.
Liberti M.V., Locasale J.W. 2016. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 41, 211–218.
Pierce G.B., Speers W.C. 1988. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 48, 1996–2004.
Weissman I.L. 2000. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 100, 157–168.
Hannen R., Bartsch J.W. 2018. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 592, 2023–2031.
Young H.E., Black A.C. 2004. Adult stem cells. J. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 276, 75–102.
Takahashi K., Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663–676.
Senga S.S., Grose R.P. 2021. Hallmarks of cancer—the new testament. Open Biol. 11, 200358.
Stevanovic M., Kovacevic-Grujicic N., Mojsin M., Milivojevic M., Drakulic D. 2021. SOX transcription factors and glioma stem cells: choosing between stemness and differentiation. World J. Stem Cells. 13, 1417–1445.
Bonnet D., Dick J.E. 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737.
Carvalho J. 2020. Cell reversal from a differentiated to a stem-like state at cancer initiation. Front. Oncol. 10, 541.
Milanovic M., Fan D.N.Y., Belenki D., Däbritz J.H.M., Zhao Z., Yu Y., Dörr J.R., Dimitrova L., Lenze D., Monteiro Barbosa I.A., Mendoza-Parra M.A., Kanashova T., Metzner M., Pardon K., Reimann M., Trumpp A., Dörken B., Zuber J., Gronemeyer H., Hummel M., Dittmar G., Lee S., Schmitt C.A. 2018. Senescence-associated reprogramming promotes cancer stemness. Nature. 553, 96–100.
Zon L.I. 2008. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 453, 306–313.
Sturmlechner I., Zhang C., Sine C.C., van Deursen E.J., Jeganathan K.B., Hamada N., Grasic J., Friedman D., Stutchman J.T., Can I., Hamada M., Lim D.Y., Lee J.H., Ordog T., Laberge R.M., Shapiro V., Baker D.J., Li H., van Deursen J.M. 2021. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science. 374, eabb3420.
Walcher L., Kistenmacher A.K., Suo H., Kitte R., Dluczek S., Strauß A., Blaudszun A.R., Yevsa T., Fricke S., Kossatz-Boehlert U. 2020. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front. Immunol. 11, 1280.
Crea F., Nur Saidy N.R., Collins C.C., Wang Y. 2015. The epigenetic/noncoding origin of tumor dormancy. Trends Molec. Med. 21, 206–211.
Barney L.E., Hall C.L., Schwartz A.D., Parks A.N., Sparages C., Galarza S., Platt M.O., Mercurio A.M., Peyton S.R. 2020. Tumor cell-organized fibronectin maintenance of a dormant breast cancer population. Sci. Adv. 6, eaaz4157.
Shiozawa Y., Berry J.E., Eber M.R., Jung Y., Yumoto K., Cackowski F.C., Yoon H.J., Parsana P., Mehra R., Wang J., McGee S., Lee E., Nagrath S., Pienta K.J., Taichman R.S. 2016. The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget. 7, 41217–41232.
Sosa M.S., Bragado P., Aguirre-Ghiso J.A. 2014. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat. Rev. Cancer. 14, 611–622.
Kobayashi A., Okuda H., Xing F., Pandey P.R., Watabe M., Hirota S., Pai S.K., Liu W., Fukuda K., Chambers C., Wilber A., Watabe K. 2011. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 208, 2641–2655.
Yang A., Qin S., Schulte B.A., Ethier S.P., Tew K.D., Wang G.Y. 2017. MYC inhibition depletes cancer stem-like cells in triple-negative breast cancer. Cancer Res. 77, 6641–6650.
Wei S.C., Yang J. 2016. Forcing through tumor metastasis: the interplay between tissue rigidity and epithelial-mesenchymal transition. Trends Cell Biol. 26, 111–120.
De Cock J.M., Shibue T., Dongre A., Keckesova Z., Reinhardt F., Weinberg RA. 2016. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 76, 6778–6784.
Paget S. 1889. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 8, 98–101.
Schofield R. 1978. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cell. 4, 7–25.
Ossowski L., Reich E. 1983. Changes in malignant phenotype of a human carcinoma conditioned by growth environment. Cell. 33, 323–333.
Fu T., Dai L.J., Wu S.Y., Xiao Y., Ma D., Jiang Y.Z., Shao Z.M. 2021. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 14, 98.
Korneva Yu.S., Ukrainets R.V. 2019. Principles of premetastatic niche formation. J. Mod. Oncol. 21, 6–9.
Peinado H., Zhang H., Matei I.R., Costa-Silva B., Hoshino A., Rodrigues G., Psaila B., Kaplan R.N., Bromberg J.F., Kang Y., Bissell M.J., Cox T.R., Giaccia A.J., Erler J.T, Hiratsuka S., Ghajar C.M., Lyden D. 2017. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer. 17, 302–317.
Ingangi V., Minopoli M., Ragone C., Motti M.L., Carriero M.V. 2019. Role of microenvironment on the fate of disseminating cancer stem cells. Front. Oncol. 9, 82.
Giles A.J., Reid C.M., Evans J.D., Murgai M., Vicioso Y., Highfill S.L., Kasai M., Vahdat L., Mackall C.L., Lyden D., Wexler L., Kaplan R.N. 2016. Activation of hematopoietic stem/progenitor cells promotes immunosuppression within the pre-metastatic niche. Cancer Res. 76, 1335–1347.
Liu Y., Cao X. 2016. Immunosuppressive cells in tumor immune escape and metastasis. J. Mol. Med. 94, 509–522.
Peinado H., Aleckovic M., Lavotshkin S., Matei I., Costa-Silva B., Moreno-Bueno G., Hergueta-Redondo M., Williams C., Garcia-Santos G., Ghajar C., Nitadori-Hoshino A., Hoffman C., Badal K., Garcia B.A., Callahan M.K., Yuan J., Martins V.R., Skog J., Kaplan R.N., Brady M.S., Wolchok J.D., Chapman P.B., Kang Y., Bromberg J., Lyden D. 2012. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 883–891.
Kaplan R.N., Riba R.D., Zacharoulis S., Bramley A.H., Vincent L., Costa C., MacDonald D.D., Jin D.K., Shido K., Kerns S.A., Zhu Z., Hicklin D., Wu Y., Port J.L., Altorki N., Port E.R., Ruggero D., Shmelkov S.V., Jensen K.K., Rafii S., Lyden D. 2005. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 438, 820–827.
Hoshino A., Costa-Silva B., Shen T.L., Rodrigues G., Hashimoto A., Tesic Mark M., Molina H., Kohsaka S., Di Giannatale A., Ceder S., Singh S., Williams C., Soplop N., Uryu K., Pharmer L., King T., Bojmar L., Davies A.E., Ararso Y., Zhang T., Zhang H., Hernandez J., Weiss J.M., Dumont-Cole V.D., Kramer K., Wexler L.H., Narendran A., Schwartz G.K., Healey J.H., Sandstrom P., Labori K.J., Kure E.H., Grandge-nett P.M., Hollingsworth M.A., de Sousa M., Kaur S., Jain M., Mallya K., Batra S.K., Jarnagin W.R., Brady M.S., Fodstad O., Muller V., Pantel K., Minn A.J., Bissell M.J., Garcia B.A., Kang Y., Rajasekhar V.K., Ghajar C.M., Matei I., Peinado H., Bromberg J., Lyden D. 2015. Tumour exosome integrins determine organotropic metastasis. Nature. 527, 329–335.
Liu Y., Gu Y., Han Y., Zhang Q., Jiang Z., Zhang X., Huang B., Xu X., Zheng J., Cao X. 2016. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell. 30, 243–256.
Akoto T., Saini S. 2021. Role of exosomes in prostate cancer metastasis. Int. J. Mol. Sci. 22, 3528.
Costa-Silva B., Aiello N.M., Ocean A.J., Singh S., Zhang H., Thakur B.K., Becker A., Hoshino A., Mark M.T., Molina H., Xiang J., Zhang T., Theilen T.M., García-Santos G., Williams C., Ararso Y., Huang Y., Rodrigues G., Shen T.L., Labori K.J., Lothe I.M., Kure E.H., Hernandez J., Doussot A., Ebbesen S.H., Grandgenett P.M., Hollingsworth M.A., Jain M., Mallya K., Batra S.K., Jarnagin W.R., Schwartz R.E., Matei I., Peinado H., Stanger B.Z., Bromberg J., Lyden D. 2015. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell. Biol. 17, 816–826.
Lin D., Chen X., Lin Z., Lin J., Liu Y., Liu D. 2021. Paper-supported co-culture system for dynamic investigations of the lung-tropic migration of breast cancer cells. Biomed. Mater. 16, 025028.
Lin R., Wang S., Zhao R.C. 2013. Exosomes from human adipose-derived mesenchymal stem cells promote migration through Wnt signaling pathway in a breast cancer cell model. Mol. Cell. Biochem. 383, 13–20.
Abravanel D.L., Belka G.K., Pan T.C., Pant D.K., Collins M.A., Sterner C.J., Chodosh L.A. 2015. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J. Clin. Invest. 125, 2484–2496.
Capulli M., Hristova D., Valbret Z., Carys K., Arjan R., Maurizi A., Masedu F., Cappariello A., Rucci N., Teti A. 2019. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer. 121, 157‒171.
Yamazaki S., Iwama A., Takayanagi S., Eto K., Ema H., Nakauchi H. 2009. TGF-beta as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood. 113, 1250–1256.
Lawson M.A., McDonald M.M., Kovacic N., Hua Khoo W., Terry R.L., Down J., Kaplan W., Paton-Hough J., Fellows C., Pettitt J.A., Neil Dear T., Van Valckenborgh E., Baldock P.A., Rogers M.J., Eaton C.L., Vanderkerken K., Pettit A.R., Quinn J.M., Zannettino A.C., Phan T.G., Croucher P.I. 2015. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 6, 8983.
Lu X., Mu E., Wei Y., Riethdorf S., Yang Q., Yuan M., Yan J., Hua Y., Tiede B.J., Lu X., Haffty B.G., Pantel K., Massagué J., Kang Y. 2011. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell. 20, 701–714.
Weilbaecher K.N., Guise T.A., McCauley L.K. 2011. Cancer to bone: a fatal attraction. Nat. Rev. Cancer. 11, 411–425.
Luo X., Fu Y., Loza A.J., Murali B., Leahy K.M., Ruhland M.K., Gang M., Su X., Zamani A., Shi Y., Lavine K.J., Ornitz D.M., Weilbaecher K.N., Long F., Novack D.V., Faccio R., Longmore G.D., Stewart S.A. 2016. Stromal-initiated changes in the bone promote metastatic niche development. Cell Rep. 14, 82–92.
Gao H., Chakraborty G., Lee-Lim A.P., Mo Q., Decker M., Vonica A., Shen R., Brogi E., Brivanlou A.H., Giancotti F.G. 2012. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell. 150, 764–779.
Ruppender N., Larson S., Lakely B., Kollath L., Brown L., Coleman I., Coleman R., Nguyen H., Nelson P.S., Corey E., Snyder L.A., Vessella R.L., Morrissey C., Lam H.M. 2015. Cellular adhesion promotes prostate cancer cells escape from dormancy. PLoS One. 10, e0130565.
Trumpp A., Essers M., Wilson A. 2010. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 10, 201–209.
Decker A.M., Jung Y., Cackowski F., Taichman R.S. 2016. The role of hematopoietic stem cell niche in prostate cancer bone metastasis. J. Bone Oncol. 5, 117–120
Kunisaki Y., Bruns I., Scheiermann C., Ahmed J., Pinho S., Zhang D., Mizoguchi T., Wei Q., Lucas D., Ito K., Mar J.C., Bergman A., Frenette P.S. 2013. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 502, 637–643.
Hoshide R., Jandial R. 2017. The role of the neural niche in brain metastasis. Clin. Exp. Metastasis. 34, 369–376.
Zeng Q., Michael I.P., Zhang P., Saghafinia S., Knott G., Jiao W., McCabe B.D., Galván J.A., Robinson H.P.C., Zlobec I., Ciriello G., Hanahan D. 2019. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 573, 526–531.
Agudo J., Park E.S., Rose S.A., Alibo E., Sweeney R., Dhainaut M., Kobayashi K.S., Sachidanandam R., Baccarini A., Merad M., Brown B.D. 2018. Quiescent tissue stem cells evade immune surveillance. Immunity. 48, 271–285.
Vera-Ramirez L., Vodnala S.K., Nini R., Hunter K.W., Green, J.E. 2018. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 9, 1944.
La Belle Flynn A., Calhoun B.C., Sharma A., Chang J.C., Almasan A., Schiemann W.P. 2019. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 10, 3668.
Aguirre-Ghiso J.A. 2018. How dormant cancer persists and reawakens. Science, 361, 1314–1315.
Fessler E., Dijkgraaf F.E., De Sousa E.M.F., Medema J.P. 2013. Cancer stem cell dynamics in tumor progression and metastasis: is the microenvironment to blame? Cancer Lett. 341, 97–104.
Carlson P., Dasgupta A., Grzelak C.A., Kim J., Barrett A., Coleman I.M., Shor R.E., Goddard E.T., Dai J., Schweitzer E.M., Lim A.R., Crist S.B., Cheresh D.A., Nelson P.S., Hansen K.C., Ghajar C.M. 2019. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat. Cell. Biol. 21, 238–250.
Price T.T., Burness M.L., Sivan A., Warner M.J., Cheng R., Lee C.H., Olivere L., Comatas K., Magnani J., Kim Lyerly H., Cheng Q., McCall C.M., Sipkins D.A. 2016. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 8, 340ra73.
Zhao L., Zhang K., He H., Yang Y., Li W., Liu T., Li J. 2021. The relationship between mesenchymal stem cells and tumor dormancy. Front. Cell. Dev. Biol. 9, 731393.
Postovit L.M., Margaryan N.V., Seftor E.A., Kirschmann D.A., Lipavsky A., Wheaton W.W., Abbott D.E., Seftor R.E., Hendrix M.J. 2008. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl. Acad. Sci. U. S. A. 105, 4329–4934.
Saad N., Alberio R., Johnson A.D., Emes R.D., Giles T.C., Clarke P., Grabowska A.M., Allegrucci C. 2018. Cancer reversion with oocyte extracts is mediated by cell cycle arrest and induction of tumour dormancy. Oncotarget. 9, 16008–16027.
Weidenfeld K., Barkan D. 2018. EMT and stemness in tumor dormancy and outgrowth: are they intertwined processes? Front. Oncol. 8, 381.
Hsu S.K., Chiu C.C., Dahms H.U., Chou C.K., Cheng C.M., Chang W.T., Cheng K.C., Wang H.D., Lin I.L. 2019. Unfolded protein response (upr) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int. J. Mol. Sci. 20, 2518.
Aguirre Ghiso J.A., Kovalski K., Ossowski L. 1999. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell. Biol. 147, 89–104.
Walker C., Mojares E., Del Rio Hernandez A. 2018. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 19, 3028.
Poltavets V., Kochetkova M., Pitson S.M., Samuel M.S. 2018. The role of the extracellular matrix and its molecular and cellular regulators in cancer cell plasticity. Fron. Oncol. 8, 431.
Talukdar S., Bhoopathi P., Emdad L., Das S., Sarkar D., Fisher P.B. 2019. Dormancy and cancer stem cells: an enigma for cancer therapeutic targeting. Adv. Cancer Res. 141, 43–84.
Malanchi I., Santamaria-Martínez A., Susanto E., Peng H., Lehr H.A., Delaloye J.F., Huelsken J. 2011. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 481, 85–89.
Oskarsson T., Batlle E., Massague J. 2014. Metastatic stem cells: sources, niches, and vital pathways. Cell Stem Cell. 14, 306–321.
Ghajar C.M., Peinado H., Mori H., Matei I.R., Evason K.J., Brazier H., Almeida D., Koller A., Hajjar K.A., Stainier D.Y., Chen E.I., Lyden D., Bissell M.J. 2013. The perivascular niche regulates breast tumour dormancy. Nat. Cell. Biol. 15, 807–817.
Locatelli F., Nazio F., Bordi M., Cianfanelli V., Cecconi F. 2019. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell. Death Differ. 26, 690–702.
Sosa M.S., Bragado P., Debnath J., Aguirre-Ghiso J.A. 2013. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv. Exp. Med. Biol. 734, 73–89.
Akkoc Y., Peker N., Akcay A., Gozuacik D. 2021. Autophagy and cancer dormancy. Front. Oncol. 11, 627023.
Domingo-Domenech J., Vidal S.J., Rodriguez-Bravo V., Castillo-Martin M., Quinn S.A., Rodriguez-Barrueco R., Bonal D.M., Charytonowicz E., Gladoun N., de la Iglesia-Vicente J., Petrylak D.P., Benson M.C., Silva J.M., Cordon-Cardo C. 2012. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 22, 373–388.
Heidel F.H., Bullinger L., Feng Z., Wang Z., Neff T.A., Stein L., Kalaitzidis D., Lane S.W., Armstrong S.A. 2012. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 10, 412–424
Steinbichler T.B., Dudás J., Skvortsov S., Ganswindt U., Riechelmann H., Skvortsova I.I. 2018. Therapy resistance mediated by cancer stem cells. Semin. Cancer Biol. 53, 156–167.
Urra H., Hetz C. 2014. A novel ER stress-independent function of the UPR in angiogenesis. Mol. Cell. 54, 542–544.
Baeriswyl V., Christofori G. 2009. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 19, 329–337.
Almog N., Ma L., Raychowdhury R., Schwager C., Erber R., Short S., Hlatky L., Vajkoczy P., Huber P.E., Folkman J., Abdollahi A. 2009. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 69, 836–844.
Naumov G.N., Folkman J., Straume O. 2009. Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin. Exp. Metastasis. 26, 51–60.
Zhao H., Wu L., Yan G., Chen Y., Zhou M., Wu Y., Li Y. 2021. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct. Target Ther. 6, 263.
Nishida J., Momoi Y., Miyakuni K., Tamura Y., Takahashi K., Koinuma D., Miyazono K., Ehata S. 2020. Epigenetic remodelling shapes inflammatory renal cancer and neutrophil-dependent metastasis. Nat. Cell Biol. 22, 465–475.
Nolan E., Bridgeman V.L., Ombrato L., Karoutas A., Rabas N., Sewneth C.A.N., Vasquez M., Rodrigues F.S., Horswell S., Faull P., Carter R., Malanchi I. 2022. Radiation exposure elicits a neutrophil-driven response in healthy lung tissue that enhances metastatic colonization. Nat. Cancer. 3, 173–187.
Barkan D., El Touny L.H., Michalowski A.M., Smith J.A., Chu I., Davis A.S., Webster J.D., Hoover S., Simpson R.M., Gauldie J., Green J.E. 2010. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 70, 5706–5716.
Cox T.R., Bird D., Baker A.M., Barker H.E., Ho M.W., Lang G., Erler J.T. 2013. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 73, 1721–1732.
Weidenfeld K., Schif-Zuck S., Abu-Tayeh H., Kang K., Kessler O., Weissmann M., Neufeld G., Barkan D. 2016. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget. 7, 71362–71377.
Albrengues J., Shields M.A., Ng D., Park C.G., Ambrico A., Poindexter M.E., Upadhyay P., Uyeminami D.L., Pommier A., Küttner V., Bružas E., Maiorino L., Bautista C., Carmona E.M., Gimotty P.A., Fearon D.T., Chang K., Lyons S.K, Pinkerton K.E., Trotman L.C., Goldberg M.S., Yeh J.T., Egeblad M. 2018. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 361, eaao4227.
Bickett T.E., Karam S.D. 2020. Tuberculosis–cancer parallels in immune response regulation. Int. J. Mol. Sci. 21, 6136.
Liu K., Sun E., Lei M., Li L., Gao J., Nian X., Wang L. 2019. BCG-induced formation of neutrophil extracellular traps play an important role in bladder cancer treatment. Clin. Immunol. 201, 4–14.
Krall J.A., Reinhardt F., Mercury O.A., Pattabiraman D.R., Brooks M.W., Dougan M., Lambert A.W., Bierie B., Ploegh H.L., Dougan S.K., Weinberg R.A. 2018. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 10, eaan3464.
Klein C.A. 2009. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer. 9, 302–312.
Buell J.F., Beebe T.M., Trofe J., Gross T.G., Alloway R.R., Hanaway M.J., Woodle E.S. 2004. Donor transmitted malignancies. Ann. Transplant. 9, 53–56.
Wang H.-F., Wang S.-S., Huang M.-C., Liang X.-H., Tang Y.-J., Tang Y.-L. 2019. Targeting immune-mediated dormancy: a promising treatment of cancer. Front. Oncol. 9, 498.
Sosa M.S., Valderas A.A., Bragrado P., Wen H.C., Aguirre-Ghiso J.A. 2011. ERK1/2 and p38α/β signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin. Cancer Res. 17, 5850–5857.
Correia A.L., Guimaraes J.C., Auf der Maur P., De Silva D., Trefny M.P., Okamoto R., Bruno S., Schmidt A., Mertz K., Volkmann K., Terracciano L., Zippelius A., Vetter M., Kurzeder C., Weber W.P., Bentires-Alj M. 2021. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature. 594, 566–571.
Yi J.S., Cox M.A., Zajac A.J. 2010. T-Cell exhaustion: characteristics, causes and conversion. Immunology. 129, 474–481
Philip M., Schietinger A. 2022. CD8+ T cell differentiation and dysfunction in cancer. Nat. Rev. Immunol. 22, 209–223. https://doi.org/10.1038/s41577-021-00574-3
Zheng L., Qin S., Si W., Wang A., Xing B., Gao R., Ren X., Wang L., Wu X., Zhang J., Wu N., Zhang N., Zheng H., Ouyang H., Chen K., Bu Z., Hu X., Ji J., Zhang Z. 2021. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 374, abe6474.
Funding
This work did not require any funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest. This article does not contain any studies involving animals or human subjects performed by any of the authors.
Additional information
Translated by T. Tkacheva
Abbreviations: MSC, mesenchymal stem cell; BM, bone marrow; EMT, epithelial-to-mesenchymal transition; MET, mesenchymal-to-epithelial transition; CTC, circulating tumor cell; ECM, extracellular matrix; DTC, disseminated tumor cell; CSC, cancer stem cell; HSC, hematopoietic stem cell; PMN, premetastatic niche; MN, metastatic niche; PVN, perivascular niche.
Rights and permissions
About this article

Cite this article
Pshennikova, E.S., Voronina, A.S. Dormancy: There and Back Again. Mol Biol 56, 735–755 (2022). https://doi.org/10.1134/S0026893322050119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0026893322050119