
Abstract
Methyltransferases (MTases) play an important role in the functioning of living systems, catalyzing the methylation reactions of DNA, RNA, proteins, and small molecules, including endogenous compounds and drugs. Many human diseases are associated with disturbances in the functioning of these enzymes; therefore, the study of MTases is an urgent and important task. Most MTases use the cofactor S‑adenosyl‑L‑methionine (SAM) as a methyl group donor. SAM analogs are widely applicable in the study of MTases: they are used in studies of the catalytic activity of these enzymes, in identification of substrates of new MTases, and for modification of the substrates or substrate linking to MTases. In this review, new synthetic analogs of SAM and the problems that can be solved with their usage are discussed.
Similar content being viewed by others


INTRODUCTION
Methyltransferases (MTases) play an important role in the functioning of living systems, catalyzing the methylation reactions of DNA, RNA, proteins, and small molecules, including endogenous compounds and drugs. DNA methylation determines the epigenetic regulation of gene expression [1]. RNA methylation is necessary for the correct course of the processes involving these molecules, primarily protein synthesis and splicing [2]. Methylation expands the functional diversity of proteins [3]. It is involved in a large number of intracellular processes, for example, histone methylation regulates gene expression [4]. Methylation of small molecules is the way to change their properties and participation in biochemical processes [5].
Disruption of MTases changes the nature of intracellular methylation processes, which, in turn, often leads to the emergence of various diseases [2, 6]; therefore, the study of MTases is an important task. Many methods and approaches utilizing analogs of methylation cofactor S-adenosyl-L-methionine (SAM, or AdoMet) have been developed to study MTases. Using SAM analogs, one can control the activity of MTases, determine substrates for newly discovered proteins of this class, and chemically modify methylation substrates.
A considerable part of this review is devoted to synthetic SAM analogs obtained over the past two decades and the problems that can be solved using these analogs.
METHYLTRANSFERASES
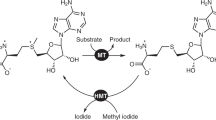
Methyltransferase [E.C. 2.1.1] represent a wide group of enzymes that catalyze methylation of various substrates and regulate such biological processes as metabolism, biosynthesis, functioning and degradation of nucleic acids, protein activity, and detoxification of exogenous compounds [7]. The overwhelming majority of known MTases utilize SAM as a donor of the methyl group, which, after cleavage of the CH3 group, is converted into S-adenosyl-L-homocysteine (SAH) (Fig. 1) [8].

Transfer of the methyl group by the SAM-dependent MTase from the cofactor to the substrate. During this reaction, SAM is converted to SAH.
Currently about 300 SAM-dependent MTases are known in the human proteome; the type of substrate (small molecules, lipids, proteins, or nucleic acids) and the type of methylated atom (usually nitrogen, oxygen, carbon, or sulfur) are known for about 120 MTases [9, 10].
Role of MTases in Biological Processes
In the living system, methylation has various functions, depending on the type of substrate. DNA methylation is responsible for the epigenetic regulation of eukaryotic gene expression; it plays an important role in the embryonic development of multicellular organisms and is involved in genome imprinting and carcinogenesis. In prokaryotic cells, methylation is used to control DNA repair and replication time [11], as well as in restriction–modification systems to protect against foreign genetic elements [12].
RNA methylation supplements the four-letter genetic code with nucleotides containing modified bases or 2'-methylhydroxy groups. It provides biochemical, biophysical, and metabolic stabilization of RNA, which are necessary for these molecules to perform specific functions, as well as quality control of RNA. Methylation is inherent to most RNAs that perform catalytic functions, including those involved in protein synthesis and splicing [13]. It is also involved in the emergence of antibiotic resistance in bacteria [14].
Protein methylation allows significant expansion of the 20-amino acid “alphabet,” introducing greater diversity into the amino acid composition of proteins and changing their functional properties. The amino group of lysine and the guanidine fragment of arginine are methylated most often; less often methylated are the hydroxyl groups of serine and tyrosine, and the carboxyl groups of the side chains of glutamic and aspartic acid [15]. Also, the N- and C-terminal groups of any amino acid residues in proteins, including the amide fragments of the side chains of glutamine and asparagine, can undergo methylation [16]. Methylation of lysine and arginine in histone molecules is the best studied and represented in cells. These modifications regulate gene transcription [17].
Methylation of small molecules is one of the biosynthesis processes for many endogenous compounds. Products of methylation by MTases include protein cofactors, signaling and protective molecules, membrane components, pigments, and many other compounds [18, 19].
Methylation Cofactor SAM
SAM, a cofactor of almost all known MTases [8], acts as a methyl group donor in the methylation reaction of N, C, O, and S-nucleophilic centers. This is the second most common organic cofactor after ATP [20]. The main feature of the SAM molecule is the trivalent sulfur atom, which ensures its high reactivity in the methylation reaction of various substrates (DNA, RNA, proteins, small molecules), usually proceeding according to the SN2 mechanism.
The asymmetrically substituted sulfonium ion in the SAM molecule is a chiral center. The S and R epimers are optically stable and can be separated. It has been shown that the methylation reaction is stereospecific, and it is preferable that only the natural S-epimer enters into the reaction, while the R-epimer, binding in the same active center, does not transfer the methyl group to the substrate [21, 22].
SAM is also used as a source of methylene groups (in the biosynthesis of cyclopropyl fatty acids), amino groups (in the synthesis of 7,8-diaminoperlagonic acid, a precursor of biotin), ribosyl moieties (in the biosynthesis of epoxykevosin, a modified tRNA nucleoside), and aminopropyl groups (in the synthesis of ethylene and polyamines) [23].
In living systems, SAM is usually formed from ATP and L-methionine with methionine adenosyltransferase (MAT, S-adenosylmethionine synthetase) as the catalyst [24, 25]. This enzymatic reaction is also used for the synthesis of SAM in preparative quantities from ATP and methionine [26, 27]. To increase conversion, inorganic pyrophosphatase is added to the reaction mixture, which utilizes the pyrophosphate formed [28]. Another variant of the reaction yielding SAM is based on the use of the SalL enzyme, which catalyzes the interaction of 5-chloradenosine (CIDA) with methionine (Fig. 2) [29]. Yet another way to obtain SAM is to isolate this product from yeast [30]. A synthetic method to produce SAM is also used, based on the interaction of a methyl halide with SAH under acidic conditions [22, 31]. In the this case, however, a mixture of S and R epimers is formed.

Enzymatic and synthetic methods to produce SAM.
In neutral and alkaline environments, SAM undergoes spontaneous degradation via two parallel pathways: degradation to methylthioadenosine (MTA) and homoserine lactone, and hydrolysis to adenine and S‑ribosylmethionine [32].
The presence of a trivalent sulfur atom determines the four main pathways of SAM metabolism in cells: (1) removal of the methyl group in the methylation reaction to form SAH; (2) cleavage of the sulfur–carbon bond of the methionine chain with the formation of MTA and homoserine lactone; (3) radical cleavage of the bond between the sulfur atom and the ribose residue, catalyzed by iron-containing enzymes, to form a reactive 5'-deoxyadenosyl radical; and (4) radical cleavage with the formation of the ACP radical and MTA (Fig. 3) [33–35].

SAM decomposition scheme. Pathway 1, MTases-catalyzed methyl group transfer; pathway 2, non-enzymatic cleavage of homoserinolactone; pathways 3 and 4, radical cleavage catalyzed by iron–sulfur enzymes. Reprinted (adapted) from [33] with the permission of the publisher.
It has been shown that the synthesis of functionally active isosteric analogs of SAM increases their stability in the main decomposition reactions (depurination, intramolecular cyclization, and sulfonium epimerization). Binding of such analogs of SAM with MTases proceeds almost identically to the initial cofactor [36–38]. Thorson and co-workers have synthesized analogs of SAM (Fig. 4), in which the nitrogen atom in position 7 of the adenine fragment was replaced by carbon, and the carboxyl group, by a tetrazole ring, which led to a significant increase in the stability of the SAM analog at pH 8. Biochemical and structural studies showed that these SAM analogs bind to the dnrK MTase (carminomycin 4-O-methyltransferase), while the enzymatic activity of the protein is completely retained [36].

Isosteric analogs of SAM. The half-decomposition time of the compounds is indicated under close to physiological conditions. tMet, tetrazole-L-methionine; 7dzAdo, S-7-deazaadenosyl. Reprinted from [36] with permission from the publisher.
Another isosteric substitution in the SAM molecule is the replacement of the carboxyl group with a phosphonium or phosphonic group (Fig. 5). This results in the prevention of SAM lactonization, which stabilizes SAM. It has been shown that such molecules are able to effectively bind to MTases [39, 40].

Phosphorus-containing isosteric analogs of SAM.
Methylation Coproduct SAH
When MTases transfer the methyl group from SAM to a substrate, the SAH molecule is formed. In most cases, SAH is a competitive inhibitor of MTases. The strength of SAH binding to MTases is often even higher than that of the SAM cofactor [33, 41]. Therefore, cellular homeostasis of SAM and SAH is very important and degradation of SAH after the methylation reaction must be precisely regulated. In eukaryotic cells, SAH decomposes with the formation of homocysteine and adenosine in a reaction catalyzed by SAH hydrolase (Fig. 6) [41]. In prokaryotic cells, SAH is cleaved to adenine and S-ribosylhomocysteine predominantly in a reaction catalyzed by MTA/SAH nucleosidase [42]. All known prokaryotic MTA/SAH nucleosidases are three-substrate enzymes capable of cleaving adenine not only from SAH, but also from MTA and 5'dAdo [43].

Pathways of enzymatic degradation of SAH.
SYNTHETIC ANALOGS OF SAM
Synthetic SAM analogs are created and used to solve several types of problems related to the study or activity control of SAM-dependent MTases. In this review, synthetic analogs of SAM are considered based on the problems they are used to solve.
The first group consists of MTase inhibitors. These compounds bind to the enzyme and block its catalytic activity. In the second group, we select SAM analogs that contain functional groups other than methyl. They are recognized by MTase, bind the enzyme, and enter the catalytic reactions, which allows the substrates to be modified with new molecular fragments. The third group of synthetic SAM analogs are compounds recognized by MTase as a cofactor, which, under the action of MTase, form an adduct with the enzyme substrate. Finally, to the fourth group, we assigned SAM analogs that are capable of binding to MTases, but do not enter further catalytic reactions. These are used to introduce functional fragments that allow the study of MTases.
Inhibitors of MTases
There are many MTase inhibitors with different mechanisms of action. They were obtained using various strategies, including virtual and high-throughput screening followed by lead compound optimization [44–46].
Some compounds of natural origin (Fig. 7) act as competitive inhibitors of DNA MTases (DNMT) [47]. These include polyphenols, flavonoids, anthraquinones, and compounds of some other classes. Among the first MTase inhibitors described were curcumin, (–)-epigallocatechin-3-gallate (EGCG), machanin, genistein, quercetin, silibinin, luteolin, quasinol Q, hypericin, boswellic acid, and lycopene [47, 48].

Some natural competitive inhibitors of DNA MTases.
Another class of inhibitors are analogs of the SAM cofactor, which are involved in competitive binding to MTases and lack specificity for a particular MTase, i.e. these are the so-called PAN inhibitors. The group includes the coproduct of methylation, SAH; the decomposition product of SAM, methylthioadenosine; and sinfungin, an antifungal agent of bacterial origin. Modification of substituents in the inhibitor molecule in accordance with the characteristics of the active site of MTase allows the selectivity of its binding to a specific enzyme to be increased (Fig. 8) [49–56].

Examples of synthetic MTase inhibitors. LLY-283 is a selective inhibitor of MTase PRMT5, which is responsible for the formation of most mono- and dimethylated arginine residues in proteins [57]; compound EPZ-5676, an inhibitor of MTase DOT1L, which is responsible for methylation of lysine-79 in histone H3 [49, 51, 52]; a potential inhibitor of MTases Nsp14 and Nsp16 of SARS-Cov-2, which are responsible for methylation of the mRNA cap [56]; bisubstrate inhibitor of nicotinamide-N-MTase (NNMT), an enzyme responsible for the formation of N-methylnicodinamide from nicotinamide [50]; N-propylsinfungin, a sinfungin derivative, selectively inhibiting MTase SETD2, which methylates lysine residues in proteins [53, 54].
The nucleoside drugs azacytidine (5-azacytidine) and decitabine (5-aza-2'-deoxycytidine), whose action is based on the inhibition of DNA MTases, in particular DNMT3A, are used in the treatment of myelodysplastic syndrome [58]. However, these drugs cannot be classified as cofactor analogs. These are classic nucleoside inhibitors, that is, substrate (DNA) analogs. Methylation of these fragments in the DNA strand leads to covalent cross-linking with the enzyme and thus irreversible blockade of its catalytic activity [59].
Analogs of SAM Used to Transfer New Functional Groups onto a Substrate
This section discusses SAM analogs and derivatives that can serve as synthetic cofactors for the introduction of new functional groups, including various labels, into MTase substrates. They are used to search for substrates of new MTases, as well as to study the functional activity of enzymes. The transferred label can be, for example, an isotopically substituted methyl group. In addition, MTases are prone to “make mistakes” and transfer more complex functional groups to substrates.
Transfer of an isotopically substituted methyl group. Using the SAM molecule isotopically substituted at the methyl group, radioactive or stable isotopes of carbon and hydrogen can be introduced into an MTase substrate by in vitro methylation reactions.
(A) Transfer of a radioactive label. To introduce a radioactive label into the MTase substrate, an in vitro methylation reaction is carried out with a recombinant MTase or cell extract [60] in the presence of a SAM molecule containing 14C or 3H atoms in the methyl group.
The methylation reaction under the action of SAM-dependent MTases is accompanied by the formation of SAH, which inhibits the reaction. Therefore, to increase the MTase turnover rate, SAH nucleosidase, an enzyme that cleaves SAH, is often added to the reaction mixture [61].
The amount of radioactive methyl group introduced into macromolecules is estimated using a scintillation counter [62] or measured by radioautographic analysis after separation in polyacrylamide gel [63]. Before calculating the radioactivity, it is necessary to separate the unreacted radioactive SAM and low molecular weight radioactive decay products from the labeled substrate. When studying DNA MTases, the use of DE-81 filter paper for DNA binding after the termination of the reaction is considered classical [64]. In the case of protein methylation, isolation of proteins and peptides from the reaction mixture using the ZipTip-C4 pipette tips containing C4 sorbent, which selectively binds peptides, was proposed [65]. When RNA methylation is carried out in vitro, the product is separated by precipitating RNA from the reaction mixture with ethanol after phenol–chloroform extraction of proteins [63]. Another variant of the detection of a labeled reaction product in the presence of an excess of radioactive SAM is called SPA (scintillation proximity assay). In this case, pre-biotinylated substrates are used, for example, modified peptides that interact with beads containing a scintillator [66].
In vitro studies of methylation processes using radioactive labels make it possible to identify and confirm the macromolecular substrates of MTases. For example, this method established that the YbeA Mtase methylates pseudouridine at position 1915 of the 23S rRNA of Escherichia coli [67], and the YfiC Mtase of E. coli is responsible for the modification of m6A37 in valine tRNA [62].
(B) Transfer of a methyl group containing stable isotopes. The isotopically substituted SAM molecule can be used to introduce the stable isotopes 2H and/or 13C into the MTase substrate. This isotopic tag allows for a quantitative assessment of the methylation product content using mass spectrometry. For example, in vitro RNA methylation with S-(5'-adenosyl)-L-methionine-d3 followed by RNA analysis in LC-MS/MS experiments was used to determine METTL14 methyltransferase activity. This protein binds the MTase partner METTL3 and, as part of the strong heterodimeric complex METTL3–METTL14, modifies m6A in mammalian mRNA [68].
In addition, the introduction of the 13C isotope opens up the possibility of studying methylation using nuclear magnetic resonance (NMR), since this carbon isotope has a nonzero spin magnetic moment and can be detected by NMR. For example, solid-state NMR was used to analyze tumor tissues grown with the addition of 13C-methionine [69]. Once in cells, labeled methionine is converted into 13C-labeled SAM, which then serves as an MTase substrate. In this work, by measuring the 13C,1H-HSQC NMR spectra of methyl groups, the differences in the distribution of 13CH3 groups in tumors and normal tissues were analyzed, which may be associated with atypical methionine metabolism, in particular, that caused by the incorrect work of MTases.
Transfer of a double-activated functional group. The SAM sulfonium center is a key fragment in the transmethylation reaction; therefore, it is this part of the molecule that is most often modified. The development of SAM analogs has led to effective synthetic cofactors for labeling substrates by transferring activated functional groups from SAM to the substrate, which can then be ligated to more complex fragments. These functional groups were called “doubly activated,” and the method was called methyltransferase-directed transfer of activated groups (mTAG) [70]. The transferred groups are amine-containing fragments that can be modified in the reaction with an N-hydroxysuccinimide ester; fragments with a terminal alkyne or azide, which can be functionalized in the reaction of azide–alkyne cycloaddition (click chemistry); or photoactivated fragments.
The mechanism of methyl group transfer from SAM to the substrate molecule has been studied in detail [71]. One of the factors determining the transfer rate is the small size of the transferred methyl group. Its replacement by ethyl or propyl substitute at the trivalent sulfur atom of SAM leads to a significant decrease in the transfer rate due to steric effects [72, 73]. In 2006, Elmar Weinhold’s group reported a new type of SAM analog, in which the methyl group was replaced by an allyl or propargyl group (Fig. 9) [74, 75]. The reaction rate (SN2) of the transfer of the functional group from such SAM analogs to the substrate is only an order of magnitude slower, despite the steric factor. The reaction proceeds due to the stabilization of the transition state involving the π-electrons of the double or triple bond. When DNA MTase from Thermus aquaticus (M.TaqI, methylates adenine residue (N6) in the TCGA site) is used, propargyl transfer from the SAM analog to DNA is quite efficient [74].

Scheme for the synthesis of SAM analogs containing propargyl and allyl groups. Reprinted (adapted) from [75] with the permission of the publisher.
Similar SAM derivatives are obtained by the reaction of nucleophilic substitution of an activated radical (allyl or propargyl) with SAH (Fig. 9). The reaction is carried out under acidic conditions, which stabilizes the resulting product and affects the regioselectivity due to protonation of amino groups [75]. During the reaction, two diastereomers are formed, which can be separated by HPLC.
Propargyl-containing analogs are of great interest as reporters of methylation, since the triple bond transferred to the substrate can be further modified with a functional label using the azide–alkyne cycloaddition (AAC) reaction [76–79].
The low stability of propargyl-SAM, associated with the rapid hydrolysis of the alkynyl group under physiological conditions (the half-life is several minutes), makes it difficult to work with (Fig. 10) [80]. Thus, for some MTases, the transmethylation reaction requires a long incubation time. During this time, propargyl-SAM undergoes hydrolysis to keto-AdoMet, which leads to low conversion of propargyl transfer to the substrate [71, 81].

Propargyl analogs of SAM and ProSeAM. The ways of decomposition of compounds and their stability are shown. Reprinted (adapted) from [81] with the permission of the publisher.
Later, the ketone analog keto-AdoMet (Fig. 11) was shown to be useful for labeling substrates, since its high reactivity and the absence of keto groups in proteins and DNA allows specific labeling. This approach was first used by Zhaohui Sunny Zhou. After the transfer of the keto group from keto-AdoMet to the substrate in the presence of thiopurine-S-methyltransferase (TPMT), it was reacted with a hydroxylamine derivative of the Alexa Fluor 647 dye. This made it possible to isolate the in vivo methylation product from the cell lysate by HPLC [82].

Using keto-AdoMet in the transmethylation reaction. Reprinted (adapted) from [82] with the permission of the publisher.
Modified propargyl-SAM analogs with increased stability were described by the Weinhold group. Pent-2-en-4-ynyl radical was transferred from the SAM analog (AdoEnYn, Fig. 12) to histone H3 in the presence of histone H3 Dim-5 lysine methyltransferase at pH 9. Histone H3 modified with a terminal alkyne can additionally bind to azide-PEG-biotin using the CuAAC reaction [83].

SAM analogs often used for labeling proteins.
The Minkui Luo group synthesized a SAM analog containing the 4-propargyloxybut-2-enyl radical (Pob-SAM, Fig. 12). The transfer of the functional fragment to the substrate did not occur in the presence of the native form of arginine methyltransferase (PRMT1). However, by rational protein engineering, mutant forms of the enzyme with changes in the SAM-binding pocket have been obtained. Thus, the Y39F/M48G-PRMT1 enzyme showed high efficiency of histone H4 modification in the presence of Pob-SAM, which at the next step was modified with a fluorescent probe in the CuAAC reaction [85]. SAM analogs with a bulky functional fragment are often inactive when using native forms of MTases, but this problem can often be solved by creating modified forms of enzymes [85–87]. In some DNA and RNA MTases, there is no significant effect of the size of the transferred fragment on the enzymatic activity. Thus, the native forms of MTases Hen1, Ecm1, M. TaqI, M. HhaI, and Dam effectively catalyze the transfer of a large fragment containing a fluorophore or biotin residue [88–93].
The Saulius Klimašauskas group investigated the stability of various alkynyl-containing SAM analogs. They have a half-life in the range 3 min to 5 h at pH 7.4 [80] and degrade along the path of alkyne hydration. The Minkui Luo group proposed replacement of the sulfur atom in SAM with selenium. For the selenium-containing derivatives of SAM (SeAM) they synthesized, no hydration of the alkynyl fragment was observed at pH 7.5. It was found that the decomposition of selenium analogs proceeds along the path of the formation of propargyl-Se-MTA and homoserine lactone (Fig. 10) [94]. The reactivity of the selenium derivative ProSeAM is higher than that of its sulfur analog, which increases the efficiency of enzymatic transfer of the functional group due to the increased activation of the selenonium center compared to the sulfonium analog [95]. The advantage of ProSeAM has been demonstrated by efficient labeling of the PKMT (protein lysine methyltransferases) series of substrates [81, 96]. The high stability of ProSeAM and its compatibility with most MTases made it possible to perform proteomic analysis of the activity of endogenous protein MTases in various cell lines [81].
Thorson and et al. studied the possibility of enzymatic synthesis of a series of SAM and SeAM derivatives from ATP and the corresponding methionine derivatives in the presence of various MAT enzymes. The catalytic subunit of human SAM synthase MATII (hMAT2A) and SAM synthase of methanogenic thermophilic archaea Methanocaldococcus jannaschii (mMAT) exhibited high substrate plasticity. It is known that the expression of the hMAT2A gene is increased in cancer cells. This may open the way for the synthesis of S/Se-AdoMet analogs as metabolic probes for studying the role of methylation in the development of tumor processes [97, 98].
The use of natural MAT enzymes in the synthesis of SAM analogs is limited to aliphatic groups at the sulfur atom of the SAM analog [99, 100]. The search for a universal SAM synthase capable of converting methionine derivatives containing an S-benzyl fragment led to the creation of the I122A/I330A form of the MAT enzyme from Cryptosporidium hominis (ChMAT). In 2020, Andrea Rentmeister’s team, through point substitutions of amino acid residues in the hydrophobic binding pocket of ChMAT, in which the methyl group of SAM is located, determined that the replacement of bulky amino acid residues with less bulky ones affects the activity of the enzyme in relation to methionine analogs containing large terminal fragments. It was found that ChMAT-I122A and ChMAT-I330A catalyzed reaction of interaction between ATP and the S-benzyl derivative of methionine with a satisfactory conversion of 5–25%. The enzyme containing a double substitution I122A/I330A (PC-ChMAT) showed a conversion of 65–70%. Comparison of X‑ray diffraction data for ChMAT and PC-ChMAT showed that these amino acid substitutions did not change the overall structure of the active site, but at the same time reduced steric hindrances in the active center. Similarly, the L147A/I351A form of thermostable SAM synthase from M. jannaschii (PC-MjMAT) was constructed; it catalyzes the conversion of sterically loaded methionine derivatives and ATP into SAM analogs at 65°C, which made it possible to use them in cascade reactions with M. TaqI, in which the optimal activity is observed at elevated temperatures [101]. In parallel, Thorson et al. investigated 38 mutant forms of hMAT2A SAM-synthases, with amino acid substitutions which affected the binding of the enzyme to the cofactor in the region of the carboxyl group of SAM. The K289L hMAT2A form exhibited a change in selectivity towards the methionine analog, L-methioninol. The resulting SAM analog exhibited increased stability [102].
Double-activated SAM analogs that do not contain a triple bond have found application in the study of MTases [70, 84, 103]. Rentmeister and colleagues, using the example of two MTases (Ecm1 and M. TaqI), demonstrated the transfer of a norbornene fragment (Fig. 13) attached via a p-xylylene linker to the sulfur atom of the SAM analog (AdoNorb and AdoNorc) to nucleic acids of different sizes. Subsequent bioconjugation with fluorophores or biotin was performed in the absence of copper (I) salts, which can be useful in a thiol-rich cell environment due to the low stability of the azides therein [104]. Benzyl fragments were transported by DNA MTase Ecm1 with catalytic efficiency higher than that for the methyl group from SAM. This demonstrates the broad applicability of the corresponding SAM analogs [105].

Norbornene SAM analogs used for labeling DNA (top) and a general diagram of the labeling process (bottom). Reprinted (adapted) from [105, Creative Commons Attribution-NonCommercial 3.0 License].
In the work of Rentmeister et al., new SAM derivatives carrying the most common photocrosslinking fragments (arylazide, diazirin, and benzophenone), attached through a benzyl linker, were obtained (Fig. 14). It was shown that the obtained photocrosslinkers can be enzymatically transferred to N7-cap-mRNA, the target of MTase Ecm1. Upon photoactivation of the modified substrate by UV radiation, the labeled RNA is crosslinked with the eIF4E cap-binding protein interacting with it [91].

SAM analogs with photocaging groups AdoBP, AdoArAz, or AdoDiaz. Reprinted (adapted) from [91] with the permission of the publisher.
Further development of bioconjugation methods has led to the creation of rewritable labels. In 2020, the Paco Fernandez-Trillo group developed a new SAM analog that has an acylhydrazone linker and a terminal azide as a substrate label (Fig. 15). The design of the label allows modification of the DNA substrates of MTases (M.TaqI and M.MpeI) and then removal of the label and followed by modification of the same substrate with functional fragments, permanently or with the possibility to be rewritten. Using the M.MpeI DNA MTase as an example, a protocol was developed for the isolation of fluorescent DNA followed by rewriting the label with biotin for isolation on streptavidin magnetic beads [106].

A SAM analog containing an acylhydrazone linker and a terminal azide (top); a general diagram of the sequential functionalization of DNA (bottom). Reprinted (adapted) from [106] with the permission of the publisher.
The relatively low stability of SAM analogs imposes restrictions on their synthesis and isolation techniques. The main problem of the classical method of synthesis arises at the last stage—the S-addition of highly electrophilic alkyl triflates or allyl bromides to the precursor of the SAM analog. This step proceeds with low yield and requires a large excess (20–200 eq) of an expensive bifunctional linker. A modification of the synthesis strategy proposed by Johan Hofkens et al. allows the synthesis of non-natural SAM analogs containing cysteine, and not homocysteine, in the amino acid portion of the molecule from the available starting compounds in high yield (Fig. 16). The proposed method changes the sequence of attachment of substituents to the sulfur atom: first, an MTA analog, modified with a functional linker or dye, is formed and then a scaffold of the SAM analog is created by attaching an amino acid moiety. It was also shown that M.TaqI MTase can transfer a large label from the unnatural SAM analog containing a PEG linker with a dye to the substrate [107].

SAM homolog, truncated by one CH2 group in the amino acid fragment and containing a fluorophore group.
The use of molecular systems with photocaging groups is an important tool in the study of interactions and functions of biomolecules [108–110]. MTases using the SAM cofactor post-synthetically methylate biomolecules, thereby modulating their activity. In many cases, this process is dynamic and reversible [111]. In 2018, SAM analogs containing photocaging groups were described for the first time (Fig. 17). They can be transferred to the substrate (DNA) in the presence of M. TaqI MTase, thereby blocking the R. TaqI restriction endonuclease binding site. Removal of the photocaging group occurs upon irradiation with UV light (365 nm) for 10–30 min. Thus, the use of these SAM analogs makes it possible to simulate natural processes involving methylation and demethylation enzymes, and to regulate these processes in time [112, 113]. Later, an enzymatic cascade containing modified SAM synthase (PC-MAT) and DNA (MTaqI) or RNA (Ecm1) MTase was proposed, which made it possible to introduce photocaging groups using ATP and methionine analogs [101, 114–118].

SAM analogs containing a photocaging group (a); and scheme of MTase-catalyzed transfer of a photoactivated group from AdoPC to DNA followed by photodegradation (b). Reprinted (adapted) from [113, Creative Commons Attribution-NonCommercial 3.0 License].
Analogs of SAM That Covalently Bind the Substrate
Another type of SAM analog are molecules capable of covalently binding to a substrate under the action of MTases. These SAM analogs most often include an aziridine ring or a Michael acceptor.
Aziridine derivatives are highly active electrophiles with a three-membered ring containing one nitrogen atom. MTA derivatives containing an aziridine group instead of a sulfur atom can react via SN2 with ring opening and the formation of a cofactor–substrate conjugate (Fig. 18). In 1998, the Weinhold group presented alkylation of a DNA substrate with an aziridine derivative of MTA in the presence of M.TaqI MTase (N6-adenine-DNA methyltransferase), while this reaction was not observed in the absence of the enzyme [119]. The described approach opened up the possibility of creating bifunctional structures containing a fluorophore or affinity label for detecting the position of a cofactor analog after conjugation [120–123].

Labeling DNA with a labeled MTA derivative, which covalently binds the substrate via an aziridine group. Reprinted (adapted) from [120] with the permission of the publisher.
Next, the 5'-N-substituted compound 1 (Fig. 19), which contains a fragment of nitrogen mustard gas and a functional group, was created [124]. Such compounds are quite stable and allow the difficulties associated with the low stability of the aziridine moiety to be avoided. The highly reactive aziridine ring is easily formed in situ upon intramolecular cyclization (Fig. 19). The transferred functional group allows further biorthogonal attachment of additional labels.

Derivative of MTA 1 and derivative of SAM 2 containing highly reactive nitrogen mustard moiety.
SAM analog 2 (Fig. 19), containing the amino acid moiety, showed a higher alkylating ability than compound 1, which is due to an increase in the binding strength of the modified cofactor with MTase [125]. The effectiveness of this approach has been shown for MTase substrates of various classes [126–128].
The influence of the amino acid portion of SAM analogs has been described by the Yongcheng Song team. A homologue of compound 2, in which the length of the amino acid moiety of the cofactor was increased by one methylene group (Fig. 20), could significantly enhance inhibition of an MTase DOT1L (methylates lysine 79 of histone H3). Thus, the IC50 value of this homologue decreased to 0.038 μM compared to 15.7 μM for compound 2. It is assumed that the homologue containing an additional CH2 group reproduces the binding of SAM to MTase to a greater extent, since two C–N bonds (~1.47 Å each) are much shorter than the C–S bonds (~1.82 Å) in SAM and SAH [51].

Compound 3 (homologue of compound 2 containing a fragment of a nitrogen mustard) inhibits the catalytic activity of the DOT1L MTase. Reprinted (adapted) from [51] with the permission of the publisher.
Aziridine derivatives are promising for use as probes for the identification of MTase substrates [129, 130]; however, a number of disadvantages limit their widespread use. First, the synthesis of such molecules is laborious: low yields at the last stages, formation of β-halide upon removal of protective groups, and low stability of the product complicate their preparation. Second, the formed bisubstrate adducts irreversibly inhibit MTases, which requires stoichiometric amounts of the enzyme for labeling. Third, the high reactivity of aziridine analogs can lead to non-specific and non-enzymatic alkylation of nucleophilic centers of DNA and protein molecules. To date, SAM aziridine analogs can only be obtained synthetically.
Despite the convenience and versatility of MTase-driven transfer of a functional group from a SAM analog to a substrate [101], in practice this approach is limited for studying the substrate–MTase interaction in situ, since substrates of other MTases present in the cell can also be labeled, which leads to loss of specificity. In this case, a direct relationship between a specific substrate–enzyme pair cannot be established.
A solution to this problem was proposed by J.K. Coward and A.E. Pegg. They developed the concept of enzyme inhibition by multisubstrate adducts [131]. The idea is based on the fact that the binding strength of a substrate and/or a cofactor with an enzyme may be low, but the binding strength of a bisubstrate consisting of covalently bound substrate and cofactor molecules is multiplied due to the loss of the entropic contribution to the free energy of binding for one of the fragments of the bisubstrate [132].
In 2016, the Zhaohui Sunny Zhou group carried out enzymatic synthesis of an analog of SAM AdoVin (S-adenosyl-L-vinthionine, Fig. 21) containing a Michael acceptor. Using thiopurine methyltransferase (TPMT) as an example, they showed that it is possible to isolate a protein from reaction mixtures in a complex with a substrate–AdoVin adduct, which made it possible to identify a new substrate of the enzyme [133].

Preparation of AdoVin from L-vinthionine and ATP. Under the action of the TPMT MTase, AdoVin forms an adduct substrate–AdoVin, which plays the role of a bisubstrate inhibitor. Reprinted (adapted) from [133] with the permission of the publisher.
MTase-Binding Analogs of SAM
For most MTases, SAH competes for the SAM binding site and often has a binding constant higher than SAM [18, 33]. SAH analogs have been proposed as agents for the selective photoinduced isolation of MTases and further study of adducts by mass spectrometry. For the first time, “hook” compounds for SAM-dependent MTases were synthesized by Weinhold’s group [134]. Such “hooks” consist of three parts: a SAH fragment for directed non-covalent binding to enzymes (Fig. 22 (1)), a photocaging group for covalent cross-linking with an enzyme (Fig. 22 (2)), and a biotin-containing fragment, which serves for the subsequent isolation of the resulting adducts using magnetic beads with immobilized streptavidin (Fig. 22 (3)). SAH was chosen as the targeting group to which the remaining fragments were attached via amino linkers at the N-6 or C-8 position of the purine residue (Fig. 22).

General structure of a trifunctional “hook” molecule [134]. 1, SAH fragment; 2, photocaging group; and 3, biotin residue.
The effectiveness of these compounds has been demonstrated by the example of various MTases acting on DNA, RNA, and proteins. These compounds formed a covalent crosslink upon irradiation and allowed for isolation and identification of SAH-linked proteins from complex mixtures, such as cell lysate. The efficiency of isolation of MTases without covalent crosslinking by the photocaging group turned out to be significantly lower [134, 135]. On the basis of this scaffold, “hooks” containing fluorescent labels have also been obtained [136, 137].
Iredia Iyamu and Rong Huang obtained an azo analog of the SAH coproduct in their 2020 work (Fig. 23). It is a bisubstrate MTase inhibitor equipped with a fluorescent dye attached via a long linker to the N-6 position of the adenine ring. The high binding constant for NNMT MTase allowed the development of a competitive assay to search for new NNMT inhibitors using fluorescence polarization (FP). Importantly, FP competitive assay allows identification of inhibitors that directly or allosterically interfere with the binding of the cofactor to the active site of NNMT. The high reproducibility of the method makes it possible to use it for high-throughput screening of NNMT inhibitors [138].

The structure of a bisubstrate NNMT inhibitor with a fluorescent label [138].
Fluorescent SAM analogs can be used to determine the binding constant of the cofactor not only with MTases, but also with other SAM-binding molecules. Thus, by measuring the fluorescence of the DAPSM analog (Fig. 24), the binding constant of SAM with an RNA riboswitch (III riboswitch RNA) was determined [139–141].

Some fluorescent derivatives of SAM. Reprinted (adapted) from [8] with the permission of the publisher.
CONCLUSION
The examples above allow us to conclude that the possibilities of using synthetic SAM analogs in the study of MTases and their substrates are vast.
Over the past two decades, a huge variety of synthetic SAM analogs have been developed, each with its own specific properties. Interest in such compounds is growing from year to year. This is facilitated by the constantly expanding range of tasks in which such molecules can be used. These tasks include the control of MTase activity using selective low-molecular-weight inhibitors, the establishment of MTase functions using unique labels, and various modifications of MTase substrates.
However, there are still many unresolved issues. For example, most SAM analogs are poorly stable under the conditions required for further reactions involving MTases. Another difficulty is that the structural differences between SAM analogs and the natural cofactor usually affect the strength of their binding to MTases. Synthetic molecules may not be well recognized by MTase; they can bind, yet not enter, the subsequent enzymatic reaction; or they can bind to the enzyme, react, and, due to the high affinity, not be released from the MTase active center after the end of the enzymatic reaction. The question of the non-specificity of MTases also remains unresolved when using SAM analogs in cellular systems (in vivo or in vitro). The identification, modification, or isolation of the substrate of the studied MTase is complicated by the presence of a large number of other MTases in the cell competing for the cofactor, which leads to a nonspecific transfer of the functional group to non-target substrates.
Nevertheless, the constant expansion of the synthetic base, as well as the continuously growing number of studies in the field of the development of new molecules that meet certain properties required for the study of MTases, give rise to the hope that many of these problems will soon be overcome.
REFERENCES
Moore L.D., Le T., Fan G. 2013. DNA methylation and its basic function. Neuropsychopharmacology. 38 (1), 23–38.
Chen X., Sun Y.Z., Liu H., Zhang L., Li J.Q., Meng J. 2017. RNA methylation and diseases: Experimental results, databases, Web servers and computational models. Brief. Bioinformatics. 20 (3), 896–917.
Clarke S. 1993. Protein methylation. Curr. Opin. Cell Biol. 5 (6), 977–983.
Lee D.Y., Teyssier C., Strahl B.D., Stallcup M.R. 2005. Role of protein methylation in regulation of transcription. Endocrine Rev. 26 (2), 147–170.
Noel J.P., Dixon R.A., Pichersky E., Zubieta C., Ferrer J.L. 2003. Chapter two: Structural, functional, and evolutionary basis for methylation of plant small molecules. Recent Adv. Phytochem. 37 (C), 37–58.
Stojković V., Fujimori D.G. 2017. Mutations in RNA methylating enzymes in disease. Curr. Opin. Chem. Biol. 41, 20–27.
Bateman A., Martin M.J., Orchard S., Magrane M., Agivetova R., Ahmad S., Alpi E., Bowler-Barnett E.H., Britto R., Bursteinas B., Bye-A-Jee H., Coetzee R., Cukura A., Silva A.Da, Denny P., et al. 2021. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49 (D1), D480–D489.
Zhang J., Zheng Y.G. 2016. SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chem. Biol. 11 (3), 583–597.
Martin J.L., McMillan F.M. 2002. SAM (dependent. I AM: The S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12 (6), 783–793.
Cornelissen N.V., Michailidou F., Muttach F., Rau K., Rentmeister A. 2020. Nucleoside-modified AdoMet analogues for differential methyltransferase targeting. Chem. Commun. 56 (14), 2115–2118.
Greenberg M.V.C., Bourc’his D. 2019. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 20 (10), 590–607.
Koonin E.V., Makarova K.S., Wolf Y.I. 2017. Evolutionary genomics of defense systems in archaea and bacteria. Annu. Rev. Microbiol. 71 (1), 233–261.
Sergiev P.V., Aleksashin N.A., Chugunova A.A., Polikanov Y.S., Dontsova O.A. 2018. Structural and evolutionary insights into ribosomal RNA methylation. Nat. Chem. Biol. 14 (3), 226–235.
Wilson D.N. 2014. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12 (1), 35–48.
Boriack-Sjodin P.A., Swinger K.K. 2016. Protein methyltransferases: A distinct, diverse, and dynamic family of enzymes. Biochemistry. 55 (11), 1557–1569.
Heurgu V., Champ S. 2002. The hemK gene in Escherichia coli encodes the N5-glutamine methyltransferase that modifies peptide release factors. EMBO J. 21 (4), 769–778.
Jambhekar A., Dhall A., Shi Y. 2019. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell. Biol. 20 (10), 625–641.
Liscombe D.K., Louie G.V., Noel J.P. 2012. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat. Prod. Rep. 29 (10), 1238–1250.
Zhang C., Sultan S.A., Rehka T., Chen X. 2021. Biotechnological applications of S-adenosyl-methionine-dependent methyltransferases for natural products biosynthesis and diversification. Bioresources Bioprocessing. 8 (1), 1–21.
Fauman E.B., Blumenthal R.M., Cheng X. 1999. Structure and evolution of AdoMet-dependent methyltransferases. S-Adenosylmethionine-Dependent Methyltransferases. 1–38.
Borchardt R.T., Wu Y.S. 1976. Potential inhibitors of S‑adenosylmethionine-dependent methyltransferases. 5. Role of the asymmetric sulfonium pole in the enzymatic binding of S-adenosyl-L-methionine. J. Med. Chem. 19 (9), 1099–1103.
de La Haba G., Jamieson G.A., Mudd S.H., Richards H.H. 1959. S-Adenosylmethionine: The relation of configuration at the sulfonium center to enzymatic reactivity. J. Am. Chem. Soc. 81 (15), 3975–3980.
Fontecave M., Atta M., Mulliez E. 2004. S-Adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 29 (5), 243–249.
Markham G.D., Pajares M.A. 2009. Structure-function relationships in methionine adenosyltransferases. Cell. Mol. Life Sci. 66 (4), 636–648.
Popadić D., Mhaindarkar D., Dang Thai M.H.N., Hailes H.C., Mordhorst S., Andexer J.N. 2021. A bicyclic: S-Adenosylmethionine regeneration system applicable with different nucleosides or nucleotides as cofactor building blocks. RSC Chem. Biol. 2 (3), 883–891.
Park J., Tai J., Roessner C.A., Scott A.I. 1996. Enzymatic synthesis of S-adenosyl-L-methionine on the preparative scale. Bioorg. Med. Chem. 4 (12), 2179–2185.
Davis T.D., Kunakom S., Burkart M.D., Eustaquio A.S. 2018. Preparation, assay, and application of chlorinase SalL for the chemoenzymatic synthesis of S-adenosyl-L-methionine and analogs. Meth. Enzymol. 604, 367–388.
Walsby C.J., Hong W., Broderick W.E., Cheek J., Ortillo D., Broderick J.B., Hoffman B.M. 2002. Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe-4S]+ cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 124 (12), 3143–3151.
Sadler J.C., Humphreys L.D., Snajdrova R., Burley G.A. 2017. A tandem enzymatic sp2-C-methylation process: coupling in situ S-adenosyl-L-methionine formation with methyl transfer. ChemBioChem. 18 (11), 992–995.
Hayakawa K., Kajihata S., Matsuda F., Shimizu H. 2015. 13C-metabolic flux analysis in S-adenosyl-L-methionine production by Saccharomyces cerevisiae. J. Biosci. Bioeng. 120 (5), 532–538.
Moriya S.S., Samejima K., Taira H., Hiramatsu K., Kawakita M. 2020. ESI-Q-TOF-MS determination of polyamines and related enzyme activity for elucidating cellular polyamine metabolism. Anal. Biochem. 607, 113831.
Matos J.R., Wong C.H. 1987. S-Adenosylmethionine: stability and stabilization. Bioorg. Chem. 15 (1), 71–80.
Mariasina S.S., Chang C.F., Petrova O.A., Efimov S.V., Klochkov V.V., Kechko O.I., Mitkevich V.A., Sergiev P.V., Dontsova O.A., Polshakov V.I. 2020. Williams–Beuren syndrome-related methyltransferase WBSCR27: Cofactor binding and cleavage. FEBS J. 287 (24), 5375–5393.
Yeates T.O. 2002. Structures of SET domain proteins: Protein lysine methyltransferases make their mark. Cell. 111 (1), 5–7.
Dixon M.M., Huang S., Matthews R.G., Ludwig M. 1996. The structure of the C-terminal domain of methionine synthase: Presenting S-adenosylmethionine for reductive methylation of B12. Structure. 4 (11), 1263–1275.
Huber T.D., Wang F., Singh S., Johnson B.R., Zhang J., Sunkara M., Van Lanen S.G., Morris A.J., Phillips G.N., Thorson J.S. 2016. Functional AdoMet isosteres resistant to classical AdoMet degradation pathways. ACS Chem. Biol. 11 (9), 2484–2491.
McKean I.J.W., Sadler J.C., Cuetos A., Frese A., Humphreys L.D., Grogan G., Hoskisson P.A., Burley G.A. 2019. S-Adenosylmethionine cofactor modifications enhance the biocatalytic repertoire of small molecule C-alkylation. Angew. Chem. Int. Ed. 58 (49), 17583–17588.
Vranken C., Fin A., Tufar P., Hofkens J., Burkart M.D., Tor Y. 2016. Chemoenzymatic synthesis and utilization of a SAM analog with an isomorphic nucleobase. Org. Biomol. Chem. 14 (26), 6189–6192.
Alferov K.V., Zhukov Y.N., Khurs E.N., Khomutov R.M. 2003. Stable organophosphorus analogues of S-adenosylmethionine and S-methylmethionine. Mendeleev Commun. 13 (6), 243–244.
Zhukov Yu.N., Khomutov A.R., Osipova T.I., Khomutov R.N. 1999. Synthesis of phosphine analogs of sulfur-containing amino acids. Izv. ASkd. Nauk, Ser. Khim., no. 7, 1360–1363
Ueland P.M. 1982. Pharmacological and biochemical aspects of S-adenosylhomocysteine and S-adenosylhomocysteine hydrolase. Pharmacol. Rev. 34 (3), 223–253.
Walker R.D., Duerre J.A. 1975. S-Adenosylhomocysteine metabolism in various species. Can. J. Biochem. 53 (3), 312–319.
Parveen N., Cornell K.A. 2011. Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 79 (1), 7–20.
Medina-Franco J.L., Méndez-Lucio O., Dueñas-González A., Yoo J. 2015. Discovery and development of DNA methyltransferase inhibitors using in silico approaches. Drug Discov. Today. 20 (5), 569–577.
Richart L., Margueron R. 2020. Drugging histone methyltransferases in cancer. Curr. Opin. Chem. Biol. 56, 51–62.
Lin Q., Jiang F., Schultz P.G., Gray N.S. 2001. Design of allele-specific protein methyltransferase inhibitors. J. Am. Chem. Soc. 123 (47), 11608–11613.
Zwergel C., Valente S., Mai A. 2016. DNA methyltransferases inhibitors from natural sources. Curr. Top. Med. Chem. 16 (7), 680–696.
Saldívar-González F.I., Gómez-García A., Chávez-Ponce De León D.E., Sánchez-Cruz N., Ruiz-Rios J., Pilón-Jiménez B.A., Medina-Franco J.L. 2018. Inhibitors of DNA methyltransferases from natural sources: A computational perspective. Front. Pharmacol. 9 (OCT), 1144.
Yu W., Chory E.J., Wernimont A.K., Tempel W., Scopton A., Federation A., Marineau J.J., Qi J., Barsyte-Lovejoy D., Yi J., Marcellus R., Iacob R.E., Engen J.R., Griffin C., Aman A., Wienholds E., et al. 2012. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 3 (1), 1–12.
Gao Y., Van Haren M.J., Moret E.E., Rood J.J.M., Sartini D., Salvucci A., Emanuelli M., Craveur P., Babault N., Jin J., Martin N.I. 2019. Bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) with enhanced activity. J. Med. Chem. 62 (14), 6597–6614.
Yao Y., Chen P., Diao J., Cheng G., Deng L., Anglin J.L., Prasad B.V.V., Song Y. 2011. Selective inhibitors of histone methyltransferase DOT1L: Design, synthesis, and crystallographic studies. J. Am. Chem. Soc. 133 (42), 16746–16749.
Rugo H.S., Jacobs I., Sharma S., Scappaticci F., Paul T.A., Jensen-Pergakes K., Malouf G.G. 2020. The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: A narrative review. Adv. Ther. 37 (7), 3059–3082.
Zheng W., Ibáñez G., Wu H., Blum G., Zeng H., Dong A., Li F., Hajian T., Allali-Hassani A., Amaya M.F., Siarheyeva A., Yu W., Brown P.J., Schapira M., Vedadi M., Min J., Luo M. 2012. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 134 (43), 18004–18014.
Tisi D., Chiarparin E., Tamanini E., Pathuri P., Coyle J.E., Hold A., Holding F.P., Amin N., Martin A.C.L., Rich S.J., Berdini V., Yon J., Acklam P., Burke R., Drouin L., et al. 2016. Structure of the epigenetic oncogene MMSET and inhibition by N-alkyl sinefungin derivatives. ACS Chem. Biol. 11 (11), 3093–3105.
Chen D., Dong C., Dong G., Srinivasan K., Min J., Noinaj N., Huang R. 2020. Probing the plasticity in the active site of protein N-terminal methyltransferase 1 using bisubstrate analogues. J. Med. Chem. 63 (15), 8419–8431.
Bobiļeva O., Bobrovs R., Kaņepe I., Patetko L., Kalniņš G., Šišovs M., Bula A.L., Grī Nberga S., Borodušķis M.R., Ramata-Stunda A., Rostoks N., Jirgensons A., Tā Rs K., Jaudzems K. 2021. Potent SARS-CoV-2 mRNA cap methyltransferase inhibitors by bioisosteric replacement of methionine in SAM cosubstrate. ACS Med. Chem. Lett. 12 (7), 1102–1107.
Bonday Z.Q., Cortez G.S., Grogan M.J., Antonysamy S., Weichert K., Bocchinfuso W.P., Li F., Kennedy S., Li B., Mader M.M., Arrowsmith C.H., Brown P.J., Eram M.S., Szewczyk M.M., Barsyte-Lovejoy D., Vedadi M., et al. 2018. LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med. Chem. Lett. 9 (7), 612–617.
Leone G., Voso M.T., Teofili L., Lübbert M. 2003. Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS. Clin. Immunol. 109 (1), 89–102.
Silverman L.R. 2004. DNA methyltransferase inhibitors in myelodysplastic syndrome. Best Pract. Res. Clin. Haematol. 17 (4), 585–594.
Shimba S., Bokar J.A., Rottman F., Reddy R. 1995. Accurate and efficient N-6-adenosine methylation in spliceosomal U6 small nucelar RNA by HeLa cell extract in vitro. Nucleic Acids Res. 23 (13), 2421–2426.
Siegrist J., Netzer J., Mordhorst S., Karst L., Gerhardt S., Einsle O., Richter M., Andexer J.N. 2017. Functional and structural characterisation of a bacterial O-methyltransferase and factors determining regioselectivity. FEBS Lett. 591 (2), 312–321.
Golovina A.Y., Sergiev P.V., Golovin A.V., Serebryakova M.V., Demina I., Govorun V.M., Dontsova O.A. 2009. The yfiC gene of E. coli encodes an adenine-N6- methyltransferase that specifically modifies A37 of tRNA1Val(cmo5UAC). RNA. 15 (6), 1134–1141.
Mendel M., Chen K.M., Homolka D., Gos P., Pandey R.R., McCarthy A.A., Pillai R.S. 2018. Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol. Cell. 71 (6), 986–1000.e11.
Rubin R.A., Modrich P. 1977. EcoRI methylase. Physical and catalytic properties of the homogeneous enzyme. J. Biol. Chem. 252 (20), 7265–7272.
Hevel J.M., Price O.M. 2020. Rapid and direct measurement of methyltransferase activity in about 30 min. Methods 175, 3–9.
Wu J., Xie N., Feng Y., Zheng Y.G. 2012. Scintillation proximity assay of arginine methylation. J. Biomol. Screening. 17 (2), 237–244.
Ero R., Peil L., Liiv A., Remme J. 2008. Identification of pseudouridine methyltransferase in Escherichia coli. RNA. 14 (10), 2223–2233.
Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., Dai Q., Chen W., He C. 2014. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10 (2), 93–95.
Morvan D., Demidem A., Guenin S., Madelmont J.C. 2006. Methionine-dependence phenotype of tumors: Metabolite profiling in a melanoma model using L-[methyl-13C]methionine and high-resolution magic angle spinning 1H-13C nuclear magnetic resonance spectroscopy. Magn. Reson. Med. 55 (5), 984–996.
Lukinavičius G., Lapiene V., Staševskij Z., Dalhoff C., Weinhold E., Klimašauskas S. 2007. Targeted labeling of DNA by methyltransferase-directed transfer of activated groups (mTAG). J. Am. Chem. Soc. 129 (10), 2758–2759.
Binda O., Boyce M., Rush J.S., Palaniappan K.K., Bertozzi C.R., Gozani O. 2011. A chemical method for labeling lysine methyltransferase substrates. ChemBioChem. 12 (2), 330–334.
Dippe M., Brandt W., Rost H., Porzel A., Schmidt J., Wessjohann L.A. 2015. Rationally engineered variants of S-adenosylmethionine (SAM) synthase: Reduced product inhibition and synthesis of artificial cofactor homologues. Chem. Commun. 51 (17), 3637–3640.
Ho D.K., Wu J.C., Santi D.V., Floss H.G. 1991. Stereochemical studies of the C-methylation of deoxycytidine catalyzed by HhaI methylase and the N-methylation of deoxyadenosine catalyzed by EcoRI methylase. Arch. Biochem. Biophys. 284 (2), 264–269.
Dalhoff C., Lukinavičius G., Klimašauskas S., Weinhold E. 2006. Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat. Chem. Biol. 2 (1), 31–32.
Dalhoff C., Lukinavičius G., Klimašauskas S., Weinhold E. 2006. Synthesis of S-adenosyl-L-methionine analogs and their use for sequence-specific transalkylation of DNA by methyltransferases. Nat. Protocols. 1 (4), 1879–1886.
Jalali E., Thorson J.S. 2021. Enzyme-mediated bioorthogonal technologies: Catalysts, chemoselective reactions and recent methyltransferase applications. Curr. Opin. Biotechnol. 69, 290–298.
Tomkuvienė M., Mickutė M., Vilkaitis G., Klimašauskas S. 2019. Repurposing enzymatic transferase reactions for targeted labeling and analysis of DNA and RNA. Curr. Opin. Biotechnol. 55, 114–123.
Muttach F., Rentmeister A. 2016. One-pot modification of 5′-capped RNA based on methionine analogs. Methods. 107, 3–9.
Deen J., Vranken C., Leen V., Neely R.K., Janssen K.P.F., Hofkens J. 2017. Methyltransferase-directed labeling of biomolecules and its applications. Angew. Chem. Int. Ed. 56 (19), 5182–5200.
Lukinavičius G., Tomkuvienè M., Masevičius V., Klimašauskas S. 2013. Enhanced chemical stability of AdoMet analogues for improved methyltransferase-directed labeling of DNA. ACS Chem. Biol. 8 (6), 1134–1139.
Bothwell I.R., Islam K., Chen Y., Zheng W., Blum G., Deng H., Luo M. 2012. Se-Adenosyl-L-selenomethionine cofactor analogue as a reporter of protein methylation. J. Am. Chem. Soc. 134 (36), 14905–14912.
Lee B.W.K., Sun H.G., Zang T., Ju-Kim B., Alfaro J.F., Zhou Z.S. 2010. Enzyme-catalyzed transfer of a ketone group from an S-adenosyl-L-methionine analogue: A tool for the functional analysis of methyltransferases. J. Am. Chem. Soc. 132 (11), 3642–3643.
Peters W., Willnow S., Duisken M., Kleine H., Macherey T., Duncan K.E., Litchfield D.W., Lüscher B., Weinhold E. 2010. Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angew. Chem. Int. Ed. 49 (30), 5170–5173.
Huber T.D., Johnson B.R., Zhang J., Thorson J.S. 2016. AdoMet analog synthesis and utilization: Current state of the art. Curr. Opin. Biotechnol. 42, 189–197.
Wang R., Zheng W., Yu H., Deng H., Luo M. 2011. Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-L-methionine analogues. J. Am. Chem. Soc. 133 (20), 7648–7651.
Islam K., Bothwell I., Chen Y., Sengelaub C., Wang R., Deng H., Luo M. 2012. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-L-methionine. J. Am. Chem. Soc. 134 (13), 5909–5915.
Islam K., Zheng W., Yu H., Deng H., Luo M. 2011. Expanding cofactor repertoire of protein lysine methyltransferase for substrate labeling. ACS Chem. Biol. 6 (7), 679–684.
Kunkel F., Lurz R., Weinhold E. 2015. A 7-deazaadenosylaziridine cofactor for sequence-specific labeling of DNA by the DNA cytosine-c5 methyltransferase M.HhaI. Molecules. 20 (11), 20805–20822.
Plotnikova A., Osipenko A., Masevičius V., Vilkaitis G., Klimašauskas S. 2014. Selective covalent labeling of miRNA and siRNA duplexes using HEN1 methyltransferase. J. Am. Chem. Soc. 136 (39), 13550–13553.
Flade S., Jasper J., Gieß M., Juhasz M., Dankers A., Kubik G., Koch O., Weinhold E., Summerer D. 2017. The N6-position of adenine is a blind spot for TAL-effectors that enables effective binding of methylated and fluorophore-labeled DNA. ACS Chem. Biol. 12 (7), 1719–1725.
Muttach F., Mäsing F., Studer A., Rentmeister A. 2017. New AdoMet analogues as tools for enzymatic transfer of photo-cross-linkers and capturing RNA–protein interactions. Chemistry—A Eur. J. 23 (25), 5988–5993.
Holstein J.M., Muttach F., Schiefelbein S.H.H., Rentmeister A. 2017. Dual 5′ cap labeling based on regioselective RNA methyltransferases and bioorthogonal reactions. Chemistry—A Eur. J. 23 (25), 6165–6173.
Holstein J.M., Anhäuser L., Rentmeister A. 2016. Modifying the 5′-cap for click reactions of eukaryotic mRNA and to tune translation efficiency in living cells. Angew. Chem. Int. Ed. 55 (36), 10899–10903.
Bothwell I.R., Luo M. 2014. Large-scale, protection-free synthesis of Se-adenosyl-L-selenomethionine analogues and their application as cofactor surrogates of methyltransferases. Organic Lett. 16 (11), 3056–3059.
Iwig D.F., Grippe A.T., McIntyre T.A., Booker S.J. 2004. Isotope and elemental effects indicate a rate-limiting methyl transfer as the initial step in the reaction catalyzed by Escherichia coli cyclopropane fatty acid synthase. Biochemistry. 43 (42), 13510–13524.
Willnow S., Martin M., Lüscher B., Weinhold E. 2012. A selenium-based click AdoMet analogue for versatile substrate labeling with wild-type protein methyltransferases. ChemBioChem. 13 (8), 1167–1173.
Singh S., Zhang J., Huber T.D., Sunkara M., Hurley K., Goff R.D., Wang G., Zhang W., Liu C., Rohr J., Van Lanen S.G., Morris A.J., Thorson J.S. 2014. Facile chemoenzymatic strategies for the synthesis and utilization of S-adenosyl-L-methionine analogues. Angew. Chem. Int. Ed. 53 (15), 3965–3969.
Niland C.N., Ghosh A., Cahill S.M., Schramm V.L. 2021. Mechanism and inhibition of human methionine adenosyltransferase 2A. Biochemistry. 60 (10), 791–801.
Wang R., Islam K., Liu Y., Zheng W., Tang H., Lailler N., Blum G., Deng H., Luo M. 2013. Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J. Am. Chem. Soc. 135 (3), 1048–1056.
Lukinavičius G., Lapinaite A., Urbanavičiute G., Gerasimaite R., Klimašauskas S. 2012. Engineering the DNA cytosine-5 methyltransferase reaction for sequence-specific labeling of DNA. Nucleic Acids Res. 40 (22), 11594–11602.
Michailidou F., Klöcker N., Cornelissen N.V., Singh R.K., Peters A., Ovcharenko A., Kümmel D., Rentmeister A. 2021. Engineered SAM synthetases for enzymatic generation of AdoMet analogs with photocaging groups and reversible DNA modification in cascade reactions. Angew. Chem. Int. Ed. 60 (1), 480–485.
Huber T.D., Clinger J.A., Liu Y., Xu W., Miller M.D., Phillips G.N., Thorson J.S. 2020. Methionine adenosyltransferase engineering to enable bioorthogonal platforms for AdoMet-utilizing enzymes. ACS Chem. Biol. 15 (3), 695–705.
van Dülmen M., Muthmann N., Rentmeister A. 2021. Chemo-enzymatic modification of the 5′ Cap maintains translation and increases immunogenic properties of mRNA. Angew. Chem. Int. Ed. 60 (24), 13280–13286.
Hong V., Presolski S.I., Ma C., Finn M.G. 2009. Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 48 (52), 9879–9883.
Muttach F., Muthmann N., Reichert D., Anhäuser L., Rentmeister A. 2017. A benzylic linker promotes methyltransferase catalyzed norbornene transfer for rapid bioorthogonal tetrazine ligation. Chem. Sci. 8 (12), 7947–7953.
Wilkinson A.A., Jagu E., Ubych K., Coulthard S., Rushton A.E., Kennefick J., Su Q., Neely R.K., Fernandez-Trillo P. 2020. Site-selective and rewritable labeling of DNA through enzymatic, reversible, and click chemistries. ACS Cent. Sci. 6 (4), 525–534.
Goyvaerts V., Van Snick S., D’Huys L., Vitale R., Helmer Lauer M., Wang S., Leen V., Dehaen W., Hofkens J. 2020. Fluorescent SAM analogues for methyltransferase based DNA labeling. Chem. Commun. 56 (22), 3317–3320.
Liu Q., Deiters A. 2014. Optochemical control of deoxyoligonucleotide function via a nucleobase-caging approach. Acc. Chem. Res. 47 (1), 45–55.
Hemphill J., Govan J., Uprety R., Tsang M., Deiters A. 2014. Site-specific promoter caging enables optochemical gene activation in cells and animals. J. Am. Chem. Soc. 136 (19), 7152–7158.
Vaníková Z., Janoušková M., Kambová M., Krásný L., Hocek, M. 2019. Switching transcription with bacterial RNA polymerase through photocaging, photorelease and phosphorylation reactions in the major groove of DNA. Chem. Sci. 10 (14), 3937–3942.
Klose R.J., Zhang Y. 2007. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8 (4), 307–318.
Heimes M., Kolmar L., Brieke C. 2018. Efficient cosubstrate enzyme pairs for sequence-specific methyltransferase-directed photolabile caging of DNA. Chem. Commun. 54(90), 12718–12721.
Anhäuser L., Muttach F., Rentmeister A. 2018. Reversible modification of DNA by methyltransferase-catalyzed transfer and light-triggered removal of photo-caging groups. Chem. Commun. 54 (5), 449–451.
Anhäuser L., Klöcker N., Muttach F., Mäsing F., Špaček P., Studer A., Rentmeister A. 2020. A benzophenone-based photocaging strategy for the N7 position of guanosine. Angew. Chem. Int. Ed. 59 (8), 3161–3165.
Park M., Patel N., Keung A.J., Khalil A.S. 2019. Engineering epigenetic regulation using synthetic read-write modules. Cell. 176 (1–2), 227–238. e20.
Xie Q., Wu T.P., Gimple R.C., Li Z., Prager B.C., Wu Q., Yu Y., Wang P., Wang Y., Gorkin D.U., Zhang C., Dowiak A.V., Lin K., Zeng C., Sui Y., et al. 2018. N6-methyladenine DNA modification in glioblastoma. Cell. 175 (5), 1228–1243.e20.
Ovcharenko A., Weissenboeck F.P., Rentmeister A. 2021. Tag-free internal RNA labeling and photocaging based on mRNA methyltransferases. Angew. Chem. Int. Ed. 60 (8), 4098–4103.
Michailidou F., Rentmeister A. 2021. Harnessing methylation and AdoMet-utilising enzymes for selective modification in cascade reactions. Org. Biomol. Chem. 19 (17), 3756–3762.
Pignot M., Siethoff C., Linscheid M., Weinhold E. 1998. Coupling of a nucleoside with DNA by a methyltransferase. Angew. Chem. Int. Ed. 37 (20), 2888–2891.
Pljevaljcic G., Pignot M., Weinhold E. 2003. Design of a new fluorescent cofactor for DNA methyltransferases and sequence-specific labeling of DNA. J. Am. Chem. Soc. 125 (12), 3486–3492.
Comstock L.R., Rajski S.R. 2005. Conversion of DNA methyltransferases into azidonucleosidyl transferases via synthetic cofactors. Nucleic Acids Res. 33 (5), 1644–1652.
Comstock L.R., Rajski S.R. 2005. Methyltransferase-directed DNA strand scission. J. Am. Chem. Soc. 127 (41), 14136–14137.
Townsend A.P., Roth S., Williams H.E.L., Stylianou E., Thomas N.R. 2009. New S-adenosyl-L-methionine analogues: Synthesis and reactivity studies. Org. Lett. 11 (14), 2976–2979.
Weller R.L., Rajski S.R. 2005. DNA methyltransferase-moderated click chemistry. Org. Lett. 7 (11), 2141–2144.
Weller R.L., Rajski S.R. 2006. Design, synthesis, and preliminary biological evaluation of a DNA methyltransferase-directed alkylating agent. ChemBioChem. 7 (2), 243–245.
Du Y., Hendrick C.E., Frye K.S., Comstock L.R. 2012. Fluorescent DNA labeling by N-mustard analogues of S-adenosyl-L-methionine. ChemBioChem. 13 (15), 2225–2233.
Zhang C., Weller R.L., Thorson J.S., Rajski S.R. 2006. Natural product diversification using a non-natural cofactor analogue of S-adenosyl-L-methionine. J. Am. Chem. Soc. 128 (9), 2760–2761.
Hymbaugh Bergman S.J., Comstock L.R. 2015. N‑mustard analogs of S-adenosyl-L-methionine as biochemical probes of protein arginine methylation. Bioorg. Med. Chem. 23 (15), 5050–5055.
Sirasunthorn N., Jailwala A., Gerber A., Comstock L.R. 2019. Evaluation of N-mustard analogues of S-adenosyl-L-methionine with eukaryotic DNA methyltransferase 1. ChemistrySelect. 4 (35), 10525–10531.
Hymbaugh S.J., Pecor L.M., Tracy C.M., Comstock L.R. 2019. Protein arginine methyltransferase 1-dependent labeling and isolation of histone H4 through N-mustard analogues of S-adenosyl-L-methionine. ChemBioChem. 20 (3), 379–384.
Coward J.K., Pegg A.E. 1987. Specific multisubstrate adduct inhibitors of aminopropyltransferases and their effect on polyamine biosynthesis in cultured cells. Adv. Enzyme Regulat. 26 (C), 107–113.
Polshakov V.I., Batuev E.A., Mantsyzov A.B. 2019. Methods of NMR spectroscopy for screening and analysis of biotarget–ligand interactions. Usp. Khim. 88 (1), 59–98.
Qu W., Catcott K.C., Zhang K., Liu S., Guo J.J., Ma J., Pablo M., Glick J., Xiu Y., Kenton N., Ma X., Duclos R.I., Zhou Z.S. 2016. Capturing unknown substrates via in situ formation of tightly bound bisubstrate adducts: S-Adenosyl-vinthionine as a functional probe for AdoMet-dependent methyltransferases. J. Am. Chem. Soc. 138 (9), 2877–2880.
Dalhoff C., Hüben M., Lenz T., Poot P., Nordhoff E., Köster H., Weinhold E. 2010. Synthesis of S-adenosyl-L-homocysteine capture compounds for selective photoinduced isolation of methyltransferases. ChemBioChem. 11 (2), 256–265.
Lenz T., Poot P., Gräbner O., Glinski M., Weinhold E., Dreger M., Köster H. 2010. Profiling of methyltransferases and other S-adenosyl-L-homocysteine-binding proteins by Capture Compound Mass Spectrometry (CCMS). J. Visualized Exp. 46(46), 2264.
Brown L.J., Baranowski M., Wang Y., Schrey A.K., Lenz T., Taverna S.D., Cole P.A., Sefkow M. 2014. Using S-adenosyl-L-homocysteine capture compounds to characterize S-adenosyl-L-methionine and S-adenosyl-L-homocysteine binding proteins. Anal. Biochem. 467, 14–21.
Hong W., Dowden J. 2011. Facile synthesis of N-6 adenosine modified analogue toward S-adenosyl methionine derived probe for protein arginine methyltransferases. Chinese Chem. Lett. 22 (12), 1439–1442.
Iyamu I.D., Huang R. 2020. Development of fluorescence polarization-based competition assay for nicotinamide N-methyltransferase. Anal. Biochem. 604, 113833.
Burgos E.S., Walters R.O., Huffman D.M., Shechter D. 2017. A simplified characterization of: S-adenosyl-L-methionine-consuming enzymes with 1-Step EZ-MTase: A universal and straightforward coupled-assay for in vitro and in vivo setting. Chem. Sci. 8 (9), 6601–6612.
Ottink O.M., Nelissen F.H.T., Derks Y., Wijmenga S.S., Heus H.A. 2010. Enzymatic stereospecific preparation of fluorescent S-adenosyl-L-methionine analogs. Anal. Biochem. 396 (2), 280–283.
Hickey S.F., Hammond M.C. 2014. Structure-guided design of fluorescent S-adenosyl-L methionine analogs for a high-throughput screen to target SAM-I riboswitch RNAs. Chem. Biol. 21 (3), 345–356.
Funding
This work was supported by the Russian Foundation for Basic Research (project no. 20-04-00318) and the Interdisciplinary Scientific and Educational School of Moscow University “Molecular Technologies of Living Systems and Synthetic Biology.”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests. The authors declare no conflicts of interest.
This article does not contain any research involving humans or animals as research objects.
Additional information
Translated by N. Onishchenko
Abbreviations: MTase, methyltransferase; NNMT, nicotinamide-N-methyltransferase; SAM, AdoMet, or S-adenosyl-L-methionine; MAT, methionine adenosyltransferase; mTAG, methyltransferase-directed transfer of activated groups; CuAAC, copper-catalyzed azide–alkyne cycloaddition; PC, photo-caging groups; PEG linker, hydrophobic polyethylene glycol fragment; FP, fluorescence polarization (fluorescence anisotropy).
Rights and permissions
About this article

Cite this article
Rudenko, A.Y., Mariasina, S.S., Sergiev, P.V. et al. Analogs of S-Adenosyl-L-Methionine in Studies of Methyltransferases. Mol Biol 56, 229–250 (2022). https://doi.org/10.1134/S002689332202011X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S002689332202011X