
Abstract
One of the latest methods in modern molecular biology is labeling genomic loci in living cells using fluorescently labeled Cas protein. The NIH Foundation has made the mapping of the 4D nucleome (the three-dimensional nucleome on a timescale) a priority in the studies aimed to improve our understanding of chromatin organization. Fluorescent methods based on CRISPR–Cas are a significant step forward in visualization of genomic loci in living cells. This approach can be used for studying epigenetics, cell cycle, cellular response to external stimuli, rearrangements during malignant cell transformation, such as chromosomal translocations or damage, as well as for genome editing. In this review, we focused on the application of CRISPR–Cas fluorescence technologies as components of multimodal imaging methods for in vivo mapping of chromosomal loci, in particular, attribution of fluorescence signal to morphological and anatomical structures in a living organism. The review discusses the approaches to the highly sensitive, high-precision labeling of CRISPR–Cas components, delivery of genetically engineered constructs into cells and tissues, and promising methods for molecular imaging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
To understand the functioning of a genome in any living organism, it is important not only to establish the linear order of genes and their regulatory elements on the chromosomes, but to decipher the spatial (3D) chromatin organization, as well as time-dependent changes in this organization. This type of research involves mapping of the 4D nucleome (i.e., 3D nucleome on the time scale) which has become one of the priorities of the World Nucleome Project and the Nucleome Project of the National Institute of Health (NIH) [1]. The 4D nucleome projects combine advanced technologies of the 3D genomics, single cell sequencing, and high-resolution visualization in order to study formation, maintenance, and rearrangement of the 3D nucleome under various conditions in different types of cells, or even in single cells. In 2022, the Nucleome Browser (http://www.nucleome.org) was launched, which is an interactive multimodal data visualization and exploration platform including 2292 genomic tracks and 732 sets of nucleome images [2].
The CRISPR–Cas9 system is one of the discoveries that have shaken the scientific world. CRISPR has received a worldwide recognition in 2012 after publication of the study headed by Emmanuelle Charpentier and Jennifer Doudna in Science [3], for which they were awarded the 2020 Nobel Prize in Chemistry (https://www.nobelprize.org/prizes/chemistry/2020/press-release/). The CRISPR–Cas9 system has been used for the development of various genome editing technologies, some of which are already tested in preclinical and clinical trials [4]. Another large area of studies implementing the CRISPR–Cas9 system is in vivo fluorescence labeling of nucleome in cells [5], since visualization of the nucleome organization in the nuclei of living organisms is extremely attractive. CRISPR–Cas9-based in vivo imaging will also promote the search for the targets in the treatment of monogenic and polygenic diseases [4, 6]. By linking genomic loci to particular morphological structures in tissues and/or organs and using currently available arsenal of molecular imaging techniques, it will become possible to advance such studies to the in vivo level.
In this review, we discuss the development of nucleome labeling techniques based on the CRISPR–Cas9 system, as well as other labels that can be used for the 3D nucleome visualization in living organisms in the real-time mode.
CRISPR–Cas SYSTEMS: HISTORY OF DISCOVERY, CLASSIFICATION, AND APPLICATION
The CRISPR–Cas system was discovered in the 1980s in Escherichia coli [7] and then in archaea [8], although its role as the acquired immunity system of bacteria was understood much later [9, 10]. Since then, the CRISPR–Cas systems have been found in the genomes of most archaea and almost in half of bacteria [11]. Besides, the CRISPR–Cas systems and their components are common in mobile genetic elements (viruses, transposons, plasmids) and often spread via horizontal gene transfer [12].
The CRISPR cassette is a set of repeating DNA sequences, the so-called clustered regularly interspaced short palindromic repeats (CRISPR). CRISPR loci are separated by spacers (DNA fragments with different nucleotide sequences), that correspond to the fragments of genomes of viruses that have ever infected this bacterium. Due to the incorporation into the bacterial genome, these fragments are transferred to the daughter cells during cell division. Adjacent to the CRISPR cassette, there is always a group of genes referred to as cas (CRISPR-associated genes) [13].
Originally, it was assumed that Cas proteins were involved in DNA repair [14]. However, the discovery that spacer sequences correspond to foreign genetic elements led to the hypothesis that CRISPR–Cas was an immune system protecting a microorganism against invasion of mobile genetic elements [15-17]. This hypothesis was soon confirmed experimentally [18]. Later, it was shown that bacterial and archaeal CRISPR–Cas systems were also involved in the regulation of many physiological processes associated with signal transduction, DNA repair, and programmed cell death. In some cases, CRISPR–Cas systems affect virulence of pathogenic microorganisms [19].
In the most general case, the mechanism of CRISPR–Cas mediated immune defense is as follows. Transcription of the CRISPR cassette yields a long RNA in which unique spacer sequences are separated by hairpin structures formed by the palindromic repeats. This RNA precursor undergoes processing with the formation of short CRISPR RNAs (crRNAs) containing individual spacers. One or several Cas proteins bind to crRNA forming the so-called effector complex. This complex binds to the protospacer, which is a region of foreign DNA or RNA complementary to the spacer in crRNA. Then, the foreign DNA or RNA is cleaved either due to the Cas protein activity itself or with the involvement of additional nucleases [9].
To ensure the cleavage of foreign genetic material, the spacers within the organism’s own CRISPR–Cas system should be different from the protospacers in the foreign DNA. For this, the CRISPR–Cas system recognizes short protospacer adjacent motifs (PAMs). PAMs are 2 to 5 bp in length; they are localized in foreign DNA close to the protospacers and are absent in the spacers [20]. Cas proteins scan long DNA regions in search for PAMs and, when the PAM is found, unwind adjacent double-stranded DNA helix. The resulting single-stranded DNA regions become available for hybridization with the crRNA spacer. The formed structure consisting of the DNA/RNA heteroduplex and the single-stranded DNA is called R-loop. The CRISPR–Cas systems recognizing foreign RNA do not require the presence of PAM [20].
Despite a considerable structural diversity between the CRISPR–Cas system, the mechanism of protective action of any system includes three functional stages: (i) adaptation (acquisition of new spacers), (ii) expression (crRNA biogenesis), and (iii) interference (elimination of foreign genetic material) [11]. The adaptation module includes the integrase Cas1 (key enzyme necessary for the insertion of new spacers) and the structural protein Cas2. Depending on the system type, this module can also include other proteins, e.g., Cas4 nuclease. The expression module is responsible for the processing of crRNA precursor. In most systems, it is represented by the Cas6 enzyme, although there are also other variants (see below). The interference module is an effector complex recognizing the target sequence and hydrolyzing foreign DNA or RNA. The structure of the effector complex is one of the main factors in the classification of CRISPR–Cas systems. Some systems can include the auxiliary module represented by genes with often unknown functions but located close to the principal genes of the CRISPR–Cas system [21].
There are no genes common for all CRISPR–Cas systems without exception, which makes classifying these systems very difficult [21, 22]. The latest classification of the CRISPR–Cas systems includes 2 classes, 6 types, and 33 subtypes [21]. This classification is based on a complex computational strategy that takes into account the most characteristic (signature) genes for each type and subtype, comparisons of genes sets and types of organization of genomic loci, and construction of phylogenetic trees based on the similarity between the genes conserved within each subtype.
Class 1 includes the CRISPR–Cas systems with the effector complex containing several Cas proteins. Class 1 systems are very common and can be divided into types I, III, and IV depending on the combination of the following proteins in the effector complex: Cas3 (sometimes fused with Cas2), Cas5, Cas6, Cas7, Cas8, Cas10, and Cas11 [21].
The effector complex in type I systems is called Cascade (CRISPR-associated complex for antiviral defense) [23, 24]. After the Cascade binds to the target DNA sequence, a fragment of the complementary DNA strand remains single-stranded and is cleaved by the Cas3 protein [25, 26]. Cas3 is a natural chimeric protein consisting of helicase and nuclease. The helicase activity is ATP-dependent and is exhibited toward the DNA/DNA and RNA/DNA duplexes; the nuclease activity does not require ATP [25, 26]. Cas3 is a signature protein of type I CRISPR–Cas systems [22].
Type III CRISPR–Cas systems are unique in their ability to recognize and eliminate transcriptionally active foreign genetic material. The effector complex recognizes the target sequence in the foreign RNA during transcription, which leads to the complex localization in the transcription bubble, followed by the degradation of the single-stranded DNA. The RNA is cleaved by Cas7; the single-stranded DNA is hydrolyzed by Cas10 [27-29]. The signature protein of type III CRISPR–Cas system is the multidomain Cas10 [22]. Type III systems are also characterized by the presence of the auxiliary module genes [21].
Type IV CRISPR–Cas systems are minimalistic: they often lack proteins of the adaptation module and nucleases of the interference module. They contain Cas5 and Cas7 that are easily detected due to the similarity of their amino acid sequences with their analogs in the other types of CRISPR–Cas systems. Type IV systems can be found almost solely in plasmids and prophages [21].
Class 2 includes CRISPR–Cas systems with the effector complex containing a single multidomain protein (Cas9, Cas2, or Cas13). They are found ten-fold less frequently than the class 1 systems [30]. Class 2 is divided into types II, V, and VI. The search for the CRISPR–Cas systems by computational technologies has recently resulted in the discovery of numerous new class 2 subtypes [21].
Cas9 is a signature protein of type II systems. Cas9 has two nuclease domains, HNH and RuvC, with the HNH amino acid sequence located within the RuvC sequence [21]. Cas9 proteins recognize G-rich PAMs and cleave the DNA target with the formation of either blunt ends or one-nucleotide-long sticky ends [31]. The functioning of both adaptation and interference modules in these systems requires an additional noncoding RNA called trans-activating CRISPR RNA (tracrRNA). It is complementary to the CRISPR repeats and, therefore, forms an RNA duplex with each of the repeats in the crRNA precursor. The binding with Cas9 stabilizes these duplexes, which are then cleaved by bacterial RNase III (enzyme not included in the CRISPR–Cas system) [32, 33]. tracrRNA is encoded close to or between the genes of the CRISPR–Cas locus [34].
The first biochemically characterized Cas9 protein was Cas9 from the bacterium Streptococcus pyogenes (SpyCas9); hence, it has been used in most genetic engineering procedures employing the CRISPR–Cas systems [35]. This large crescent-shaped protein (1368 amino acid residues) is ~100 Å × 100 Å × 50 Å in size [36]. It is composed of the recognition (REC) and nuclease (NUC) lobes connected by two linkers; one of the linkers is a helix enriched in arginine residues and the other is disordered. The recognition lobe consists of three alpha-helical domains (REC1, REC2, and REC3) and is responsible for binding the guide RNA (gRNA) and DNA. The HNH domain in the nuclease lobe cleaves the DNA strand complementary to the gRNA, while RuvC hydrolyzes the noncomplementary DNA strand. The RuvC and HNH domains are not homologous; their active sites are located ~25 Å from each other. The active site of the HNH domain is disordered in the absence of nucleic acids [36]. The C-terminal region of the nuclease lobe contains the PAM-interacting (PI) domain that provides the binding with PAM [37]. The SpyCas9 protein recognizes the PAM sequence 5′-NGG-3′ (and, with a lesser efficiency, 5′-NAG-3′) [3, 32].
The signature protein of type V CRISPR–Cas systems is Cas12. It is a multidomain protein containing the recognition and nuclease lobes (similarly to Cas9). The characteristic feature of Cas12 is the presence of the nuclease domain RuvC capable of introducing double-strand breaks into foreign DNA in the absence of the HNH domain. The requirements for the presence of additional RNAs depend on the system subtype. Thus, the effector complex consisting of Cas12a and crRNA is fully functional without additional RNAs. Cas12b requires tracrRNA for the crRNA maturation and target DNA cleavage (similarly to Cas9) [38]. In the case of Cas12c and Cas12d proteins, both the processing of the crRNA precursor and hydrolysis of target DNA require a molecule of short-complementarity untranslated RNA (scoutRNA), which is a recently discovered third variant of short RNA encoded in the CRISPR–Cas system [39]. In most cases, Cas12 proteins recognize T-rich PAM sequences and cleave the target DNA forming 5′ sticky ends of 5 nucleotides (Cas12a), 7 nucleotides (Cas12b), or 9 nucleotides (Cas12d) in length [31, 39]. Depending on the system subtype, type V CRISPR–Cas systems target double-stranded DNA, single-stranded DNA, or single-stranded RNA [21].
The signature protein of type VI CRISPR–Cas systems is Cas13. These systems target the RNA transcripts of foreign genomes [21]. Cas13 acts as both expression and interference modules; it is involved both in the crRNA processing and hydrolysis of target RNA. Interestingly, these two functions are based on two different ribonuclease activities. The hydrolysis of single-stranded target RNA requires interaction between the HEPN1 and HEPN2 domains in the Cas13 molecule, while crRNA processing requires interaction between HEPN2 and Helical-1 domain [40].
The ability of CRISPR–Cas systems to recognize the target nucleotide sequences in long genomic DNAs with a high specificity is a feature which is difficult to overestimate in biotechnology and medicine. CRISPR–Cas systems have been used to create technologies for precise editing of eukaryotic genomes [3, 41]. For easy use, crRNA and tracrRNA can be fused in a single molecule – the so-called single guide RNA (sgRNA) [3]. Therefore, a minimal system for genome editing consists of only two elements – the multidomain Cas9 protein and chimeric sgRNA. The target region in DNA is determined by the spacer sequence in sgRNA. The choice of the target is limited by the necessity of a PAM sequence presence adjacent to the target sequence (to be recognized by Cas9). Cas proteins from different CRISPR–Cas systems and organisms recognize different PAMs. However, the affinity of a Cas protein to a particular PAM sequence can be changed by mutagenesis [42].
At present, CRISPR–Cas9 and CRISPR–Cas12a are the most commonly used systems for precise editing of eukaryotic genomes [43, 44]. They introduce double-strand breaks into DNA, which are then repaired through homologous recombination or nonhomologous DNA end joining [45]. Many research groups work on the improvement of CRISPR–Cas systems to further increase their specificity [46]. Beside its application in medicine [47], genome editing is used for modification of microorganisms [48] and plants in agriculture and horticulture [49-54]. Recently, the first works on the eukaryotic genome editing using type I CRISPR–Cas systems have been published [55-57].
CRISPR–Cas systems are also employed in various analytical methods for DNA detection [58, 59], including SARS-CoV-2 diagnostics [60]. It should be emphasized that the diagnostics of infections caused by RNA-containing viruses uses the RNA-recognizing CRISPR–Cas13 system [61]. In terms of molecular diagnostics, CRISPR–Cas systems have several substantial advantages, such as high specificity, high sensitivity, simplicity of use, and low cost. At present, various pathogens can be detected with the corresponding CRISPR–Cas system, including Zika virus (Cas9); tuberculosis, human papilloma virus, HIV-1, hepatitis B, and SARS-CoV-2 viruses (Cas12); and SARS-CoV-2, Dengue, and Zika viruses (Cas13). Any of these diagnostic tests takes less than 3 h [62]. CRISPR–Cas can also be used for the construction of biosensors for detecting molecules other than nucleic acids [63, 64].
Point mutations interfering with the Cas9 ability to hydrolyze DNA (D10A in the RuvC domain and H840A in the HNH domain) do not impair the binding of this protein with the target sequence [65]. This catalytically inactive protein was called dCas9 (dead Cas9). Analogous catalytically inactive variants were obtained for other Cas proteins, e.g., dCas12a [66], dCas12b [67], and dCas13 [68]. These mutants have considerably extended the areas of the CRISPR–Cas application, as any functionally active domain fused with dCas can be delivered to the specified genomic loci. The functionally active domains can be transcription regulators, chromatin remodeling factors, enzymes which modify heterocyclic bases, fluorescent labels, etc. [69]. Thus, it has become possible to speak of epigenome editing [70, 71].
Type I CRISPR–Cas systems can also be used for the programmed gene repression without genome editing. For this purpose, the nuclease Cas3 gene is deleted [72]. However, such application is considerably limited by the fact that the Cascade complex consists of several proteins in the type I systems.
Since Cas9 and dCas9 are very large proteins that are encoded by large genes, their size should be decreased in order to facilitate their delivery into the cells. It has been shown that dCas9 from S. pyogenes maintained its DNA-recognizing activity in vitro and in vivo even after deletion of about one-third of its amino acid sequence (four regions in the REC2, REC3, HNH, and RuvC domains) [73]. Another type of Cas proteins that would be suitable for easy delivery is miniature proteins from the recently discovered Cas12f group. Despite their relatively small size (422-603 amino acid residues), these proteins cleave double-stranded DNA in the PAM-dependent manner [74, 75].
The orthologs of dCas9 from various organisms recognize different PAM sequences and, therefore, can be used for targeting several DNA sequences simultaneously. These dCas9 proteins also differ in size, and this fact can facilitate their delivery into the cells. The specificity of dCas9 orthologs to particular PAM sequences can be changed by introducing modifications into the amino acid sequence of the PI domain. Moreover, the recognition of the PAM sequence is not always strict and, therefore, the binding specificity can vary depending on the selected variable nucleotide in the PAM sequence. Due to these properties, dCas9 orthologs are used for the simultaneous labeling of different chromatin loci in one cell [76].
MODERN APPROACHES TO THE 3D GENOME STRUCTURE INVESTIGATION in vitro AND in vivo
Currently existing approaches to the investigation of the 3D genome architecture and visualization of chromatin loci can be divided into two large groups that have been developed independently and are used separately despite their common purposes. The first group includes the methods requiring cell fixation, such as (1) 2D- and 3D-FISH, (2) 3C (3C, 4C, 5C, Hi-C)-based techniques, and (3) CASFISH. The second group includes the methods using living cells and proteins of genome editing systems capable of binding to DNA without introducing breaks into it. These methods utilize (1) TALE (transcription activator-like effector) or ZFN (zinc finger) protein domains fused with fluorescent proteins (FPs), (2) dCas9 fused with FP, (3) dCas9 fused with the SunTag polypeptide, (4) chimeric sgRNAs containing FP-binding RNA aptamers, (5) Casilio system, and (6) FP fragments providing fluorescence complementation. The methods of the first group had been developed earlier and therefore have a broad range of applications [77]; the methods of the second group represent developments of genome editing techniques.
Chromatin labeling in fixed cells. Fluorescent hybridization in situ (FISH). Therefore, we will first discuss the methods based on the FISH technique, which has more than a half-a-century history and numerous applications [78]. Although FISH allows to label only a few genomic loci at the same time, its application has led to the discovery of chromosome territories and dynamic changes in the positions of genomic loci relative to the nuclear compartments during cell differentiation [77].
At present, there are several FISH modifications. All of them require cell fixation, DNA denaturation by special reagents, and DNA hybridization (annealing) with fluorescent probes for further visualization under a fluorescent microscope. In 2D-FISH, the nucleus spread is obtained by cell swelling in a hypotonic solution, followed by its fixation in methanol and acetic acid. As a consequence, the labeling of chromatin loci is limited to two dimensions only, because the spatial positions of the loci are assessed in the spread-out (flattened) nucleus. In 3D-FISH, the cells are fixed with formaldehyde to maintain their shape. Therefore, 3D-FISH makes it possible to measure the distances between several genomic loci and to determine the variation of these distances in a cell population [79, 80].
Although 3D-FISH is more difficult to implement than 2D-FISH, it is often used to confirm the results obtained by other methods (e.g., those based on 3C; see the next section), because it studies cell preparations, in which the state of the nuclei is close to the native one. We believe that comparing the results obtained by 2D-FISH and 3D-FISH is not exactly correct; however, 3D-FISH has undeniable advantages, since the use of several different fluorophores allows not only to visualize DNA loci, but also to relate their positions to the chromatin or nucleus morphology.
Recently developed technique of multiplexed imaging by sequential hybridization in situ [81] has made it possible to visualize hundreds of genomic loci and to obtain high-resolution images for the entire chromosome. In the same article, the authors described the method of multiplexed error-robust fluorescence in situ hybridization (MERFISH) that allowed to simultaneously visualize more than a thousand genomic loci and nascent RNA transcripts and to characterize chromatin domains, nuclear compartments, and interactions between different chromosomes and transcription processes in a single cell in situ.
3C-based techniques. The methods based on the chromosome conformation capture (3C) are used for various purposes [82-84]. They include cell fixation (most often, with formaldehyde), genome fragmentation by restriction endonucleases, and ligation of spatially close DNA fragments. The further stages vary depending on the technique used. 3C-based techniques are used for identifying the contacts between two chromosomal loci, between a locus and the entire genome, or even between multiple genomic loci.
The 3C technique [85] involves chromatin fixation followed by the chromatin cleavage with restriction endonucleases for the formation of sticky ends and further ligation of the resulting DNA fragments by the sticky ends. The mixture of DNA fragments for the ligation reaction is diluted to ensure mainly intramolecular ligations (i.e., within covalently cross-linked fragments) [86]. Next, polymerase chain reaction (PCR) is performed using the primers complementary to the sequences present in the studied genomic regions, and the amplification products are analyzed by gel electrophoresis. The efficiency of amplification for different primer pairs is compared to estimate the frequency of ligation of the respective DNA regions, which represents the frequency of interaction between these regions in the genome [86]. The use of primers is a disadvantage, as the 3C technique can be applied for detecting the mutual positions of preselected DNA regions only. Hence, the efficiency of the 3C technique can be described as “one-to-one”, which limits its application. In addition, it can detect contacts only within a limited distance (less than several hundreds of kilobases). Several 3C-based methods with a higher throughput capacity have been developed [82].
(i) 4C (circular chromosome conformation capture) technique [87] includes creation of small circular DNA molecules. For this, DNA templates obtained by ligation (as described above for the 3C technique) are cleaved by the second restriction endonuclease and then ligated again. This procedure is followed by inverse PCR using the primers to the studied sequence to amplify all interacting sequences. The resulting amplification products are analyzed by microarrays or next-generation sequencing (NGS). This advanced method allows to assess the interactions between one particular genomic locus and all other genomic loci (“one-versus-all”) [86, 88].
(ii) 5C (chromosome conformation capture carbon copy) technique [89] assumes hybridization of DNA templates obtained by the 3C technique with a mixture of oligonucleotides, each of oligonucleotides partially overlapping with a certain restriction site in the studied locus. The pairs of oligonucleotides corresponding to the interacting genomic regions are brought close together by annealing on the DNA templates and therefore, can be ligated. Each of these oligonucleotides carries one of two additional universal sequences at its 5′ end, which allows to simultaneously amplify all ligation products by the multiplexed PCR. The resulting DNA fragments are analyzed by microarrays or sequencing [86]. The 5C technique implements the “many-versus-many” principle, which makes possible simultaneous detection of millions of interactions using thousands of primers in one analysis. A great scientific achievement ensured by the 5C technique was the discovery of topologically associating domains (TADs), genomic regions whose DNA sequences preferentially contact each other [90].
(iii) Hi-C (high-throughput chromosome conformation capture) technique [91] differs from 3C in the fact that after the cleavage by a restriction endonuclease, the sticky ends are filled with biotin-labeled nucleotides. Next, the mixture is diluted and the blunt ends are ligated, so the ligation sites are labeled with biotin. The resulting DNA is cut and the biotinylated fragments are isolated with streptavidin and analyzed by NGS. This strategy implements the “all-versus-all” principle [77, 86].
One more method that is similar to those listed above but is not considered as a 3C-based technique is ChIA-PET (chromatin interaction analysis with paired-end tag) [92]. Chromatin is fixed by formaldehyde and fragmented by sonication; the fragments with the bound protein of interest are precipitated using the antibodies against this protein. Spatially close DNA ends are ligated and the ligation products are analyzed by sequencing. Therefore, the ChIA-PET technique allows to study only chromatin interactions determined by a particular DNA-binding protein [86].
To answer the question to what extent the results of FISH and 3C-based techniques can be compared, it is important to take into account the similarities and the differences of these methods. Both groups of techniques are based on the chemical cross-linking by formaldehyde, which is more efficient in the case of protein–protein interactions (due to the presence of lysine, tryptophan, and cysteine residues) than in the case of DNA–protein interactions. Although the cross-linking reaction occurs within a range of 2-3 Å, more distant DNA loci can be cross-linked due to the existence of protein–protein scaffolds. Both groups of techniques require permeability of the cell nuclei and accessibility of chromatin to fluorescent probes or restriction endonucleases. The specific requirement of the 3D-FISH technique is that the cross-linked chromatin should be slightly denatured to enable probe hybridization with the target DNA. This is achieved by heating a sample in the presence of formamide, which decreases the melting temperature of the double-stranded DNA, since the “regular” thermal denaturation could lead to the changes in the nuclear and chromatin organization. Unlike 3D-FISH, the 3C-based techniques require genome fragmentation by restriction endonucleases. The main problem when comparing the results of these methods is that the 3D-distances between the interacting loci are determined by microscopy in FISH and by sequencing in Hi-C. Another problem is that the distance within which two genomic loci can be cross-linked with formaldehyde is still unknown. However, at present, FISH remains the most reliable method for confirming the results obtained by Hi-C [93].
Since both FISH and 3C-based techniques use covalent cross-linking, they have a common disadvantage: none of these techniques allows to study the temporal dynamics which underlies the variability of chromosome conformations.
CASFISH. Another promising technique is CASFISH. It is a modification of FISH, in which the target DNA sequences are labeled by catalytically inactive protein dCas9 [94]. The cells are fixed with a methanol–acetic acid mixture to avoid denaturation of genomic DNA and, at the same time, to introduce the protein–nucleic acid probes into the nucleus. The probes are the complexes of dCas9 labeled with a fluorescent dye via the Halo-tag and sgRNA carrying a fluorescent dye of another color.
The CASFISH technique has several advantages compared to the methods described above. First, the optimized labeling procedure using protein–nucleic acid probes takes much less time than the labeling procedure in FISH which is based on nucleic probes only. Second, the mild conditions of CASFISH provide better retention of cell morphology and DNA structure. Hence, CASFISH in combination with superhigh-resolution imaging of single molecules is a useful tool for studying genome organization. Third, the two-component nature of CASFISH probes provides a great potential for multiplexing, i.e., simultaneous application of several fluorescent labels. However, this technique has its disadvantages, as only a limited number of genomic loci can be labeled. It also does not allow to study the behavior of chromatin loci in its dynamics [94].
Chromatin labeling in living cells. Using ZFN and TALE domains for labeling chromatin loci. The first endonucleases used for genome editing contained the zinc finger (ZFN) domain fused with the endonuclease domain [95]. The ZFN domain consists of several zinc finger motifs, fragments of about 30 amino acid residues long capable of binding zinc ions. There are several types of such motifs [96]. The “classical” Cys2–His2 type used in genome editing consists of an alpha-helix and a beta-hairpin, with a zinc ion coordinated by two cysteine and two histidine residues [96]. Each zinc finger motif forms contacts with 3-4 base pairs in the DNA major groove. It is possible to construct a domain consisting of several zinc finger motifs that will be able to recognize a longer DNA sequence and to interact specifically with a target DNA [97].
In a fusion protein, the ZFN domain is responsible for the specificity, while the catalytic domain derived from restriction endonuclease FokI provides DNA hydrolysis [98]. In FokI nuclease, the recognition of a specific DNA sequence and hydrolysis of the double-stranded DNA are the functions of two different structural domains. When these domains are separated by proteolysis, the FokI catalytic domain cleaves DNA nonspecifically [99]. The first chimeric endonuclease was constructed by fusing the FokI catalytic domain with the Ultrabithorax homeodomain protein of Drosophila [100]. The chimeric proteins consisting of the FokI catalytic domain and ZFN domains were soon constructed [98, 101]. The FokI catalytic domain exhibits its activity only after dimerization [102], so all chimeric proteins created based on this domain also require dimerization to become functional. Therefore, the double-stranded DNA is cleaved only where two ZFN domains bind with the opposite DNA strands, which limits the off-target effects of this construct.
Another nuclease used for genome editing is TALEN (transcription activator-like effector nuclease), a protein composed of bacterial TALE protein and the catalytic domain of FokI endonuclease. Similar to the ZFN domains, the TALE domain consists of repeats 33-35 amino acid residues in length. Each repeat recognizes one base pair in DNA, and the specificity of recognition is determined by two hypervariable amino acid residues [97, 103]. The activity of each repeat has no effect on the binding specificity of adjacent repeats, which considerably simplifies the construction of the TALEN nuclease compared to the ZFN nucleases [97].
Since the labeling of genomic loci does not require the introduction of breaks into DNA, the chimeric proteins created for this purpose are composed of the DNA-recognizing ZFN or TALE domains with the attached FP (instead of the endonuclease domain). Such constructs have been successfully used to label repeating DNA sequences [104-108]. In some cases, TALE proteins form aggregates. To avoid aggregation and to increase the contrast of imaging in living cells, fluorescently labeled TALE protein can be fused with thioredoxin [109].
Labeling of chromatin loci in living cells using the CRISPR–Cas9 system. The discovery of the catalytically inactive dCas9 protein has stimulated the development of the CRISPR–dCas9-based techniques for non-invasive visualization of genomic loci in living cells. It should be emphasized that the fluorescent label can be inserted into either protein (dCas9) or nucleic acid (sgRNA) component of the probe.
In the case of dCas9 fused with any FP, the signal from a single molecule is too weak; therefore, successive visualization is possible only if the target is a repeating DNA sequence (e.g., as in telomeres) [110]. The SunTag system developed to intensify the fluorescence signal [111] involves the coexpression of the antibody fused with the FP and dCas9 fused with a polypeptide containing numerous repeats of the epitope recognized by this antibody (SunTag). As the dCas9–sgRNA complex recognizes a unique target sequence in the genomic DNA, the SunTag polypeptide linked to it provides the binding of numerous fluorescently labeled antibodies with a single dCas9 molecule, which significantly increases the fluorescence signal [111, 112].
An alternative to the application of chimeric dCas9-FP constructs is visualization of genomic loci using modified sgRNAs capable of recruiting fluorescently labeled proteins specific for a given RNA sequence. As an example, we can mention chimeric sgRNA containing numerous repeats of a unique RNA aptamer which is capable of specific binding to a fluorescently labeled effector protein [113-115]. The most commonly used aptamer is MS2, which is an RNA loop of the MS2 bacteriophage RNA capable of binding to the MS2 bacteriophage coat protein (MCP) with high affinity and specificity [116]. During the coexpression of such chimeric sgRNA with dCas9 and MCP-FP, each dCas9–sgRNA complex is labeled by numerous FP molecules due to the interactions between the MS2 aptamer and MCP. Because of the rapid exchange of the effector protein molecules bound by the aptamer, the photobleaching is less pronounced than in the case of labeling with CRISPR–Cas9 [113].
The Casilio system [117] combines CRISPR–Cas9 and conserved RNA-binding domain of the Pumilio/FBF (PUF) proteins that can be programmed to bind to a specific 8-nucleotide RNA sequence (PUF-binding site, PBS). When fused with a FP, the PUF domain maintains the ability to recognize its target RNA. The use of sgRNA containing the tandem repeats of PBS makes it possible to label the dCas9–sgRNA complex using several chimeric PUF-FP proteins.
Bimolecular fluorescence complementation (BIFC) [118] uses two fragments of the Venus yellow fluorescent protein that can interact forming the full-size functional protein. The dCas9 fused to the Venus C-terminal fragment is coexpressed with the MCP fused to the Venus N-terminal fragment, and with a chimeric sgRNA containing the MS2 aptamer. Such coexpression results in the assembly of the dCas9–sgRNA complex and MCP binding by the MS2 aptamer, leading to the convergence of two Venus fragments and formation of the FP. The signal-to-noise ratio can be substantially improved by the introduction of the SunTag to the above system. In such modified system, dCas9–SunTag protein is coexpressed with a SunTag-recognizing antibody fused to the C-terminal fragment of the Venus protein, along with MCP fused to the N-terminal fragment of the Venus protein, and with the chimeric sgRNA containing the MS2 aptamer [118]. An analogous fluorescence complementation system was developed that used green fluorescent protein (GFP) expressed as three separate fragments [119].
The main characteristics of techniques for studying the 3D genome structure and visualizing chromatin loci are listed in Table 1. It should be emphasized that despite their high resolution and specificity, the techniques using cell fixation, DNA spreading, and denaturation cannot be applied for the in vivo visualization of genomic loci in intact nuclei retaining their 3D structure. The major drawback of the TALE-based and ZFN-based systems is the necessity for the time- and labor-consuming development of specific DNA-recognizing domains for each genomic locus under investigation. The CRISPR–Cas9 technology allows to omit these procedures, because the specificity of DNA recognition is provided by the complementary interaction of the RNA component with a particular nucleotide sequence in the genome, which substantially facilitates creation of the effector complexes. Taking into account all the above mentioned, the use of the CRISPR–Cas9 system with the chimeric dCas9-FP protein seems to be the optimal approach for the labeling of chromatin loci in living cells.
Problems in using the CRISPR–Cas9 systems and how to solve them. It is still poorly understood how the specificity of chromatin labeling, as well as the localization and transport of chimeric dCas9-FP protein, depend on the components of this protein and their combination. Other disadvantages of this system include a high background signal and retention of the chimeric protein in various cell compartments [120]. Below, we list the major problems encountered by researchers using dCas9-FP for genome labeling and possible solution to these problems.
1) Off-target binding. The dCas9 ortholog from S. pyogenes, which is most often used for labeling of chromatin loci, has a short PAM sequence (5′-NGG-3′). Although it provides a lot of freedom in choosing target DNA sequences, it is also characterized by frequent off-target binding, resulting in false-positive signals during visualization [120]. This problem can be solved by using dCas9 orthologs from other bacterial species with different PAM sequences. The orthologs recognizing long PAM sequences are preferable; although this limits the choice of the target site, but also reduces the off-target binding [121]. In addition, the variants of Cas9 from S. pyogenes with a higher specificity have been developed, as well as the variant with expanded PAM (xCas9) [122], which can be modified to visualize genomic loci with a high specificity. Another protein, Cas12a, can be used for the recognition of AT-rich PAM sequences.
2) Target site accessibility. Some DNA regions are coated with DNA-binding proteins (e.g., shelterins that protect centromeres and telomeres) and, therefore, are inaccessible for the binding with the dCas9–sgRNA complex. There are also DNA regions with a high level of topological inaccessibility, which also prevents interactions with the dCas9–sgRNA complex. The choice of the target can be improved by using other methods, such as ChIP-seq [123] and 3C.
3) Target binding selectivity. When using the Cas9–sgRNA system, the length and the nucleotide sequence of the spacer can influence the efficiency of complex binding with the target site. For example, the binding of sgRNA to the coding DNA strand is more efficient than to the template strand. This can impede visualization of genomic loci containing insufficient number of PAM sequences in the template strand. In addition, the strength of Cas9 binding with sgRNA may vary, as the protein preferably binds with gRNAs containing purine bases as the last four nucleotides of the spacer. The target binding efficiency is also affected if the gRNA contains a small number of GC dinucleotides [124].
4) Background fluorescence is a common problem in all techniques using fluorescent microscopy. One of the approaches to improve the signal/background ratio is using the fluorescence complementation technique described above [118, 119].
5) Visualization of nonrepetitive sequences. While visualization of repetitive DNA sequences requires only one sgRNA, visualizing non-repetitive elements is more complicated, as it needs several unique sgRNAs. Multiple sgRNAs can be cloned into a single plasmid by the Golden Gate assembly [125] under optimized conditions, which significantly simplifies the transfection procedure and increases its efficiency. However, simultaneous expression of several sgRNAs in the same cell may be non-synchronized, because the transcription rates of different sgRNAs can vary significantly. In order to overcome this problem, one potential strategy may be encoding different sgRNAs in one transcript, with every two sgRNAs linked by a sequence that can be excised by ribonucleases. Even in case of successful simultaneous expression of several sgRNAs, imaging of nonrepetitive regions may be challenging, because different sgRNAs can compete with each other for the binding to dCas9. The competition between different sgRNAs can be reduced by using multiple dCas9 orthologs [120].
At present, the most common approach is expression of dCas9 from S. pyogenes fused with the enhanced green fluorescence protein (eGFP) in combination with one or several sgRNAs [110]. sgRNAs can also be labeled with fluorescent molecules [126, 127]. However, both strategies have the disadvantages described above. The use of fluorescently labeled RNAs allows to label up to six DNA loci simultaneously without using multiple dCas9 orthologs [127], while fluorescent labeling of proteins provides lower background signal but is limited by the number of available dCas9-FP constructs [94]. Chimeric proteins containing dCas9 orthologs (NmdCas9 and StdCas9) have been used only in a few studies [128], none of which has attempted to optimize the chimeric proteins by combining different orthologs and FPs. In [129], dCas9-FP was fused with the target sequences for the respective sgRNAs labeled with a fluorescent peptide to increase the signal/background ratio [129]. The results obtained in almost all studies using these methods were verified by FISH [110]. In most studies, the repetitive sequences were telomeres [128] or centromeres [94], while MUC1 and MUC4 were chosen as nonrepetitive loci [110].
DELIVERY OF THE CRISPR–Cas SYSTEM COMPONENTS TO THE TARGET CELLS
The proper functioning of the CRISPR–Cas9 system, whose components are unstable, prone to degradation, and hard to penetrate into the cells, requires selection of the optimal method for the delivery into the target cells. At present, there are three main strategies to achieve this goal: physical methods and the use of viral and nonviral vectors.
Physical methods are used quite often, both in vitro and in vivo, due to their simplicity and efficiency. They can be divided into two classical and two new techniques.
Microinjection is the introduction of various molecules into the cells or cell compartments (e.g., nucleus) through a microcapillary pipette. The main advantage of this method is that it can be used in any cell, since the injection procedure does not depend on the cell type. The microinjections of CRISPR system components into the nuclei of single rapidly dividing cells have been used to generate genetic knockouts and transgenic animals. Although this method is very efficient, it has a significant drawback, namely, the microinjection should be performed in each cell [130].
In electroporation, the application of a high voltage causes formation of pores in the cell membranes, through which various molecules can enter the cells both ex vivo and in vivo. This method has been used to obtain B cells expressing therapeutic proteins after their differentiation [131]. Electroporation can cause a significant damage to the cell membrane resulting in its permanent permeabilization.
In transmembrane internalization assisted by membrane filtration (TRIAMF), the cells are passed through the pores in a filter membrane. The diameter of the pores is smaller than the cell diameter, resulting in the formation of transient pores in the cell membrane. This method was used to deliver ribonucleoproteins into hematopoietic stem and progenitor cells (HSPCs), which normally absorb exogenous molecules very poorly and need more direct transfection methods. The efficiency of TRIAMF is comparable with that of electroporation but its damaging effect on the cells is significantly lower [132].
In induced transduction by osmocytosis and propanebetaine (iTOP), a hypertonic sodium chloride solution containing propanebetaine is added to the cells. These conditions promote protein uptake from the extracellular space via macropinocytosis and induce macropinosome vesicle leakage to release proteins into the cytosol [133].
Viral methods. The process of DNA transfer between the cells using viruses is called transduction. Due to their high specificity and minimal cytotoxicity, viral vectors were one of the first vehicles used for delivering components of the CRISPR–Cas editing system. Viral vectors should comply with certain requirements:
1) the vector must be replication-defective (capable of only one cycle of infection and integration into the genome);
2) when integrated into the cell genome, the viral genome must be capable of expressing foreign genes but unable to form new viral particles that could further infect other cells;
3) cis- and trans-acting genomic elements should be separated. The trans-acting elements (auxiliary genes or proteins) are removed and replaced by a transgene. The trans-factors are delivered using trans-complementing vectors or viral particles produced by packaging cells.
The three commonly used types of viral vectors used for the delivery of nucleic acids are retroviral, adenoviral, and adeno-associated viral vectors (Table 2).
Historically, the first used viral particles were of the mouse leukemia virus (MLV), which belongs to gammaretroviruses. Their main disadvantage is that incorporation of MLV nucleic acid into the host genome is completely random, and therefore, can cause mutations.
In contrast to gammaretroviruses, lentiviruses can infect nonproliferating cells. The choice of lentiviruses for delivering the components of genome editing systems was also promoted by the fact that their penetration into the nucleus occurs without stimulation of cell proliferation and, hence, is independent of the activity of cell oncogenes. However, lentiviruses can acquire mutations both in their own nucleic acids and in the carried transgenes. If a transgene contains a sequence that interferes with the viral replication, this transgene will be underrepresented in the target cells.
Lentiviral vectors are based on the human immunodeficiency virus (HIV-1). The main genes necessary for the retrovirus survival and functioning are gag encoding structural proteins, pol encoding enzymes necessary for the reverse transcription and integration into the host genome, and env encoding the viral envelope glycoprotein. Currently, there are three generations of lentiviral vectors, each next generation being safer while maintaining high efficiency of genetic material (transgene) delivery to the target cells.
Lentiviral vectors of the first generation were represented by three independent plasmids: the envelope plasmid (carrying the env gene), packaging plasmid (carrying gag-pol, rev, rre, and genes for regulatory/accessory proteins), and transfer vector (carrying the transgene). For the lentiviral particle assembly, human embryonic kidney HEK293T cells were transiently cotransfected with all three plasmids.
Originally, the envelope plasmid contained a fragment of the env gene coding for incomplete g120 glycoprotein in order to increase the system safety, but these viral particles were inefficient in infecting the host cells. The use of heterologous genes encoding outer membrane glycoproteins of other viruses not only enhanced the system safety, but increased the efficiency and selectivity of the construct delivery to the target cells. In the second- and third-generation lentiviruses, the env gene in the envelope plasmid was replaced by the vsv-g gene encoding the vesicular stomatitis virus surface glycoprotein (VSV-G). This glycoprotein can be incorporated into the membrane of any virus; thus facilitating the vector penetration into a cell by endocytosis and reducing the need for accessory envelope proteins [135]. The use of VSV-G has increased the tropism of lentiviral particles and allowed transduction of almost all cell types.
Both the packaging plasmid and the envelope plasmid have been optimized in several steps. The first-generation packaging plasmids contained all HIV-1 genes (vif, vpr, vpu, nef, gag, pol, tat, rev), except for the env and rre genes, as well as the main 5′ donor splice site. The viral 5′ long terminal repeat (5′-LTR) was replaced by a heterologous promoter, such as the constitutive human cytomegalovirus gene enhancer/promoter or the Rous sarcoma virus LTR promoter; the 3′-terminal repeat (3′-LTR) was replaced by the polyadenylation (polyA) signal for the simian virus 40 (SV-40) or the polyA signal of the human ins gene. The viral RNA packaging signal and the primer-binding site were completely deleted.
The second-generation packaging plasmids no longer had the genes for the NEF, VIF, VPR, and VPU accessory proteins responsible for the virulence, cytotoxicity, and viral replication in vitro, which increased the safety of the lentiviral vectors. Further modification of the packaging plasmid by deleting the tat gene and transferring the rev gene into a separate independent plasmid has led to the creation of the third-generation packaging plasmids, which are considered to be the safest ones.
The transfer vectors have been optimized simultaneously with modifications of the packaging plasmids. The first generation of the transgene-carrying plasmids contained intact 5′- and 3′-LTRs and depended on the Tat-controlled transcription. To increase the system safety, the U3 element was deleted from the 3′-LTR (∆U3). In this case, restoration of the U3 element in the 5′-LTR during DNA synthesis on the full-length vector RNA template is impossible, which prevents vector mobilization, i.e., formation of new full-length vector RNA molecules and viral particles in transduced cells during superinfection with the wild-type HIV-1, after provirus incorporation into the cell chromosomal DNA. Such vectors are referred to as the second-generation self-inactivating (SIN) vectors.
Third-generation lentiviral vectors contain the 5′-LTR, in which the U3 element was replaced by the strong constitutive human cytomegalovirus promoter. Due to deletion of the tat gene in the third-generation packaging plasmids, such hybrid 5′-LTR became Tat-independent [136].
Since the original virus for creating the lentiviral vectors was HIV-1, the three potential sources of danger were the appearance of the wild-type virus in the preparations, vector mobilization in the transduced cells infected with the wild-type HIV-1, and insertional mutagenesis. The first problem was solved by creating the third-generation vectors; the second problem was solved only partially, and the third one still has no solution.
The long-term expression of Cas can increase the ratio between the intended and off-target effects of genome editing. Beside using self-inactivating vectors, transient expression Cas9 is now provided by the self-inactivating transgene system. Apart from the gene coding for Cas9, the lentiviral vector contains the genes for two sgRNAs (one directed against a genomic target and the other one directed against the Cas9-encoding gene) [137].
Adenoviral vectors (AVs) can easily include all elements of a genome editing system in a single plasmid due to their high packaging capacity. They can deliver not only the genome editing system genes, but also large donor DNA sequences to ensure targeted homologous repair. The advantage of AV-mediated gene delivery is that the sgRNA and Cas protein are expressed in a cell at a fixed ratio. Since AVs do not integrate into the host genome, Cas expression in proliferating cells is transient. AVs have been successfully used in the editing of the mouse genome in vivo, although their use was associated with some immunogenicity-related toxicity [138].
Recombinant adeno-associated viruses (AAVs) have been widely used for delivery of genetic constructs because of their low immunogenicity, low toxicity, high transduction efficiency, long-term expression of incorporated genes, and ability to transfect both proliferating and resting cells. In addition, AAVs can integrate into specific genome sites, which prevents unwanted mutations [139]. The main disadvantage of AAVs is their low carrying capacity (the maximum size of the insert is less than 5 kb), which significantly limits their application for the delivery of large molecules. In case of the CRISPR–Cas system, the capacity of the AAV cassette is insufficient for delivering chimeric Cas9-based proteins, because the nucleotide sequence of these proteins per se is approximately 5 kb. This problem can be solved by splitting SpyCas9 (Cas9 derived from Streptococcus pyogenes) into two fragments that can recombine inside the cell, so that the truncated genes will be suitable for the delivery by an AAV vector. However, this approach reduces the delivery efficiency, as well as the efficiency of target DNA hydrolysis [140].
Nonviral delivery systems, such as liposomes, polymers, and nanoparticles, are safer and easier to assemble than viral vectors and have a greater carrying capacity. However, the efficiency of delivery by nonviral vectors is low, which might be improved by their further development [141, 142].
Lipid vectors are the most commonly used nonviral delivery system. Lipid-assisted introduction of molecules into cells is called transfection. To fuse with the cell membrane and to provide the transport of nucleic acids, lipid particles should be neutral or cationic. Neutral lipids are often used as accessory molecules to enhance the transfection activity of liposomes. Recent development of liposomal systems has resulted in the emergence of lipid nanoparticles (LNPs) based on ionizable cationic lipids that acquire positive charge at low pH values (typical of late endosomes) due to the presence of tertiary amino groups. Biodegradable cationic LNPs were used to deliver the Cas–sgRNA complex into the cells to induce a gene knockout [143]. The presence of a disulfide bond in the lipid can act as a releasing mechanism, leading to the particle degradation in the cells.
Polymer-based particles can also be used for the CRISPR–Cas delivery. Polymers most commonly used for transfection are polyethylenimine (PEI) and poly(amidoamine) (PAMAM). Similar to cationic lipids, cationic polymers (e.g., PEI) form complexes with nucleic acids that are uptaken by the cells and then released intracellularly. PAMAM dendrimers consist of the central core molecules surrounded by the layers of repetitively branching units that have cationic groups (primary amines) on their surface and form complexes with nucleic acids. PAMAM dendrimers have low cytotoxicity and ensure high transfection efficiency [144].
Another type of vectors used for the RNA and DNA delivery into cells are modified nanoparticles, both virus-like and magnetic ones. They are assembled based on LNPs but with certain modifications. In the case of virus-like particles, proteins and glycoproteins of the viral membrane are incorporated into the particle lipids, so that the particles can deliver nucleic acids to the target cells in a cell-specific manner by interacting with the respective receptors on the cell membrane. Magnetic nanoparticles are used when a viral particle cannot for some reasons be delivered to the cells through the body. In this case, the viral vector is coated with a thin layer of iron nanoparticles, and a thin beam of magnetic field directs it to the target cell. For example, otherwise easy-to-use baculoviral vectors are inactivated by the serum complement system. When coated with magnetic nanoparticles, the baculoviral vector was able to successfully bypass the complement system and enter the targeted tissue, where it penetrated into the cells and transferred the genetic material [145]. Because baculoviruses cannot replicate independently in mammalian cells, this system is extremely safe.
MOLECULAR IMAGING OF NUCLEOME USING CRISPR–Cas9. REQUIREMENTS FOR in vivo VISUALIZATION
Many genome functions are regulated at the level of 3D chromatin packaging, also referred to as the higher-order chromatin organization. The elements of chromatin architecture include chromosome territories (CTs), which in turn are subdivided into A/B compartments, TADs, and CCCTC-binding factor (CTCF)-mediated chromatin loops. The higher-order chromatin organization varies between different cells, tissues, and species; it depends on the developmental stage and/or environmental conditions. Spatial and temporal changes in the chromatin structure are studied by the 4D genomics [146]. The term “4D nucleome” used in scientific literature describes a general organization of the intranuclear space in a cell [147].
Real-time chromatin visualization answers the fundamental questions about the mechanisms involved in the spatial genome organization [148]. Multiple studies have indicated that changes in the 3D nucleome structure are directly related to the development of many human diseases. For example, TADs have been shown to affect local interactions between gene enhancers and promoters, leading to changes in the gene expression, including oncogene activation [149, 150].
The term “imaging” is often used to define protein and chromatin visualization in the cells by the high-resolution microscopy. According to the traditional understanding of this term, molecular imaging is a set of techniques used for the real-time visualization of molecular events in live organisms [151]. In this review, we will refer to molecular imaging (or simply, imaging) as imaging in vivo and in situ at the whole-organism level.
The importance of using in vivo imaging for nucleome studies can be demonstrated by the following example. Up to the present, there is no consensus on the nucleosome stacking. It is still debated whether the stacking of the 10-nm DNA fiber (“beads-on-a-string”) into the 30-nm fiber occurs in vivo or it is only observed in vitro [152]. We believe that molecular imaging will be able to answer this question.
The methods successfully used for genome visualization in cells have not yet evolved into technologies for genome imaging in animals. A particular attention has been given to the modularity and multiplexity of developed methods in order to facilitate the labeling of new targets with the minimum replacement of the labeling system components. Since the labels and imaging techniques are intended for the application in biological organisms, the labels should display low cytotoxicity and meet the biosafety requirements. Fluorescence imaging techniques should be sufficiently sensitive; a combination of several coupled imaging methods should provide the association of the fluorescence signal with a specific site in a tissue/organ.
Fluorescence methods for visualization of labeled chromatin loci. 3D imaging of fluorescently labeled chromatin regions is based on the methods of super-resolution microscopy [153]. To establish the higher-order chromatin structure in vivo, the microscopy methods are used in a combination with the biochemical and 3C-based techniques [92]. Since the early 2000s, high-resolution microscopy with various FISH probes has been used to observe the 3D genome structure [154]. High-resolution localization microscopy was used for the nanostructural analysis of FISH-labeled chromatin domains with the average localization accuracy of 20 nm [155]. The method of optical reconstruction of chromatin architecture (ORCA) has allowed to visualize chromatin within small regions (100-700 kb long) and to identify the interactions between regulatory elements in Drosophila genome with a resolution up to 2 kb [156]. Combining FISH staining and interferometric photoactivation and localization microscopy (iPALM) with a special reconstruction algorithm made it possible to visualize CTCF-mediated chromatin loops in human lymphoblastoid cells with an accuracy of 2-22 nm using an oligonucleotide probe [152]. At present, new data have been obtained on the visualization of chromatin dynamics. The role of replication stress and the role of functional loss by the key regulators of histone dynamics in the emergence of global epigenetic changes, including the development of precancerous conditions, are being studied [157-159].
The main difficulties encountered in the imaging of higher-order chromatin organization in situ are related to specific DNA labeling and the limited resolution of microscopy [153]. Some problems are caused by the optical properties of biological tissues (e.g., increase in the background signal due to the tissue autofluorescence). There are also numerous artifacts caused by light scattering, photobleaching, uneven sample illumination, light path length, or variations in the excitation intensity [160]. Some of these limitations are absent in the fluorescence lifetime imaging (FLIM) [160]. The main advantage of FLIM is that the fluorescence lifetime does not depend on the fluorophore concentration and remains the same at different device settings (excitation intensity, detector sensitivity, optical path length, etc.) [161].
There are FLIM modifications providing detailed information on the photophysical phenomena that are difficult or impossible to observe when measuring the fluorescence intensity. For example, the degree of chromatin compaction can be calculated based on the inverse quadratic relationships between the fluorescence lifetime of DNA-incorporated probes and their local refractive index that depends on the DNA compaction [162, 163].
The FLIM modification most commonly used in biology is the Förster resonance energy transfer (FRET) mechanism in which there is a radiation-free energy transfer from a fluorescence donor to an acceptor. For this, both fluorophores should be in a close proximity to each other (less than 10 nm) causing depopulation of excited electronic states of the donor. As a result, the fluorescence lifetime of the donor decreases and the fluorescence lifetime of the acceptor increases, allowing detection of the spatial association between the fluorophores [160, 164]. FRET has been well established as a method for studying the structural and dynamic changes in nucleosomes, both for their ensembles and for individual molecules [165]. FLIM-FRET was used to demonstrate the difference in the compaction of euchromatin and heterochromatin [163]. Using H2B histone expressed as a fusion protein with eGFP and mCherry in HeLa cells, FLIM-FRET was employed to assess the dynamics of chromatin compaction at the nucleosome level during the DNA damage response [166].
We should emphasize the advantages of imaging using genetically encoded FPs, whose contribution to the studies of molecular interactions in biology and biomedicine has been invaluable [167, 168]. A large variety of imaging sensors have been designed based on the FRET pairs of colored FPs [159, 169].
We have successfully applied such approach to visualize caspase-3 activity in tumor cells and subcutaneous xenografts of human tumors in nude mice using a FRET sensor. The caspase activity sensor was based on TagRFP as a fluorescence donor and chromoprotein KFP (kindling fluorescent protein) as an acceptor. In vivo caspase-3 activation in response to the antitumor therapy was monitored noninvasively over a long period of time (about 30 days) [170-172].
Fluorescently labeled genetically encoded CRISPR–Cas9 can be visualized by the FRET-based approach as well. With the involvement of mathematical calculations, FLIM-FRET technique also makes it possible to estimate the distance between the molecules [161].
The main disadvantages of fluorescence lifetime measurements include the long data collection time, which can impede visualization of fast events, and special requirements for the accuracy of instrument response. Besides, fluorescence lifetime is sensitive to the changes in temperature, pH, and viscosity, making it difficult to interpret the data. Some authors used the higher-performance FLIM for imaging the dynamics of fast molecular processes [173-175]. In order to reduce tissue autofluorescence and to increase the depth of penetration, electromagnetic radiation in the red and near-infrared (NIR) spectral regions can be used, since photons with the respective energies (those inducing fluorescence and those being emitted) are poorly absorbed by live tissues [176, 177].
A combination of fluorescence imaging with other visualization techniques (including magnetic resonance imaging) can ensure the correspondence of the detected fluorescent signals to a particular morphological region in a tissue or an organ. Different imaging techniques can successfully complement each other in solving various biological tasks.
Multimodal imaging for nucleome visualization. In addition to optical imaging, molecular visualization techniques include single photon emission computed tomography (SPECT), positron emission tomography (PET), magnetic resonance imaging (MRI), X-ray computed tomography (CT), and ultrasound imaging (sonography). However, these methods significantly differ in their sensitivity and cost-efficiency [178, 179]. CT, MRI, and optical methods complement each other well and are successfully implemented in preclinical studies [180]. Combining magnetic resonance and optical markers allowed to obtain macroscopic MRI images with a ~50-µm spatial resolution [179]. Fluorescence imaging in vitro can provide detailed microscopic information at the subcellular level, while MRI contrast agents can be directly labeled with fluorescent dyes, thus allowing a bimodal visualization [181]. MRI labels range from the low-molecular-weight T1 and T2 contrast agents to the bimodal probes and multifunctional nanoparticles based on composite nanomaterials [182, 183].
High-sensitivity images with a high spatial resolution in the real-time mode can be obtained by combining activated fluorescence (via the fluorogenic reaction) and activated MRI (via self-assembly in situ) and used for localization of molecular events. For example, a P-CyFF-Gd probe was activated by an endogenous alkaline phosphatase overexpressed on cell membrane, leading to the nanoparticles accumulation in the membrane, which could be directly visualized in living cells and in mice [184]. This strategy can be used for the development of other activated probes for the bimodal visualization, including CRISPR–Cas9 imaging.
A valuable finding is the fact that some low-molecular-weight X-ray or magnetic resonance contrast agents can have an effect of optical clearing, as well as enhance the fluorescence of deep molecular markers expressed in vivo.
Optical clearing methods. Detection of low-intensity fluorescence in living tissues is challenging. Prolonging the exposure time does not solve the problem, as it leads to the proportional increase in the signal/background ratio. In this case, optical clearing is a simple but effective tool for increasing the fluorescence image contrast.
Most biological tissues are optically opaque because they absorb and scatter light. The main absorption regions in the UV spectrum are as follows: 200 and 230 nm (proteins); 260 nm (DNA and RNA); 275 and 345 nm (oxidized hemoglobin); 275 and 360 nm (reduced hemoglobin) [185]. Although one of the standard methods for measuring the concentrations of purified proteins is measuring absorbance at 280 nm (tryptophan, tyrosine, and phenylalanine residues), proteins absorb mostly at 200-230 nm due to the presence of peptide bonds. Therefore, this region is considered to be “protein-associated” in the context of tissue optical transparency. The main absorption regions of biological tissues in the visible and infrared ranges are as follows: 970, 1180, 1450, 1775, 1930, and 1975 nm (water); approximately 760, 830, 920, 1040, 1210, 1430, 1730, 1760, and 1900-2600 nm (lipids); 420 and 550 nm (reduced hemoglobin); 410, 540, and 575 nm (oxidized hemoglobin) [185]. There are five “windows” between the absorption regions: 350-400 nm (I), 625-975 nm (II), 1100-1350 nm (III), 1600-1870 nm (IV), and 2100-2300 nm (V) [186].
While light absorption is an inevitable consequence of the chemical composition of tissues, light scattering in biological samples can be reduced. Optical clearing methods are used for this purpose; they are based on three main mechanisms [187, 188]: reduction of the difference between the refractive indices of different tissue components, tissue dehydration with a clearing agent, and changing the structure of collagen fibers. The easiest clearing procedure is placing a thin tissue section into an immersion liquid with a high refractive index. Many substances are known to act as clearing agents, including formamide, glycerol, glucose, sucrose, dimethyl sulfoxide (DMSO), and various polyethylene glycols. Thus, the treatment of muscle tissue with 60% aqueous solution of glycerol adds two more optical windows (at 230 and 300 nm) to those mentioned above [185].
Most studies using optical clearing are performed ex vivo, i.e., in tissue samples obtained from sacrificed animals or human biopsies. At the same time, in vivo studies on the in vivo visualization of tissues and organs are very promising. However, they require the use of nontoxic clearing agents that would produce only transient effects or have the minimal long-term effects in living tissues.
Some of the low-molecular-weight medical contrast agents are capable of the optical clearing. These are X-ray contrast agents iohexol (Omnipaque) and iodixanol (Visipaque) along with magnetic resonance contrast agents gadobutrol (Gadovist), gadopentetic acid (Magnevist), and gadoteric acid (Dotarem) [189-191]. These compounds make it possible to obtain a high-contrast optical image and synchronize it with an MRI image of the same area of tissue/organ. Typically, these agents are applied to the skin surface for 10-15 min. However, the clearing effect was also achieved when Gadovist was injected intravenously [192]. Therefore, optical clearing expands the possibilities of the multimodal studying of tumors, e.g., by combining fluorescence laser imaging and optical coherence tomography (OCT) with magnetic resonance and computed tomography (CT) [191].
It should me mentioned that fluorescence is very sensitive to the fluorophore microenvironment, including solvent polarity. Hence, tissue clearing can significantly change the properties of fluorophores. For example, optical clearing with a mixture of benzyl alcohol and benzyl benzoate not only changed the fluorescence intensity of DAPI and Alexa Fluor dyes, but also shifted the positions of the maxima in their absorption and emission spectra up to several tens of nm [193]. This effect should be taken into account when selecting the optical filters for working with biological samples to avoid signal mixing. The same mixture of benzyl alcohol and benzyl benzoate caused the quenching of eGFP fluorescence to the background level [193]. In contrast, optical clearing with gadobutrol increased the fluorescence intensity of TagRFP 1.5-fold [194].
One more detail to be considered when working with optical clearing agents is that the decrease in light scattering not only facilitates the penetration of the excitation photons into the tissue, but also promotes the release of the emitted photons from this tissue. Hence, excessive clearing caused by a high concentration of the clearing agent or excessively long treatment can lead to a drop in the recorded signal [188].
All of the above should be taken into account during development of multimodal approaches for in vivo visualization.
Requirements for nontoxicity and biosafety. In addition to the above-mentioned requirements for the chromosomal loci visualization methods (sensitivity at the molecular level and correspondence to the morphological structures), the components for chromatin labeling should be low-toxic and satisfy the ADME parameters.
ADME stands for adsorption (A), distribution (D), metabolism (M), and excretion (E). Typically, ADME parameters are used to characterize pharmaceutical products. However, when used in gene therapy, such assessment should be extended, because the classical ADME parameters are applicable only to some components of the gene editing systems, such as fluorogenic substrates and labels. Similar to the requirements for the gene transfer vectors in gene therapy [195], ADME standards for the systems providing stable expression of genetically engineered products in mammalian cells need a broader interpretation. The ADME data essential for the proper risk assessment have been obtained in laboratory studies and clinical trials on the use of viral vectors in human gene therapy [196]. For example, potential virus shedding after intravenous injection of various viral vectors into rodents was evaluated in [197]. The authors assessed the limit of detection for a third-generation lentivirus, a recombinant AAV, and an E1-deleted AV tested directly from the animal stocks and after their application onto the cage plastic and bedding. No evidence of virus amplification was found in the blood, urine, and fecal samples as well as at the site of injection or in soiled bedding.
The recombinant adeno-associated virus, which has no known human pathogenicity, revealed to be the most safe vector.
It was concluded that the commonly used replication-deficient viral vectors posed the minimal exposure risk by 72 hours after the inoculation. Animal biosafety level 2 precautions are warranted during the initial administration; however, after changing the cage, level 1 safety measures may be sufficient.
Designing CRISPR–cas9-based in vivo visualization systems: from in vitro to in vivo. The preferable probes for the application in live organisms are the so-called theranostic (simultaneously diagnostic and therapeutic) probes. The CRISPR–Cas9 system can be considered as a theranostic probe because it ensures its own targeted delivery as well as the therapeutic response (DNA editing) [4, 179]. The CRISPR–Cas9 system is most efficient in the treatment of monogenic diseases, such as the Huntington’s disease, cystic fibrosis, thalassemia, and sickle cell anemia. However, CRISPR–Cas9 can be potentially used against multifactorial diseases, including cancer, diabetes, and cardiovascular disorders [4, 6, 198]. Currently, several gene-editing therapeutic agents based on the CRISPR–Cas9 system components are tested in preclinical and clinical trials. In 2021, researchers reported a successful use of a new drug NTLA-2001 in six patients suffering from hereditary transthyretin amyloidosis with polyneuropathy. The drug was based on lipid nanoparticles encapsulating mRNA for Cas9 from S. pyogenes and sgRNA [199].
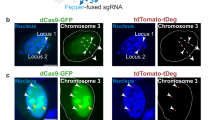
Another application of the CRISPR–Cas9 system is associated with its further modification for genome and nucleome labeling and imaging. By 2013, a new technique for the CRISPR–Cas9 delivery named RGEN (RNA-guided endonuclease) had been developed and demonstrated in cultured human cells [200]. Due to its advantages, RGEN has taken the first place in the list of gene-editing approaches (ZFN, TALEN, etc.). The RGEN technology has a simple design, as the choice of the target site is determined solely by the complementarity of its nucleotides to the sgRNA spacer; no individual proteins should be constructed for each new target site. Another important advantage of RGEN technology is the possibility of multiplexing, i.e., combining Cas9 expression with the delivery of several sgRNAs. In 2013, this system was used to visualize the telomeres functioning and to determine the intranuclear location of the membrane mucin gene loci (MUC4) [110]. In the pioneer work [5], several dCas9 orthologs were tested with sgRNAs specific to the telomere sequences. Fluorescently labeled dCas9 proteins were efficiently directed to correct target sequences. The authors succeeded in labeling two different pairs of chromosomes using sgRNAs targeting specific sequences on chromosomes 9 and 13. Next, they turned their attention to mapping pairs of loci on the same chromosome. Using dual-color pairs of dCas9 and cognate sgRNAs, the authors identified the loci located at 75 and 2 Mb from each other; the calculated fluorescence-based distances correlated with the linear distances on the chromosome’s physical map. When comparing two pairs of loci with very similar distances between them (~2 Mb), the authors were able to discriminate different degrees of chromatin compaction over even such a short distance. This work has presented the first mapping of loci within the same chromosome.
Although combined application of CRISPR–Cas9 and superresolution microscopy can improve image resolution, it does not solve the problems of background signal and low sensitivity of the method. One of the possible solutions is based on SunTag [111], a polypeptide scaffold, to which many FP molecules can bind simultaneously. According to the authors’ estimate, at least 150-200 fluorescent protein molecules are needed to obtain a detectable signal. This approach increases the fluorescence signal by orders of magnitude, so that the irradiating light intensity can be reduced, thus decreasing the photobleaching and phototoxicity.
The possibility of in vivo imaging using LiveFISH (CRISPR-based fluorescent in situ hybridization in living cell using fluorescent oligonucleotides) has been demonstrated in [201]. The complexes of chemically synthesized fluorescent sgRNAs and dCas proteins can provide fast, reliable, and scalable genomic DNA and RNA visualization in live cells, including primary cells (cells with a limited lifespan isolated from body tissues).
Let us consider the main and most promising types of molecules and approaches that can be used for the chromatin visualization in vivo. Once again, it should be emphasized that such approaches should allow multiplexing, rapid substitution of labeling system components (universality), multimodal visualization, as well as they should have low toxicity. The low-molecular-weight components of CRISPR–Cas9 systems (e.g., fluorogenic substrates) should have good pharmacokinetics and pharmacodynamics parameters.
The most frequently used and relatively simple solution is intracellular expression of DNA-binding proteins fused with fluorescent proteins (e.g., dCas9-FP) in combination with relevant sgRNAs. Two major components of these constructs have been already optimized for transcription in animal cells. Thus, Cas proteins (originally prokaryotic) can contain peptide signals for their transport into the nucleus and carry different FP sets and different FP copy numbers. Earlier, such constructs were transfected into the cells leading to unregulated constitutive expression of the protein products [5, 126, 201, 202]. However, despite the fact that the toxicity of FPs in cultured cells and in vivo is low [203, 204], the FP-containing chimeric proteins can accumulate in the cytoplasm with the development of endoplasmic reticulum stress [205-207], which limits the possibility of long-term experiments in living cells and tissues.
DNA/RNA-binding proteins based on several dCas9 orthologs from different microbial species can be used for the two-color/polychromatic labeling of intranuclear structures in animal cells. On one hand, the use of orthologs increases the number of possible PAM sequences when selecting the target site. On the other hand, it provides an opportunity for multiplexing, since it allows creation of multiple variants of chimeric proteins composed of different combinations of orthologs with different FPs [5, 127]. However, polychromatic fluorescence alone is not sufficient to visualize the mutual positioning of DNA-bound proteins and requires further probe engineering and optimization of FP pairs [208, 209]. The proper FRET-pair selection makes it possible to estimate the distance between the fluorescent probes associated with spatially convergent elements within the chromatin loops.
FPs are able to fluoresce immediately after their maturation/folding regardless of their location in the cell. Their alternative can be dCas9 proteins with enzyme tags, whose catalytic activity in the nucleus can be detected using special fluorescent quasi-substrates. Because the quasi-substrates for the Halo, SNAP, and CLIP tags can penetrate cell membrane, they can be used for protein labeling in live cells [210]. The substrates for SNAP (a product of human O6-methylguanosine transferase mutagenesis) and CLIP (O2-benzylcytosine transferase, a product of SNAP mutagenesis) are commercially available. The SNAP-catalyzed reaction proceeds at a rate an order of magnitude higher than the CLIP-catalyzed reaction. Despite this fact, coexpression of SNAP/Halo or SNAP/CLIP pairs of chimeric proteins allows to monitor two products of the labeling reaction. In other words, enzymatic reactions catalyzed by these pairs of enzyme tags are mutually orthologous, i.e., capable of proceeding simultaneously and independently of each other.
Cas9 endonucleases and their catalytically inactive dCas9 mutants are able to form functional fusion proteins with FPs [5, 127, 211], base-editing enzymes (e.g., APOBEC deaminase) [212], and ascorbate peroxidase capable of biotinylating DNA-bound proteins [213]. However, the use of enzyme tags for obtaining fluorescent pairs of dCas9 orthologs for direct enzyme-mediated labeling with fluorescent substrates has not yet been investigated. We found the only mentioning of the Cas9 chimera with SNAP in the study [214], describing the synthesis of covalent Cas9 complexes with oligonucleotides aimed to increase the probability of DNA repair via homologous recombination of insertions and deletions in animal cells.
Finally, another promising approach in the development of in vivo imaging systems is the synthesis of sgRNAs containing short aptamers able to specifically interact with molecules that fluoresce only after binding to the aptamer. The synthesis of these fluorescent probes and especially their delivery to live cells are challenging tasks, since the only aptamer ligands used so far were oligonucleotides with the fluorophore/quencher pairs [215, 216]. Cell transfection as the delivery method also limits the application of such oligonucleotides in animals.
Using non-fluorescent molecules which become fluorescent after their binding to the corresponding aptamers is of particular interest as a method for working with tissues of living organisms. Original fluorogenic RNA aptamers were obtained for detecting Malachite Green dye in solutions and in living tissues [217, 218]. However, they could not be used in vivo because of the high phototoxicity of Malachite Green in live cells. Later, selection of aptamers on the basis of their binding to fluorophores coupled with fluorescence-activated cell sorting (FACS) produced the tBroccoli aptamer. Despite its low affinity (KD of ~360 nM) to the ligands (substituted hydroxybenzylidenes, e.g., DFHBI-1T), tBroccoli creates a unique environment and switches on the fluorescence of GFP-like fluorophores. The extinction coefficient of the tBroccoli complex with DFHBI-1T was 29,600 M–1cm–1, which was comparable to that of FPs and its fluorescence was higher compared to other RNA aptamers (e.g., Spinach2) [219].
An interesting feature of new fluorogenic RNA aptamers is that they can fold and form fluorescent complexes with DFHBI-1T-like ligands in the presence of low magnesium concentrations and after fusion with the 3′ end of any small RNA. Once these short aptamers (49 bp in length) are folded, they activate the fluorescence of DFHBI-1T due to changes in the microenvironment that stabilizes the fluorophore excited state. This increases the quantum yield of DFHBI-1T fluorescence 1000-fold [219].
A recent study of G-quadruplex-forming aptamers has identified a family of short RNA aptamers (Mango I to Mango IV) that can bind with a high affinity the derivatives of thiazole orange (TO) dye with an extended carbomethine bridge (TO3) [220, 221]. Such aptamers can be fused to the cellular RNAs. When such recombinant RNAs were expressed, the Mango IV formed complexes with TO3 which were easily detected in mammalian cells. TO3 emits fluorescence in the red spectral region as a result of bathochromic shifts of the absorption and emission peaks the TO spectrum [222]. This approach was used for the purification of ribonucleoproteins [223] and optical imaging of live cells, since TO derivatives are nontoxic, can penetrate through biological membranes, and almost do not fluoresce in the free state (unbound to RNA) [224]. The complex of the tBroccoli-type aptamer (or its dimeric form) with DFHBI-1T (donor) can form a FRET pair with Mango IV–TO3 (acceptor) [221].
Despite the importance of genome mapping for genome editing, the aforementioned approaches have not yet been tested for visualization of close interactions in a pair of ribonucleoprotein probes in in vivo experiments. We believe that the use of fluorogenic RNA aptamers and nontoxic low-molecular-weight fluorogenic ligands, which, in turn, can be linked to paramagnetic labels (MRI contrast agents) [181], is one of the most promising approaches to the CRISPR–Cas9-mediated imaging in vivo.
CONCLUSIONS
Multicolor labeling of several genomic loci in live cells using the CRISPR–Cas9 system, a method developed at Thoru Pederson’s laboratory [5], is one of the newest applications of fluorescently labeled Cas9 protein in molecular biology. It advances the 4D nucleome mapping, as well as our understanding how the nuclear organization changes during the cell lifetime under normal and pathologic conditions. Modern methods of optical imaging (in particular, cell microscopy) allow to observe various cell phenomena at the level of chromatin compaction in an individual cell and to monitor the interactions of chromatin-binding proteins in the nuclei of live cells.
Acquisition and analysis of images reflecting mutual spatial positioning of individual genome elements is an important research topic both in terms of solving fundamental scientific problems of functional genomics and molecular biophysics and practical necessity of genome editing in somatic cells in their natural tissue microenvironment. The importance of in vivo studies is confirmed by a number of recent works, in which the data on the nucleome stacking in vitro significantly differ from there results obtained from in vivo observations.
Contributions. G.A., L.M., and S.B. wrote “Modern Approaches to Studying the 3D Genome Structure in vitro and in vivo” and Delivery of the CRISPR–Cas System Components to the Target cells” sections. A.R. wrote “The CRISPR–Cas SYSTEMS: History of Discovery, Classification, and Application” section. V.Zh. wrote Introduction, “Molecular Imaging of Nucleome Using CRISPR–Cas9. Requirements for in vivo Visualization”, Conclusion sections. Concept and finalize by Zh.V., L.G., A.R. All authors have read and agreed to the published version of the manuscript.
Abbreviations
- 3D:
-
three-dimensional, spatial
- 3C:
-
chromosome conformation capture
- AAV:
-
adeno-associated virus
- ADME:
-
absorption, distribution, metabolism, and excretion
- AV:
-
adenovirus
- cas :
-
CRISPR-associated genes
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- crRNA:
-
CRISPR RNA
- dCas9:
-
dead Cas9
- FISH:
-
fluorescent hybridization in situ
- FLIM:
-
fluorescence lifetime imaging
- FP:
-
fluorescent protein
- FRET:
-
Förster resonance energy transfer
- HIV:
-
human immunodeficiency virus
- LTR:
-
long terminal repeat
- PAM:
-
protospacer adjacent motifs
- gRNA:
-
guide RNA
- sgRNA:
-
single guide RNA
- TALE:
-
transcription activator-like effector
- tracrRNA:
-
trans-activating CRISPR RNA
- MRI:
-
magnetic resonance imaging
- ZFN:
-
zinc finger
References
Dekker, J., Belmont, A. S., Guttman, M., Leshyk, V. O., Lis, J. T., Lomvardas, S., Mirny, L. A., O’Shea, C. C., Park, P. J., Ren, B., et al. (2017) The 4D nucleome project, Nature, 549, 219-226, https://doi.org/10.1038/nature23884.
Zhu, X., Zhang, Y., Wang, Y., Tian, D., Belmont, A. S., Swedlow, J. R., and Ma, J. (2022) Nucleome Browser: an integrative and multimodal data navigation platform for 4D Nucleome, Nat. Methods, 19, 911-913, https://doi.org/10.1038/s41592-022-01559-3.
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity, Science, 337, 816-821, https://doi.org/10.1126/science.1225829.
Kotagama, O. W., Jayasinghe, C. D., and Abeysinghe, T. (2019) Era of genomic medicine: a narrative review on CRISPR technology as a potential therapeutic tool for human diseases, BioMed Res. Int., 2019, 1369682, https://doi.org/10.1155/2019/1369682.
Ma, H., Naseri, A., Reyes-Gutierrez, P., Wolfe, S. A., Zhang, S., and Pederson, T. (2015) Multicolor CRISPR labeling of chromosomal loci in human cells, Proc. Natl. Acad. Sci. USA, 112, 3002-3007, https://doi.org/10.1073/pnas.1420024112.
Xu, X., Hulshoff, M. S., Tan, X., Zeisberg, M., and Zeisberg, E. M. (2020) CRISPR/Cas derivatives as novel gene modulating tools: possibilities and in vivo applications, Int. J. Mol. Sci., 21, 3038, https://doi.org/10.3390/ijms21093038.
Ishino, Y., Shinagawa, H., Makino, K., Amemura, M., and Nakata, A. (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product, J. Bacteriol., 169, 5429-5433, https://doi.org/10.1128/jb.169.12.5429-5433.1987.
Mojica, F. J., Juez, G., and Rodríguez-Valera, F. (1993) Transcription at different salinities of Haloferax mediterranei sequences adjacent to partially modified PstI sites, Mol. Microbiol., 9, 613-621, https://doi.org/10.1111/j.1365-2958.1993.tb01721.x.
Sorek, R., Lawrence, C. M., and Wiedenheft, B. (2013) CRISPR-mediated adaptive immune systems in bacteria and archaea, Annu. Rev. Biochem., 82, 237-266, https://doi.org/10.1146/annurev-biochem-072911-172315.
Lander, E. S. (2016) The heroes of CRISPR, Cell, 164, 18-28, https://doi.org/10.1016/j.cell.2015.12.041.
Mohanraju, P., Makarova, K. S., Zetsche, B., Zhang, F., Koonin, E. V., and van der Oost, J. (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems, Science, 353, aad5147, https://doi.org/10.1126/science.aad5147.
Koonin, E. V., and Makarova, K. S. (2017) Mobile genetic elements and evolution of CRISPR-Cas systems: all the way there and back, Genome Biol. Evol., 9, 2812-2825, https://doi.org/10.1093/gbe/evx192.
Jansen, R., Embden, J. D., Gaastra, W., and Schouls, L. M. (2002) Identification of genes that are associated with DNA repeats in prokaryotes, Mol. Microbiol., 43, 1565-1575, https://doi.org/10.1046/j.1365-2958.2002.02839.x.
Makarova, K. S., Aravind, L., Grishin, N. V., Rogozin, I. B., and Koonin, E. V. (2002) A DNA repair system specific for thermophilic Archaea and bacteria predicted by genomic context analysis, Nucleic Acids Res., 30, 482-496, https://doi.org/10.1093/nar/30.2.482.
Bolotin, A., Quinquis, B., Sorokin, A., Ehrlich, S. D. (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin, Microbiology (Reading), 151, 2551-2561, https://doi.org/10.1099/mic.0.28048-0.
Mojica, F. J., Díez-Villaseñor, C., García-Martínez, J., and Soria, E. (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements, J. Mol. Evol., 60, 174-182, https://doi.org/10.1007/s00239-004-0046-3.
Makarova, K. S., Grishin, N. V., Shabalina, S. A., Wolf, Y. I., and Koonin, E. V. (2006) A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action, Biol. Direct, 1, 7, https://doi.org/10.1186/1745-6150-1-7.
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A., and Horvath, P. (2007) CRISPR provides acquired resistance against viruses in prokaryotes, Science, 315, 1709-1712, https://doi.org/10.1126/science.1138140.
Faure, G., Makarova, K. S., and Koonin, E. V. (2019) CRISPR-Cas: Complex functional networks and multiple roles beyond adaptive immunity, J. Mol. Biol., 431, 3-20, https://doi.org/10.1016/j.jmb.2018.08.030.
Gleditzsch, D., Pausch, P., Müller-Esparza, H., Özcan, A., Guo, X., Bange, G., and Randau, L. (2019) PAM identification by CRISPR-Cas effector complexes: diversified mechanisms and structures, RNA Biol., 16, 504-517, https://doi.org/10.1080/15476286.2018.1504546.
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J. J., Charpentier, E., Cheng, D., Haft, D. H., Horvath, P., et al. (2020) Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants, Nat. Rev. Microbiol., 18, 67-83, https://doi.org/10.1038/s41579-019-0299-x.
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., Barrangou, R., Brouns, S. J., Charpentier, E., Haft, D. H., et al. (2015) An updated evolutionary classification of CRISPR-Cas systems, Nat. Rev. Microbiol., 13, 722-736, https://doi.org/10.1038/nrmicro3569.
Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., Snijders, A. P., Dickman, M. J., Makarova, K. S., Koonin, E. V., and van der Oost, J. (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes, Science, 321, 960-964, https://doi.org/10.1126/science.1159689.
Jore, M. M., Lundgren, M., van Duijn, E., Bultema, J. B., Westra, E. R., Waghmare, S. P., Wiedenheft, B., Pul, U., Wurm, R., Wagner, R., et al. (2011) Structural basis for CRISPR RNA-guided DNA recognition by Cascade, Nat. Struct. Mol. Biol., 18, 529-536, https://doi.org/10.1038/nsmb.2019.
Sinkunas, T., Gasiunas, G., Fremaux, C., Barrangou, R., Horvath, P., and Siksnys, V. (2011) Cas3 is a single-stranded DNA nuclease and ATP-dependent helicase in the CRISPR/Cas immune system, EMBO J., 30, 1335-1342, https://doi.org/10.1038/emboj.2011.41.
Xiao, Y., Luo, M., Dolan, A. E., Liao, M., and Ke, A. (2018) Structure basis for RNA-guided DNA degradation by Cascade and Cas3, Science, 361, eaat0839, https://doi.org/10.1126/science.aat0839.
Taylor, D. W., Zhu, Y., Staals, R. H., Kornfeld, J. E., Shinkai, A., van der Oost, J., Nogales, E., and Doudna, J. A. (2015) Structural biology. Structures of the CRISPR-Cmr complex reveal mode of RNA target positioning, Science, 348, 581-585, https://doi.org/10.1126/science.aaa4535.
Mogila, I., Kazlauskiene, M., Valinskyte, S., Tamulaitiene, G., Tamulaitis, G., and Siksnys, V. (2019) Genetic dissection of the type III-A CRISPR-Cas system Csm complex reveals roles of individual subunits, Cell Rep., 26, 2753-2765.e4, https://doi.org/10.1016/j.celrep.2019.02.029.
You, L., Ma, J., Wang, J., Artamonova, D., Wang, M., Liu, L., Xiang, H., Severinov, K., Zhang, X., and Wang, Y. (2019) Structure studies of the CRISPR-Csm complex reveal mechanism of co-transcriptional interference, Cell, 176, 239-253.e16, https://doi.org/10.1016/j.cell.2018.10.052.
Crawley, A. B., Henriksen, J. R., and Barrangou, R. (2018) CRISPRdisco: an automated pipeline for the discovery and analysis of CRISPR-Cas systems, CRISPR J., 1, 171-181, https://doi.org/10.1089/crispr.2017.0022.
Garcia-Doval, C., and Jinek, M. (2017) Molecular architectures and mechanisms of Class 2 CRISPR-associated nucleases, Curr. Opin. Struct. Biol., 47, 157-166, https://doi.org/10.1016/j.sbi.2017.10.015.
Deltcheva, E., Chylinski, K., Sharma, C. M., Gonzales, K., Chao, Y., Pirzada, Z. A., Eckert, M. R., Vogel, J., and Charpentier, E. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III, Nature, 471, 602-607, https://doi.org/10.1038/nature09886.
Chylinski, K., Le Rhun, A., and Charpentier, E. (2013) The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems, RNA Biol., 10, 726-737, https://doi.org/10.4161/rna.24321.
Nethery, M. A., and Barrangou, R. (2019) Predicting and visualizing features of CRISPR-Cas systems, Methods Enzymol., 616, 1-25, https://doi.org/10.1016/bs.mie.2018.10.016.
Mali, P., Esvelt, K. M., Church, G. M. (2013) Cas9 as a versatile tool for engineering biology, Nat. Methods, 10, 957-963, https://doi.org/10.1038/nmeth.2649.
Jinek, M., Jiang, F., Taylor, D. W., Sternberg, S. H., Kaya, E., Ma, E., Anders, C., Hauer, M., Zhou, K., Lin, S., et al. (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation, Science, 343, 1247997, https://doi.org/10.1126/science.1247997.
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S. I., Dohmae, N., Ishitani, R., Zhang, F., and Nureki, O. (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA, Cell, 156, 935-949, https://doi.org/10.1016/j.cell.2014.02.001.
Shmakov, S., Abudayyeh, O. O., Makarova, K. S., Wolf, Y. I., Gootenberg, J. S., Semenova, E., Minakhin, L., Joung, J., Konermann, S., Severinov, K., et al. (2015) Discovery and functional characterization of diverse Class 2 CRISPR-Cas systems, Mol. Cell, 60, 385-397, https://doi.org/10.1016/j.molcel.2015.10.008.
Harrington, L. B., Ma, E., Chen, J. S., Witte, I. P., Gertz, D., Paez-Espino, D., Al-Shayeb, B., Kyrpides, N. C., Burstein, D., Banfield, J. F., et al. (2020) A scoutRNA is required for some type V CRISPR-Cas systems, Mol. Cell, 79, 416-424.e5, https://doi.org/10.1016/j.molcel.2020.06.022.
O’Connell, M. R. (2019) Molecular mechanisms of RNA targeting by Cas13-containing type VI CRISPR-Cas systems, J. Mol. Biol., 431, 66-87, https://doi.org/10.1016/j.jmb.2018.06.029.
Gasiunas, G., Barrangou, R., Horvath, P., and Siksnys, V. (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria, Proc. Natl. Acad. Sci. USA, 109, E2579-2586, https://doi.org/10.1073/pnas.1208507109.
Leenay, R. T., and Beisel, C. L. (2017) Deciphering, communicating, and engineering the CRISPR PAM, J. Mol. Biol., 429, 177-191, https://doi.org/10.1016/j.jmb.2016.11.024.
Swarts, D. C., and Jinek, M. (2018) Cas9 versus Cas12a/Cpf1: Structure-function comparisons and implications for genome editing, Wiley Interdiscip. Rev. RNA, 9, e1481, https://doi.org/10.1002/wrna.1481.
Khan, S., Sallard, E. (2022) Current and prospective applications of CRISPR-Cas12a in pluricellular organisms, Mol. Biotechnol., 65, 196-205, https://doi.org/10.1007/s12033-022-00538-5..
Feng, Y., Liu, S., Chen, R., and Xie, A. (2021) Target binding and residence: a new determinant of DNA double-strand break repair pathway choice in CRISPR/Cas9 genome editing, J. Zhejiang Univ. Sci. B, 22, 73-86, https://doi.org/10.1631/jzus.B2000282.
Huang, X., Yang, D., Zhang, J., Xu, J., and Chen, Y. E. (2022) Recent advances in improving gene-editing specificity through CRISPR-Cas9 nuclease engineering, Cells, 11, 2186, https://doi.org/10.3390/cells11142186.
Zhou, W., Yang, J., Zhang, Y., Hu, X., and Wang, W. (2020) Current landscape of gene-editing technology in biomedicine: applications, advantages, challenges, and perspectives, MedComm. (2020), 3, e155, https://doi.org/10.1002/mco2.155.
Ramachandran, G., and Bikard, D. (2019) Editing the microbiome the CRISPR way, Philos. Trans. R. Soc. Lond. B Biol. Sci., 374, 20180103, https://doi.org/10.1098/rstb.2018.0103.
Sirohi, U., Kumar, M., Sharma, V. R., Teotia, S., Singh, D., Chaudhary, V., Priya, and Yadav, M. K. (2022) CRISPR/Cas9 system: A potential tool for genetic improvement in floricultural crops, Mol. Biotechnol., 64, 1303-1318, https://doi.org/10.1007/s12033-022-00523-y.
Negi, C., Vasistha, N. K., Singh, D., Vyas, P., and Dhaliwal, H. S. (2022) Application of CRISPR-mediated gene editing for crop improvement, Mol. Biotechnol., 64, 1198-1217, https://doi.org/10.1007/s12033-022-00507-y.
Rasheed, A., Barqawi, A. A., Mahmood, A., Nawaz, M., Shah, A. N., Bay, D. H., Alahdal, M. A., Hassan, M. U., and Qari, S. H. (2022) CRISPR/Cas9 is a powerful tool for precise genome editing of legume crops: a review, Mol. Biol. Rep., 49, 5595-5609, https://doi.org/10.1007/s11033-022-07529-4.
Chaudhary, M., Mukherjee, T. K., Singh, R., Gupta, M., Goyal, S., Singhal, P., Kumar, R., Bhusal, N., and Sharma, P. (2022) CRISPR/Cas technology for improving nutritional values in the agricultural sector: an update, Mol. Biol. Rep., 49, 7101-7110, https://doi.org/10.1007/s11033-022-07523-w.
Zegeye, W. A., Tsegaw, M., Zhang, Y., and Cao, L. (2022) CRISPR-based genome editing: advancements and opportunities for rice improvement, Int. J. Mol. Sci., 23, 4454, https://doi.org/10.3390/ijms23084454.
Kumar, D., Yadav, A., Ahmad, R., Dwivedi, U. N., and Yadav, K. (2022) CRISPR-based genome editing for nutrient enrichment in crops: a promising approach toward global food security, Front. Genet., 13, 932859, https://doi.org/10.3389/fgene.2022.932859.
Osakabe, K., Wada, N., Murakami, E., Miyashita, N., and Osakabe, Y. (2021) Genome editing in mammalian cells using the CRISPR type I-D nuclease, Nucleic Acids Res., 49, 6347-6363, https://doi.org/10.1093/nar/gkab348.
Hou, Z., Hu, C., Ke, A., and Zhang, Y. (2022) Introducing large genomic deletions in human pluripotent stem cells using CRISPR-Cas3, Curr. Protoc., 2, e361, https://doi.org/10.1002/cpz1.361.
Tan, R., Krueger, R. K., Gramelspacher, M. J., Zhou, X., Xiao, Y., Ke, A., Hou, Z., and Zhang, Y. (2022) Cas11 enables genome engineering in human cells with compact CRISPR-Cas3 systems, Mol. Cell, 82, 852-867.e5, https://doi.org/10.1016/j.molcel.2021.12.032.
Bonini, A., Poma, N., Vivaldi, F., Kirchhain, A., Salvo, P., Bottai, D., Tavanti, A., and Di Francesco, F. (2021) Advances in biosensing: the CRISPR/Cas system as a new powerful tool for the detection of nucleic acids, J. Pharm. Biomed. Anal., 192, 113645, https://doi.org/10.1016/j.jpba.2020.113645.
Fapohunda, F. O., Qiao, S., Pan, Y., Wang, H., Liu, Y., Chen, Q., and Lü, P. (2022) CRISPR Cas system: a strategic approach in detection of nucleic acids, Microbiol. Res., 259, 127000, https://doi.org/10.1016/j.micres.2022.127000.
Li, X., Zhang, H., Zhang, J., Song, Y., Shi, X., Zhao, C., and Wang, J. (2022) Diagnostic accuracy of CRISPR technology for detecting SARS-CoV-2: a systematic review and meta-analysis, Expert Rev. Mol. Diagn., 22, 655-663, https://doi.org/10.1080/14737159.2022.2107425.
Xue, Y., Chen, Z., Zhang, W., and Zhang, J. (2022) Engineering CRISPR/Cas13 system against RNA viruses: from diagnostics to therapeutics, Bioengineering (Basel), 9, 291, https://doi.org/10.3390/bioengineering9070291.
Lou, J., Wang, B., Li, J., Ni, P., Jin, Y., Chen, S., Xi, Y., Zhang, R., and Duan, G. (2022) The CRISPR-Cas system as a tool for diagnosing and treating infectious diseases, Mol. Biol. Rep., 49, 11301-11311, https://doi.org/10.1007/s11033-022-07752-z.
Xie, S., Ji, Z., Suo, T., Li, B., and Zhang, X. (2021) Advancing sensing technology with CRISPR: from the detection of nucleic acids to a broad range of analytes – A review, Anal. Chim. Acta, 1185, 338848, https://doi.org/10.1016/j.aca.2021.338848.
Cheng, X., Li, Y., Kou, J., Liao, D., Zhang, W., Yin, L., Man, S., Ma, L. (2022) Novel non-nucleic acid targets detection strategies based on CRISPR/Cas toolboxes: A review, Biosens. Bioelectron., 215, 114559, https://doi.org/10.1016/j.bios.2022.114559.
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., and Lim, W. A. (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression, Cell, 152, 1173-1183, https://doi.org/10.1016/j.cell.2013.02.022.
Xu, X., and Qi, L. S. (2019) A CRISPR-dCas toolbox for genetic engineering and synthetic biology, J. Mol. Biol., 431, 34-47, https://doi.org/10.1016/j.jmb.2018.06.037.
Pan, C., Wu, X., Markel, K., Malzahn, A. A., Kundagrami, N., Sretenovic, S., Zhang, Y., Cheng, Y., Shih, P. M., and Qi, Y. (2021) CRISPR-Act3.0 for highly efficient multiplexed gene activation in plants, Nat. Plants, 7, 942-953, https://doi.org/10.1038/s41477-021-00953-7.
Singh, V., and Jain, M. (2022) Recent advancements in CRISPR-Cas toolbox for imaging applications, Crit. Rev. Biotechnol., 42, 508-531, https://doi.org/10.1080/07388551.2021.1950608.
Brezgin, S., Kostyusheva, A., Kostyushev, D., and Chulanov, V. (2019) Dead Cas systems: types, principles, and applications, Int. J. Mol. Sci., 20, 6041, https://doi.org/10.3390/ijms20236041.
Enríquez, P. (2016) CRISPR-mediated epigenome editing, Yale J. Biol. Med., 89, 471-486.
Xie, N., Zhou, Y., Sun, Q., and Tang, B. (2018) Novel epigenetic techniques provided by the CRISPR/Cas9 system, Stem Cells Int., 2018, 7834175, https://doi.org/10.1155/2018/7834175.
Luo, M. L., Mullis, A. S., Leenay, R. T., and Beisel, C. L. (2015) Repurposing endogenous type I CRISPR-Cas systems for programmable gene repression, Nucleic Acids Res., 43, 674-681, https://doi.org/10.1093/nar/gku971.
Shams, A., Higgins, S. A., Fellmann, C., Laughlin, T. G., Oakes, B. L., Lew, R., Kim, S., Lukarska, M., Arnold, M., Staahl, B. T., et al. (2021) Comprehensive deletion landscape of CRISPR-Cas9 identifies minimal RNA-guided DNA-binding modules, Nat. Commun., 12, 5664, https://doi.org/10.1038/s41467-021-25992-8.
Karvelis, T., Bigelyte, G., Young, J. K., Hou, Z., Zedaveinyte, R., et al. (2020) PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage, Nucleic Acids Res., 48, 5016-5023, https://doi.org/10.1093/nar/gkaa208.
Bigelyte, G., Young, J. K., Karvelis, T., Budre, K., Zedaveinyte, R., Budre, K., Paulraj, S., Djukanovic, V., Gasior, S., Silanskas, A., et al. (2021) Miniature type V-F CRISPR-Cas nucleases enable targeted DNA modification in cells, Nat. Commun., 12, 6191, https://doi.org/10.1038/s41467-021-26469-4.
Ribeiro, L. F., Ribeiro, L. F. C., Barreto, M. Q., and Ward, R. J. (2018) Protein engineering strategies to expand CRISPR-Cas9 applications, Int. J. Genomics, 2018, 1652567, https://doi.org/10.1155/2018/1652567.
Kempfer, R., and Pombo, A. (2020) Methods for mapping 3D chromosome architecture, Nat. Rev. Genet., 21, 207-226, https://doi.org/10.1038/s41576-019-0195-2.
De Jong, H. (2003) Visualizing DNA domains and sequences by microscopy: a fifty-year history of molecular cytogenetics, Genome, 46, 943-946, https://doi.org/10.1139/g03-107.
Cui, C., Shu, W., and Li, P. (2016) Fluorescence in situ hybridization: cell-based genetic diagnostic and research applications, Front. Cell Dev. Biol., 4, 89, https://doi.org/10.3389/fcell.2016.00089.
Szabo, Q., Cavalli, G., and Bantignies, F. (2021) Higher-order chromatin organization using 3D DNA fluorescent in situ hybridization, Methods Mol. Biol., 2157, 221-237, https://doi.org/10.1007/978-1-0716-0664-3_13.
Su, J. H., Zheng, P., Kinrot, S. S., Bintu, B., and Zhuang, X. (2020) Genome-scale imaging of the 3D organization and transcriptional activity of chromatin, Cell, 182, 1641-1659.e26, https://doi.org/10.1016/j.cell.2020.07.032.
Goel, V. Y., and Hansen, A. S. (2021) The macro and micro of chromosome conformation capture, Wiley Interdiscip. Rev. Dev. Biol., 10, e395, https://doi.org/10.1002/wdev.395.
Mohanta, T. K., Mishra, A. K., and Al-Harrasi, A. (2021) The 3D genome: from structure to function, Int. J. Mol. Sci., 22, 11585, https://doi.org/10.3390/ijms222111585.
Bouwman, B. A. M., Crosetto, N., and Bienko, M. (2022) The era of 3D and spatial genomics, Trends Genet., 38, 1062-1075, https://doi.org/10.1016/j.tig.2022.05.010.
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002) Capturing chromosome conformation, Science, 295, 1306-1311, https://doi.org/10.1126/science.1067799.
De Wit, E., and de Laat, W. (2012) A decade of 3C technologies: insights into nuclear organization, Genes Dev., 26, 11-24, https://doi.org/10.1101/gad.179804.111.
Zhao, Z., Tavoosidana, G., Sjölinder, M., Göndör, A., Mariano, P., Wang, S., Kanduri, C., Lezcano, M., Sandhu, K. S., Singh, U., et al. (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions, Nat. Genet., 38, 1341-1347, https://doi.org/10.1038/ng1891.
Giorgetti, L., and Heard, E. (2016) Closing the loop: 3C versus DNA FISH, Genome Biol., 17, 215, https://doi.org/10.1186/s13059-016-1081-2.
Dostie, J., Richmond, T. A., Arnaout, R. A., Selzer, R. R., Lee, W. L., Honan, T.A., Rubio, E. D., Krumm, A., Lamb, J., Nusbaum, C., et al. (2006) Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements, Genome Res., 16, 1299-1309, https://doi.org/10.1101/gr.5571506.
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., Hu, M., Liu, J. S., and Ren, B. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions, Nature, 485, 376-380, https://doi.org/10.1038/nature11082.
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., Amit, I., Lajoie, B. R., Sabo, P. J., Dorschner, M. O., et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science, 326, 289-293, https://doi.org/10.1126/science.1181369.
Fullwood, M. J., Liu, M. H., Pan, Y. F., Liu, J., Xu, H., Mohamed, Y. B., Orlov, Y. L., Velkov, S., Ho, A., Mei, P. H., et al. (2009) An oestrogen-receptor-alpha-bound human chromatin interactome, Nature, 462, 58-64, https://doi.org/10.1038/nature08497.
Kong, S., and Zhang, Y. (2019) Deciphering Hi-C: from 3D genome to function, Cell Biol. Toxicol., 35, 15-32, https://doi.org/10.1007/s10565-018-09456-2.
Deng, W., Shi, X., Tjian, R., Lionnet, T., and Singer, R. H. (2015) CASFISH: CRISPR/Cas9-mediated in situ labeling of genomic loci in fixed cells, Proc. Natl. Acad. Sci. U. S. A., 112, 11870-11875, https://doi.org/10.1073/pnas.1515692112.
Harrison, P. T., and Hart, S. (2018) A beginner’s guide to gene editing, Exp. Physiol., 103, 439-448, https://doi.org/10.1113/EP086047.
Krishna, S. S., Majumdar, I., and Grishin, N. V. (2003) Structural classification of zinc fingers: survey and summary, Nucleic Acids Res., 31, 532-550, https://doi.org/10.1093/nar/gkg161.
Gaj, T., Gersbach, C. A., and Barbas, C. F., 3rd (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering, Trends Biotechnol., 31, 397-405, https://doi.org/10.1016/j.tibtech.2013.04.004.
Kim, Y. G., Cha, J., and Chandrasegaran, S. (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain, Proc. Natl. Acad. Sci. USA, 93, 1156-1160, https://doi.org/10.1073/pnas.93.3.1156.
Li, L., Wu, L. P., and Chandrasegaran, S. (1992) Functional domains in FokI restriction endonuclease, Proc. Natl. Acad. Sci. USA, 89, 4275-4279, https://doi.org/10.1073/pnas.89.10.4275.
Kim, Y. G., and Chandrasegaran, S. (1994) Chimeric restriction endonuclease, Proc. Natl. Acad. Sci. USA, 91, 883-887, https://doi.org/10.1073/pnas.91.3.883.
Huang, B., Schaeffer, C. J., Li, Q., and Tsai, M. D. (1996) Splase: a new class IIS zinc-finger restriction endonuclease with specificity for Sp1 binding sites, J. Protein Chem., 15, 481-489, https://doi.org/10.1007/BF01886856.
Bitinaite, J., Wah, D. A., Aggarwal, A. K., and Schildkraut, I. (1998) FokI dimerization is required for DNA cleavage, Proc. Natl. Acad. Sci. USA, 95, 10570-10575, https://doi.org/10.1073/pnas.95.18.10570.
Moore, R., Chandrahas, A., and Bleris, L. (2014) Transcription activator-like effectors: a toolkit for synthetic biology, ACS Synth. Biol., 3, 708-716, https://doi.org/10.1021/sb400137b.
Lindhout, B. I., Fransz, P., Tessadori, F., Meckel, T., Hooykaas, P. J., and van der Zaal, B. J. (2007) Live cell imaging of repetitive DNA sequences via GFP-tagged polydactyl zinc finger proteins, Nucleic Acids Res., 35, e107, https://doi.org/10.1093/nar/gkm618.
Ma, H., Reyes-Gutierrez, P., and Pederson, T. (2013) Visualization of repetitive DNA sequences in human chromosomes with transcription activator-like effectors, Proc. Natl. Acad. Sci. USA, 110, 21048-21053, https://doi.org/10.1073/pnas.1319097110.
Miyanari, Y., Ziegler-Birling, C., and Torres-Padilla, M. E. (2013) Live visualization of chromatin dynamics with fluorescent TALEs, Nat. Struct. Mol. Biol., 20, 1321-1324, https://doi.org/10.1038/nsmb.2680.
Thanisch, K., Schneider, K., Morbitzer, R., Solovei, I., Lahaye, T., Bultmann, S., and Leonhardt, H. (2014) Targeting and tracing of specific DNA sequences with dTALEs in living cells, Nucleic Acids Res., 42, e38, https://doi.org/10.1093/nar/gkt1348.
Fujimoto, S., Sugano, S. S., Kuwata, K., Osakabe, K., and Matsunaga, S. (2016) Visualization of specific repetitive genomic sequences with fluorescent TALEs in Arabidopsis thaliana, J. Exp. Bot., 67, 6101-6110, https://doi.org/10.1093/jxb/erw371.
Ren, R., Deng, L., Xue, Y., Suzuki, K., Zhang, W., Yu, Y., Wu, J., Sun, L., Gong, X., Luan, H., et al. (2017) Visualization of aging-associated chromatin alterations with an engineered TALE system, Cell Res., 27, 483-504, https://doi.org/10.1038/cr.2017.18.
Chen, B., Gilbert, L. A., Cimini, B. A., Schnitzbauer, J., Zhang, W., Li, G. W., Park, J., Blackburn, E. H., Weissman, J. S., Qi, L. S., et al. (2013) Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system, Cell, 155, 1479-1491, https://doi.org/10.1016/j.cell.2013.12.001.
Tanenbaum, M. E., Gilbert, L. A., Qi, L. S., Weissman, J. S., and Vale, R. D. (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging, Cell, 159, 635-646, https://doi.org/10.1016/j.cell.2014.09.039.
Ye, H., Rong, Z., and Lin, Y. (2017) Live cell imaging of genomic loci using dCas9-SunTag system and a bright fluorescent protein, Protein Cell, 8, 853-855, https://doi.org/10.1007/s13238-017-0460-0.
Shao, S., Zhang, W., Hu, H., Xue, B., Qin, J., Sun, C., Sun, Y., Wei, W., and Sun, Y. (2016) Long-term dual-color tracking of genomic loci by modified sgRNAs of the CRISPR/Cas9 system, Nucleic Acids Res., 44, e86, https://doi.org/10.1093/nar/gkw066.
Wang, S., Su, J. H., Zhang, F., and Zhuang, X. (2016) An RNA-aptamer-based two-color CRISPR labeling system, Sci. Rep., 6, 26857, https://doi.org/10.1038/srep26857.
Fu, Y., Rocha, P. P., Luo, V. M., Raviram, R., Deng, Y., Mazzoni, E. O., and Skok, J. A. (2016) CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci, Nat. Commun., 7, 11707, https://doi.org/10.1038/ncomms11707.
Johansson, H. E., Liljas, L., and Uhlenbeck, O. C. (1997) RNA recognition by the MS2 phage coat protein, Semin. Virol., 8, 176-185, https://doi.org/10.1006/smvy.1997.0120.
Cheng, A. W., Jillette, N., Lee, P., Plaskon, D., Fujiwara, Y., Wang, W., Taghbalout, A., and Wang, H. (2016) Casilio: a versatile CRISPR-Cas9-Pumilio hybrid for gene regulation and genomic labeling, Cell Res., 26, 254-257, https://doi.org/10.1038/cr.2016.3.
Hong, Y., Lu, G., Duan, J., Liu, W., and Zhang, Y. (2018) Comparison and optimization of CRISPR/dCas9/gRNA genome-labeling systems for live cell imaging, Genome Biol., 19, 39, https://doi.org/10.1186/s13059-018-1413-5.
Chaudhary, N., Nho, S. H., Cho, H., Gantumur, N., Ra, J. S., Myung, K., and Kim, H. (2020) Background-suppressed live visualization of genomic loci with an improved CRISPR system based on a split fluorophore, Genome Res., 30, 1306-1316, https://doi.org/10.1101/gr.260018.119.
Wu, X., Mao, S., Ying, Y., Krueger, C. J., and Chen, A. K. (2019) Progress and challenges for live-cell imaging of genomic loci using CRISPR-based platforms, Genom. Proteom. Bioinform., 17, 119-128, https://doi.org/10.1016/j.gpb.2018.10.001.
Lee, C. M., Cradick, T. J., and Bao, G. (2016) The Neisseria meningitidis CRISPR-Cas9 system enables specific genome editing in mammalian cells, Mol. Ther., 24, 645-654, https://doi.org/10.1038/mt.2016.8.
Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L., Sun, N., Zeina, C. M., Gao, X., Rees, H. A., Lin, Z., et al. (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity, Nature, 556, 57-63, https://doi.org/10.1038/nature26155.
Kim, T. H., and Dekker, J. (2018) ChIP-seq, Cold Spring Harbor protocols, 2018, https://doi.org/10.1101/pdb.prot082644.
Wang, T., Wei, J. J., Sabatini, D. M., and Lander, E. S. (2014) Genetic screens in human cells using the CRISPR-Cas9 system, Science, 343, 80-84, https://doi.org/10.1126/science.1246981.
Engler, C., and Marillonnet, S. (2014) Golden gate cloning, Methods Mol. Biol., 1116, 119-131, https://doi.org/10.1007/978-1-62703-764-8_9.
Chen, B., and Huang, B. (2014) Imaging genomic elements in living cells using CRISPR/Cas9, Methods Enzymol., 546, 337-354, https://doi.org/10.1016/B978-0-12-801185-0.00016-7.
Ma, H., Tu, L. C., Naseri, A., Huisman, M., Zhang, S., Grunwald, D., and Pederson, T. (2016) Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow, Nat. Biotechnol., 34, 528-530, https://doi.org/10.1038/nbt.3526.
Ma, D., Peng, S., Huang, W., Cai, Z., and Xie, Z. (2018) Rational design of mini-Cas9 for transcriptional activation, ACS Synth. Biol., 7, 978-985, https://doi.org/10.1021/acssynbio.7b00404.
Chen, B., Hu, J., Almeida, R., Liu, H., Balakrishnan, S., Covill-Cooke, C., Lim, W. A., and Huang, B. (2016) Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci, Nucleic Acids Res., 44, e75, https://doi.org/10.1093/nar/gkv1533.
Xu W. (2019) Microinjection and micromanipulation: a historical perspective, Methods Mol. Biol., 1874, 1-16, https://doi.org/10.1007/978-1-4939-8831-0_1.
Hung, K. L., Meitlis, I., Hale, M., Chen, C. Y., Singh, S., Jackson, S. W., Miao, C. H., Khan, I. F., Rawlings, D. J., and James, R. G. (2018) Engineering protein-secreting plasma cells by homology-directed repair in primary human B cells, Mol. Ther., 26, 456-467, https://doi.org/10.1016/j.ymthe.2017.11.012.
Yen, J., Fiorino, M., Liu, Y., Paula, S., Clarkson, S., Quinn, L., Tschantz, W. R., Klock, H., Guo, N., Russ, C., et al. (2018) TRIAMF: a new method for delivery of Cas9 ribonucleoprotein complex to human hematopoietic stem cells, Sci. Rep., 8, 16304, https://doi.org/10.1038/s41598-018-34601-6.
D’Astolfo, D. S., Pagliero, R. J., Pras, A., Karthaus, W. R., Clevers, H., Prasad, V., Lebbink, R. J., Rehmann, H., and Geijsen, N. (2015) Efficient intracellular delivery of native proteins, Cell, 161, 674-690, https://doi.org/10.1016/j.cell.2015.03.028.
Supotnitskiy, M. V. (2011) Genotherapeutic vector systems based on viruses [In Russian], Biopreparats (Biopharmaceuticals), 3, 15-26.
Spirin, P. V., Vilgelm, A. E., and Prassolov, V. S. (2008) Lentiviral vectors, Mol. Biol., 42, 814-825, https://doi.org/10.1134/S002689330805018X.
Sakuma, T., Barry, M. A., and Ikeda, Y. (2012) Lentiviral vectors: basic to translational, Biochem. J., 443, 603-618, https://doi.org/10.1042/BJ20120146.
Merienne, N., Vachey, G., de Longprez, L., Meunier, C., Zimmer, V., Perriard, G., Canales, M., Mathias, A., Herrgott, L., Beltraminelli, T., et al. (2017) The self-inactivating KamiCas9 system for the editing of CNS disease genes, Cell Rep., 20, 2980-2991, https://doi.org/10.1016/j.celrep.2017.08.075.
Zetsche, B., Volz, S. E., and Zhang, F. (2015) A split-Cas9 architecture for inducible genome editing and transcription modulation, Nat. Biotechnol., 33, 139-142, https://doi.org/10.1038/nbt.3149.
Somia, N., and Verma, I. M. (2000) Gene therapy: trials and tribulations, Nat. Rev. Genet., 1, 91-99, https://doi.org/10.1038/35038533.
Wang, Q., Chen, S., Xiao, Q., Liu, Z., Liu, S., Hou, P., Zhou, L., Hou, W., Ho, W., Li, C., et al. (2017) Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection, Retrovirology, 14, 51, https://doi.org/10.1186/s12977-017-0375-0.
Li, L., He, Z. Y., Wei, X. W., Gao, G. P., and Wei, Y. Q. (2015) Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors, Hum. Gene Ther., 26, 452-462, https://doi.org/10.1089/hum.2015.069.
Li, L., Hu, S., and Chen, X. (2018) Non-viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities, Biomaterials, 171, 207-218, https://doi.org/10.1016/j.biomaterials.2018.04.031.
Wang, M., Zuris, J. A., Meng, F., Rees, H., Sun, S., Deng, P., Han, Y., Gao, X., Pouli, D., Wu, Q., et al. (2016) Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles, Proc. Natl. Acad. Sci. USA, 113, 2868-2873, https://doi.org/10.1073/pnas.1520244113.
Kretzmann, J. A., Ho, D., Evans, C. W., Plani-Lam, J., Garcia-Bloj, B., Mohamed, A. E., O'Mara, M. L., Ford, E., Tan, D., Lister, R., et al. (2017) Synthetically controlling dendrimer flexibility improves delivery of large plasmid DNA, Chem. Sci., 8, 2923-2930, https://doi.org/10.1039/c7sc00097a.
Zhu, H., Zhang, L., Tong, S., Lee, C. M., Deshmukh, H., and Bao, G. (2019) Spatial control of in vivo CRISPR-Cas9 genome editing via nanomagnets, Nat. Biomed. Eng., 3, 126-136, https://doi.org/10.1038/s41551-018-0318-7.
Kumar, S., Kaur, S., Seem, K., Kumar, S., and Mohapatra, T. (2021) Understanding 3D genome organization and its effect on transcriptional gene regulation under environmental stress in plant: a chromatin perspective, Front. Cell Dev. Biol., 9, 774719, https://doi.org/10.3389/fcell.2021.774719.
Li, G. (2020) The 3D organization of genome in the nucleus: from the nucleosome to the 4D nucleome, Sci. China Life Sci., 63, 791-794, https://doi.org/10.1007/s11427-020-1723-y.
Park, T. L., Lee, Y., and Cho, W. K. (2021) Visualization of chromatin higher-order structures and dynamics in live cells, BMB Rep., 54, 489-496, https://doi.org/10.5483/BMBRep.2021.54.10.098.
Hnisz, D., Weintraub, A. S., Day, D. S., Valton, A. L., Bak, R. O., Li, C. H., Goldmann, J., Lajoie, B. R., Fan, Z. P., Sigova, A. A., et al. (2016) Activation of proto-oncogenes by disruption of chromosome neighborhoods, Science, 351, 1454-1458, https://doi.org/10.1126/science.aad9024.
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., Horn, D., Kayserili, H., Opitz, J. M., Laxova, R., et al. (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions, Cell, 161, 1012-1025, https://doi.org/10.1016/j.cell.2015.04.004.
Weissleder, R., Pittet, M. J. (2008) Imaging in the era of molecular oncology, Nature, 452, 580-589, https://doi.org/10.1038/nature06917.
Parteka-Tojek, Z., Zhu, J. J., Lee, B., Jodkowska, K., Wang, P., Aaron, J., Chew, T. L., Banecki, K., Plewczynski, D., and Ruan, Y. (2022) Super-resolution visualization of chromatin loop folding in human lymphoblastoid cells using interferometric photoactivated localization microscopy, Sci. Rep., 12, 8582, https://doi.org/10.1038/s41598-022-12568-9.
Boettiger, A., and Murphy, S. (2020) Advances in chromatin imaging at kilobase-scale resolution, Trends Genet., 36, 273-287, https://doi.org/10.1016/j.tig.2019.12.010.
Esa, A., Edelmann, P., Kreth, G., Trakhtenbrot, L., Amariglio, N., Rechavi, G., Hausmann, M., and Cremer, C. (2000) Three-dimensional spectral precision distance microscopy of chromatin nanostructures after triple-colour DNA labelling: a study of the BCR region on chromosome 22 and the Philadelphia chromosome, J. Microsc., 199, 96-105, https://doi.org/10.1046/j.1365-2818.2000.00707.x.
Weiland, Y., Lemmer, P., and Cremer, C. (2011) Combining FISH with localisation microscopy: Super-resolution imaging of nuclear genome nanostructures, Chromosome Res., 19, 5-23, https://doi.org/10.1007/s10577-010-9171-6.
Mateo, L. J., Murphy, S. E., Hafner, A., Cinquini, I. S., Walker, C. A., and Boettiger, A. N. (2019) Visualizing DNA folding and RNA in embryos at single-cell resolution, Nature, 568, 49-54, https://doi.org/10.1038/s41586-019-1035-4.
Maxouri, S., Taraviras, S., and Lygerou, Z. (2018) Visualizing the dynamics of histone variants in the S-phase nucleus, Genome Biol., 19, 182, https://doi.org/10.1186/s13059-018-1556-4.
Clément, C., Orsi, G. A., Gatto, A., Boyarchuk, E., Forest, A., Hajj, B., Miné-Hattab, J., Garnier, M., Gurard-Levin, Z. A., Quivy, J. P., et al. (2018) High-resolution visualization of H3 variants during replication reveals their controlled recycling, Nat. Commun., 9, 3181, https://doi.org/10.1038/s41467-018-05697-1.
Stepanov, A. I., Besedovskaia, Z. V., Moshareva, M. A., Lukyanov, K. A., and Putlyaeva, L. V. (2022) Studying chromatin epigenetics with fluorescence microscopy, Int. J. Mol. Sci., 23, 8988, https://doi.org/10.3390/ijms23168988.
Becker, W. (2015) Advanced Time-Correlated Single Photon Counting Applications, Springer Berlin, Heidelberg, https://doi.org/10.1007/978-3-319-14929-5.
Datta, R., Heaster, T. M., Sharick, J. T., Gillette, A. A., and Skala, M. C. (2020) Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications, J. Biomed. Opt., 25, 071203, https://doi.org/10.1117/1.JBO.25.7.071203.
Pliss, A., and Prasad, P. N. (2020) High resolution mapping of subcellular refractive index by Fluorescence Lifetime Imaging: a next frontier in quantitative cell science? Methods Appl. Fluoresc., 8, 032001, https://doi.org/10.1088/2050-6120/ab8571.
Levchenko, S. M., Pliss, A., Peng, X., Prasad, P. N., and Qu, J. (2021) Fluorescence lifetime imaging for studying DNA compaction and gene activities, Light Sci. Appl., 10, 224, https://doi.org/10.1038/s41377-021-00664-w.
Sherrard, A., Bishop, P., Panagi, M., Villagomez, M. B., Alibhai, D., and Kaidi, A. (2018) Streamlined histone-based fluorescence lifetime imaging microscopy (FLIM) for studying chromatin organization, Biol. Open, 7, bio031476, https://doi.org/10.1242/bio.031476.
Chanou, A., and Hamperl, S. (2021) Single-molecule techniques to study chromatin, Front. Cell Dev. Biol., 9, 699771, https://doi.org/10.3389/fcell.2021.699771.
Lou, J., Scipioni, L., Wright, B. K., Bartolec, T. K., Zhang, J., Masamsetti, V. P., Gaus, K., Gratton, E., Cesare, A. J., and Hinde, E. (2019) Phasor histone FLIM-FRET microscopy quantifies spatiotemporal rearrangement of chromatin architecture during the DNA damage response, Proc. Natl. Acad. Sci. USA, 116, 7323-7332, https://doi.org/10.1073/pnas.1814965116.
Labas, Y. A., Gurskaya, N. G., Yanushevich, Y. G., Fradkov, A. F., Lukyanov, K. A., Lukyanov, S. A., and Matz, M. V. (2002) Diversity and evolution of the green fluorescent protein family, Proc. Natl. Acad. Sci. USA, 99, 4256-4261, https://doi.org/10.1073/pnas.062552299.
Chudakov, D. M., Lukyanov, S., and Lukyanov, K. A. (2005) Fluorescent proteins as a toolkit for in vivo imaging, Trends Biotechnol., 23, 605-613, https://doi.org/10.1016/j.tibtech.2005.10.005.
Hochreiter, B., Garcia, A. P., and Schmid, J. A. (2015) Fluorescent proteins as genetically encoded FRET biosensors in life sciences, Sensors (Basel), 15, 26281-26314, https://doi.org/10.3390/s151026281.
Rusanov, A. L., Ivashina, T. V., Vinokurov, L. M., Fiks, I. I., Orlova, A. G., Turchin, I. V., Meerovich, I. G., Zherdeva, V. V., and Savitsky, A. P. (2010) Lifetime imaging of FRET between red fluorescent proteins, J. Biophotonics, 3, 774-783, https://doi.org/10.1002/jbio.201000065.
Savitsky, A. P., Rusanov, A. L., Zherdeva, V. V., Gorodnicheva, T. V., Khrenova, M. G., and Nemukhin, A. V. (2012) FLIM-FRET imaging of caspase-3 activity in live cells using pair of red fluorescent proteins, Theranostics, 2, 215-226, https://doi.org/10.7150/thno.3885.
Zherdeva, V., Kazachkina, N. I., Shcheslavskiy, V., and Savitsky, A. P. (2018) Long-term fluorescence lifetime imaging of a genetically encoded sensor for caspase-3 activity in mouse tumor xenografts, J. Biomed. Opt., 23, 035002, https://doi.org/10.1117/1.JBO.23.3.035002.
Giacomelli, M. G., Sheikine, Y., Vardeh, H., Connolly, J. L., and Fujimoto, J. G. (2015) Rapid imaging of surgical breast excisions using direct temporal sampling two photon fluorescent lifetime imaging, Biomed. Opt. Express, 6, 4317-4325, https://doi.org/10.1364/BOE.6.004317.
Ryu, J., Kang, U., Kim, J., Kim, H., Kang, J. H., Kim, H., Sohn, D. K., Jeong, J. H., Yoo, H., and Gweon, B. (2018) Real-time visualization of two-photon fluorescence lifetime imaging microscopy using a wavelength-tunable femtosecond pulsed laser, Biomed. Opt. Express, 9, 3449-3463, https://doi.org/10.1364/BOE.9.003449.
Bower, A. J., Li, J., Chaney, E. J., Marjanovic, M., Spillman, D. R., Jr., and Boppart, S. A. (2018) High-speed imaging of transient metabolic dynamics using two-photon fluorescence lifetime imaging microscopy, Optica, 5, 1290-1296, https://doi.org/10.1364/OPTICA.5.001290.
Nothdurft, R., Sarder, P., Bloch, S., Culver, J., and Achilefu, S. (2012) Fluorescence lifetime imaging microscopy using near-infrared contrast agents, J. Microsc., 247, 202-207, https://doi.org/10.1111/j.1365-2818.2012.03634.x.
Qiao, H., Wu, J., Zhang, X., Luo, J., Wang, H., and Ming, D. (2021) The advance of CRISPR-Cas9-based and NIR/CRISPR-Cas9-based imaging system, Front. Chem., 9, 786354, https://doi.org/10.3389/fchem.2021.786354.
Räty, J. K., Liimatainen, T., Kaikkonen, M. U., Gröhn, O., Airenne, K. J., and Ylä-Herttuala, S. (2007) Non-invasive imaging in gene therapy, Mol. Ther., 15, 1579-1586, https://doi.org/10.1038/sj.mt.6300233.
Niu, G., and Chen, X. (2009) The role of molecular imaging in drug delivery, Drug Deliv. (Lond.), 3, 109-113.
Auletta, L., Gramanzini, M., Gargiulo, S., Albanese, S., Salvatore, M., and Greco, A. (2017) Advances in multimodal molecular imaging, Q. J. Nucl. Med. Mol. Imaging, 61, 19-32, https://doi.org/10.23736/S1824-4785.16.02943-5.
Kircher, M. F., Mahmood, U., King, R. S., Weissleder, R., and Josephson, L. (2003) A multimodal nanoparticle for preoperative magnetic resonance imaging and intraoperative optical brain tumor delineation, Cancer Res., 63, 8122-8125.
Pan, D., Caruthers, S. D., Chen, J., Winter, P. M., SenPan, A., Schmieder, A. H., Wickline, S. A., and Lanza, G. M. (2010) Nanomedicine strategies for molecular targets with MRI and optical imaging, Future Med. Chem., 2, 471-490, https://doi.org/10.4155/fmc.10.5.
Polimeni, J. R., and Wald, L. L. (2018) Magnetic resonance imaging technology-bridging the gap between noninvasive human imaging and optical microscopy, Curr. Opin. Neurobiol., 50, 250-260, https://doi.org/10.1016/j.conb.2018.04.026.
Yan, R., Hu, Y., Liu, F., Wei, S., Fang, D., Shuhendler, A. J., Liu, H., Chen, H. Y., and Ye, D. (2019) Activatable NIR fluorescence/MRI bimodal probes for in vivo imaging by enzyme-mediated fluorogenic reaction and self-assembly, J. Am. Chem. Soc., 141, 10331-10341, https://doi.org/10.1021/jacs.9b03649.
Carneiro, I., Carvalho, S., Henrique, R., Oliveira, L., and Tuchin, V. (2019) Moving tissue spectral window to the deep-ultraviolet via optical clearing, J. Biophotonics, 12, e201900181, https://doi.org/10.1002/jbio.201900181.
Bashkatov, A. N., Berezin, K. V., Dvoretskiy, K. N., Chernavina, M. L., Genina, E. A., Genin, V. D., Kochubey, V. I., Lazareva, E. N., Pravdin, A. B., Shvachkina, M. E., et al. (2018) Measurement of tissue optical properties in the context of tissue optical clearing, J. Biomed. Opt., 23, 091416, https://doi.org/10.1117/1.JBO.23.9.091416.
Genina, E. A., Bashkatov, A. N., and Tuchin, V. V. (2010) Tissue optical immersion clearing, Expert Rev. Med. Devices, 7, 825-842, https://doi.org/10.1586/erd.10.50.
Sdobnov, A. Y., Lademann, J., Darvin, M. E., and Tuchin, V. V. (2019) Methods for optical skin clearing in molecular optical imaging in dermatology, Biochemistry (Moscow), 84, S144-S158, https://doi.org/10.1134/S0006297919140098.
Sdobnov, A. Y., Darvin, M. E., Schleusener, J., Lademann, J., and Tuchin, V. V. (2019) Hydrogen bound water profiles in the skin influenced by optical clearing molecular agents-Quantitative analysis using confocal Raman microscopy, J. Biophotonics, 12, e201800283, https://doi.org/10.1002/jbio.201800283.
Tuchina, D. K., Meerovich, I. G., Sindeeva, O. A., Zherdeva, V. V., Savitsky, A. P., Bogdanov, A. A., Jr., and Tuchin, V. V. (2020) Magnetic resonance contrast agents in optical clearing: prospects for multimodal tissue imaging, J. Biophotonics, 13, e201960249, https://doi.org/10.1002/jbio.201960249.
Tuchina, D. K., Meerovich, I. G., Sindeeva, O. A., Zherdeva, V. V., Kazachkina, N. I., Solov’ev, I., Savitsky, A. P., Bogdanov, A. A., and Tuchin, V. V. (2021) Prospects for multimodal visualisation of biological tissues using fluorescence imaging, Quantum Electronics, 51, 104-117, https://doi.org/10.1070/QEL17512.
Kazachkina, N. I., Zherdeva, V. V., Meerovich, I. G., Saydasheva, A. N., Solovyev, I. D., Tuchina, D. K., Savitsky, A. P., Tuchin, V. V., and Bogdanov, A. A., Jr. (2022) MR and fluorescence imaging of gadobutrol-induced optical clearing of red fluorescent protein signal in an in vivo cancer model, NMR Biomed., 35, e4708, https://doi.org/10.1002/nbm.4708.
Eliat, F., Sohn, R., Renner, H., Kagermeier, T., Volkery, S., Brinkmann, H., Kirschnick, N., Kiefer, F., Grabos, M., Becker, K., et al. (2022) Tissue clearing may alter emission and absorption properties of common fluorophores, Sci. Rep., 12, 5551, https://doi.org/10.1038/s41598-022-09303-9.
Kazachkina, N. I., Zherdeva, V. V., Saydasheva, A. N., Meerovich, I. G., Tuchin, V. V., Savitsky, A. P., and Bogdanov, A. A., Jr. (2021) Topical gadobutrol application causes fluorescence intensity change in RFP-expressing tumor-bearing mice, J. Biomed. Photonics Engineering, 020301, 1-7, https://doi.org/10.18287/JBPE21.07.020301.
Ylä-Herttuala S. (2017) The pharmacology of gene therapy, Mol. Ther., 25, 1731-1732, https://doi.org/10.1016/j.ymthe.2017.07.007.
Collins, D. E., Reuter, J. D., Rush, H. G., and Villano, J. S. (2017) Viral vector biosafety in laboratory animal research, Comp. Med., 67, 215-221.
Reuter, J. D., Fang, X., Ly, C. S., Suter, K. K., and Gibbs, D. (2012) Assessment of hazard risk associated with the intravenous use of viral vectors in rodents, Comp. Med., 62, 361-370.
Gunay, M. S., Ozer, A. Y., and Chalon, S. (2016) Drug delivery systems for imaging and therapy of Parkinson’s disease, Curr. Neuropharmacol., 14, 376-391, https://doi.org/10.2174/1570159x14666151230124904.
Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M. L., Seitzer, J., O'Connell, D., Walsh, K. R., Wood, K., et al. (2021) CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis, N. Engl. J. Med., 385, 493-502, https://doi.org/10.1056/NEJMoa2107454.
Mussolino, C., and Cathomen, T. (2013) RNA guides genome engineering, Nat. Biotechnol., 31, 208-209, https://doi.org/10.1038/nbt.2527.
Wang, H., Nakamura, M., Abbott, T. R., Zhao, D., Luo, K., Yu, C., Nguyen, C. M., Lo, A., Daley, T. P., La Russa, M., et al. (2019) CRISPR-mediated live imaging of genome editing and transcription, Science, 365, 1301-1305, https://doi.org/10.1126/science.aax7852.
Anton, T., Bultmann, S., Leonhardt, H., and Markaki, Y. (2014) Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system, Nucleus, 5, 163-172, https://doi.org/10.4161/nucl.28488.
Rex, T. S., Peet, J. A., Surace, E. M., Calvert, P. D., Nikonov, S. S., Lyubarsky, A. L., Bendo, E., Hughes, T., Pugh, E. N., Jr., and Bennett, J. (2005) The distribution, concentration, and toxicity of enhanced green fluorescent protein in retinal cells after genomic or somatic (virus-mediated) gene transfer, Mol. Vis., 11, 1236-1245.
Fink, D., Wohrer, S., Pfeffer, M., Tombe, T., Ong, C. J., and Sorensen, P. H. (2010) Ubiquitous expression of the monomeric red fluorescent protein mCherry in transgenic mice, Genesis, 48, 723-729, https://doi.org/10.1002/dvg.20677.
Tamura, K., Yamada, K., Shimada, T., and Hara-Nishimura, I. (2004) Endoplasmic reticulum-resident proteins are constitutively transported to vacuoles for degradation, Plant J., 39, 393-402, https://doi.org/10.1111/j.1365-313X.2004.02141.x.
Liu, M., Hodish, I., Rhodes, C. J., and Arvan, P. (2007) Proinsulin maturation, misfolding, and proteotoxicity, Proc. Natl. Acad. Sci. USA, 104, 15841-15846, https://doi.org/10.1073/pnas.0702697104.
Tortorella, L. L., Pipalia, N. H., Mukherjee, S., Pastan, I., Fitzgerald, D., and Maxfield, F. R. (2012) Efficiency of immunotoxin cytotoxicity is modulated by the intracellular itinerary, PLoS One, 7, e47320, https://doi.org/10.1371/journal.pone.0047320.
Lam, A. J., St-Pierre, F., Gong, Y., Marshall, J. D., Cranfill, P. J., Baird, M. A., McKeown, M. R., Wiedenmann, J., Davidson, M. W., Schnitzer, M. J., et al. (2012) Improving FRET dynamic range with bright green and red fluorescent proteins, Nat. Methods, 9, 1005-1012, https://doi.org/10.1038/nmeth.2171.
Bajar, B. T., Wang, E. S., Lam, A. J., Kim, B. B., Jacobs, C. L., Howe, E. S., Davidson, M. W., Lin, M. Z., and Chu, J. (2016) Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting, Sci. Rep., 6, 20889, https://doi.org/10.1038/srep20889.
Grimm, J. B., English, B. P., Chen, J., Slaughter, J. P., Zhang, Z., Revyakin, A., Patel, R., Macklin, J. J., Normanno, D., Singer, R. H., et al. (2015) A general method to improve fluorophores for live-cell and single-molecule microscopy, Nat. Methods, 12, 244-250, https://doi.org/10.1038/nmeth.3256.
Maloshenok, L. G., Abushinova, G. A., Bogdanov, A. A., and Zherdeva, V. V. (2023) Tet-regulated expression and optical clearing for in vivo visualization of genetically encoded chimeric dCas9/fluorescent protein probes, Materials, 16, 940, https://doi.org/10.3390/ma16030940.
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage, Nature, 533, 420-424, https://doi.org/10.1038/nature17946.
Gao, X. D., Rodríguez, T. C., and Sontheimer, E. J. (2019) Adapting dCas9-APEX2 for subnuclear proteomic profiling, Methods Enzymol., 616, 365-383, https://doi.org/10.1016/bs.mie.2018.10.030.
Savić, N., Ringnalda, F. C., Berk, C., Bargsten, K., Hall, J., Jinek, M., and Schwank, G. (2019) In vitro generation of CRISPR-Cas9 complexes with covalently bound repair templates for genome editing in mammalian cells, Bio Protoc., 9, e3136, https://doi.org/10.21769/BioProtoc.3136.
Mao, S., Ying, Y., Wu, X., Krueger, C. J., and Chen, A. K. (2019) CRISPR/dual-FRET molecular beacon for sensitive live-cell imaging of non-repetitive genomic loci, Nucleic Acids Res., 47, e131, https://doi.org/10.1093/nar/gkz752.
Wu, X., Mao, S., Yang, Y., Rushdi, M. N., Krueger, C. J., and Chen, A. K. (2018) A CRISPR/molecular beacon hybrid system for live-cell genomic imaging, Nucleic Acids Res., 46, e80, https://doi.org/10.1093/nar/gky304.
Baugh, C., Grate, D., and Wilson, C. (2000) 2.8 A crystal structure of the malachite green aptamer, J. Mol. Biol., 301, 117-128, https://doi.org/10.1006/jmbi.2000.3951.
Stojanovic, M. N., and Kolpashchikov, D. M. (2004) Modular aptameric sensors, J. Am. Chem. Soc., 126, 9266-9270, https://doi.org/10.1021/ja032013t.
Filonov, G. S., Moon, J. D., Svensen, N., and Jaffrey, S. R. (2014) Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution, J. Am. Chem. Soc., 136, 16299-16308, https://doi.org/10.1021/ja508478x.
Panchapakesan, S. S., Jeng, S. C., and Unrau, P. J. (2015) RNA complex purification using high-affinity fluorescent RNA aptamer tags, Ann. N. Y. Acad. Sci., 1341, 149-155, https://doi.org/10.1111/nyas.12663.
Trachman, R. J., 3rd, Cojocaru, R., Wu, D., Piszczek, G., Ryckelynck, M., Unrau, P. J., and Ferré-D’Amaré, A. R. (2020) Structure-guided engineering of the homodimeric Mango-IV fluorescence turn-on aptamer yields an RNA FRET pair, Structure, 28, 776-785.e3, https://doi.org/10.1016/j.str.2020.04.007.
Holzhauser, C., Rubner, M. M., and Wagenknecht, H. A. (2013) Energy-transfer-based wavelength-shifting DNA probes with “clickable” cyanine dyes, Photochem. Photobiol. Sci., 12, 722-724, https://doi.org/10.1039/c2pp25366f.
Panchapakesan, S., Ferguson, M. L., Hayden, E. J., Chen, X., Hoskins, A. A., Unrau, P. J. (2017) Ribonucleoprotein purification and characterization using RNA Mango, RNA, 23, 1592-1599, https://doi.org/10.1261/rna.062166.117.
Abdolahzadeh, A., Dolgosheina, E. V., and Unrau, P. J. (2019) RNA detection with high specificity and sensitivity using nested fluorogenic Mango NASBA, RNA, 25, 1806-1813, https://doi.org/10.1261/rna.072629.119.
Acknowledgments
The authors are grateful to Professor A. A. Bogdanov, Jr. (UMass Chan Medical School, Worcester) for his valuable advice and critical comments when preparing the review.
Funding
This work was supported by the Russian Science Foundation (project no. 22-14-00205, https://rscf.ru/project/22-14-00205/ [in Russian]).
Author information
Authors and Affiliations
Contributions
G.A., L.M., and S.B. wrote “Modern Approaches to Studying the 3D Genome Structure in vitro and in vivo” and Delivery of the CRISPR–Cas System Components to the Target cells” sections. A.R. wrote “The CRISPR–Cas SYSTEMS: History of Discovery, Classification, and Application” section. V.Zh. wrote Introduction, “Molecular Imaging of Nucleome Using CRISPR–Cas9. Requirements for in vivo Visualization”, Conclusion sections. Concept and finalize by Zh.V., L.G., A.R. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
The authors declare no conflicts of interest. This article does not contain description of studies with the involvement of humans or animal subjects performed by any of the authors.
Additional information
Translated from Uspekhi Biologicheskoi Khimii, 2023, Vol. 63, pp. 245-300.
Rights and permissions
About this article

Cite this article
Maloshenok, L.G., Abushinova, G.A., Ryazanova, A.Y. et al. Visualizing the Nucleome Using the CRISPR–Cas9 System: From in vitro to in vivo. Biochemistry Moscow 88 (Suppl 1), S123–S149 (2023). https://doi.org/10.1134/S0006297923140080
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297923140080