





Abstract
The cystatin CRES (cystatin-related epididymal spermatogenic; Cst8) is the defining member of a reproductive subgroup of family 2 cystatins of cysteine protease inhibitors and is present in the epididymis and spermatozoa, suggesting roles in sperm maturation and fertilization. To elucidate the role of CRES in reproduction, mice lacking the Cst8 gene were generated and their fertility examined. Although both male and female Cst8−/− mice generated offspring in vivo, spermatozoa from Cst8−/− mice exhibited a profound fertility defect in vitro. Compared to spermatozoa from Cst8+/+ mice, spermatozoa from Cst8−/− mice were unable to undergo a progesterone-stimulated acrosome reaction and had decreased levels of protein tyrosine phosphorylation, suggesting a defect in the ability of Cst8−/− spermatozoa to capacitate. Incubation of Cst8−/− spermatozoa with dibutyryl cAMP and 3-isobutyl-1-methylxanthine rescued the fertility defect, including the capacity for sperm protein tyrosine phosphorylation. Both untreated Cst8+/+ and Cst8−/− spermatozoa, however, exhibited similar increased total levels of cAMP and protein kinase A (PKA) activity throughout the capacitation time course compared to spermatozoa incubated under noncapacitating conditions. Taken together, these results suggest that mice lacking CRES may have altered local levels of cAMP/PKA activity, perhaps because of improper partitioning or tethering of these signaling molecules, or that the CRES defect does not directly involve cAMP/PKA but other signaling pathways that regulate protein tyrosine phosphorylation and capacitation.
Introduction
The cystatins are a superfamily of cysteine protease inhibitors consisting of the intracellular stefins (family 1) and the extracellular cystatins (family 2) and kininogens (family 3). The cystatin CRES (cystatin-related epididymal spermatogenic; Cst8) is a secreted family 2 cystatin with unique properties and, thus, defines a new subgroup [1, 2]. In particular, the CRES subgroup consists of eight members that are distinct from the typical family 2 cystatins, such as the ubiquitously expressed cystatin C, by their lack of consensus sites for cysteine protease inhibition and reproductive-specific expression. Specifically, CRES is synthesized and secreted by the proximal region of the mouse caput epididymidis and is present in testicular germ cells and mature spermatozoa, suggesting roles in sperm maturation and fertilization [1, 3]. In vitro studies have demonstrated that CRES, unlike cystatin C, does not inhibit cysteine proteases but inhibits a serine protease, prohormone convertase 2, suggesting a role in the regulation of proprotein processing [4].
Cystatins are presumed to function in vivo as mediators of proteolytic activity, but their biological roles are unknown. However, roles in inflammation, prohormone processing, bone resorption, and autocrine/paracrine functions have been proposed. [5–7]. The cystatin C gene knockout mouse has an unremarkable phenotype with no obvious deficiencies other than mild lethargy, suggesting some redundancy among family 2 members [8]. More profound phenotypes have been observed in other cystatin gene knockout models, including altered skin cornification in mice lacking the cystatin M/E gene [9] and myoclonic epilepsy in mice lacking the cystatin B gene [10]. Several studies of cystatin C have focused on its role in neurodegenerative diseases, because cystatin C is present in the amyloid β plaques associated with Alzheimer disease [11], and both in vitro and in vivo studies have demonstrated that cystatin C inhibits amyloid β fibril formation and deposition, suggesting a protective role [12, 13]. Cystatin C and other cystatins are themselves amyloidogenic and form aggregates in vitro and in vivo [14]. A mutant L68Q form of human cystatin C, which is highly aggregation prone, causes hereditary cystatin C amyloid angiopathy (Icelandic type), in which patients die in their early 30s as a result of cerebral hemorrhage [15].
To gain insight regarding the biological role of CRES in reproduction, mutant mice lacking the CRES gene (Cst8−/−) were produced and their fertility examined. Although Cst8−/− mice generated offspring and, thus, exhibited normal fertility, in vitro spermatozoa exhibited a profound defect in their ability to fertilize. The reduced fertility in the Cst8−/− mice was rescued by exposure of spermatozoa before fertilization to dibutyryl cAMP (dbcAMP) and the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX). Our observation that components which increase the intracellular levels of cAMP can circumvent the CRES fertility defect suggests that CRES may participate in cAMP-mediated signaling events critical for fertilization.
Materials and Methods
Media
All reagents used were from Sigma-Aldrich. Unless otherwise indicated, the medium used for all experiments was Krebs-Ringer bicarbonate medium (KRB; capacitating medium) containing 119.4 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO, 1.2 mM MgSO4, 25 mM sodium lactate, 1 mM sodium pyruvate, 5.6 mM glucose, 25.1 mM NaHCO3, 28 μM phenol red, 1.7 mM CaCl2, and 0.3% bovine serum albumin (BSA; embryo tested, A3311; Sigma). KRB with Hepes (KRB-Hepes) also contained 21 mM Hepes, and NaHCO3 was reduced to 4 mM. Noncapacitating medium was KRB except that an equal amount of NaCl was substituted for NaHCO3 and 0.3% polyvinyl alcohol (PVA) was added in place of BSA. Incomplete media (without CaCl2 and BSA/PVA) were stored in aliquots at −80°C. For an experiment, incomplete KRB was preequilibrated overnight at 37°C in a humidified water-jacketed incubator under 5% CO2. Then, CaCl2 and BSA/PVA were added, the pH adjusted to 7.4, and the medium sterile-filtered and placed in the CO2 incubator with the cap loose to allow gas exchange.
Animals
CD1 and C57BL/6 male and female mice were purchased from Charles River Laboratories. The Cst8 129SvEv/B6 gene knockout and wild-type mice were bred in house. Mice were maintained under a constant 12L:12D photoperiod with food and water ad libitum. All animal studies were conducted in accordance with the NIH Guidelines for the Care and Use of Experimental Animals.
Generation of CRES Gene Knockout Mice
The CRES (Cst8) gene knockout mice were generated by inGenious Targeting Laboratories, Inc. Briefly, a 14-kb mouse genomic DNA fragment was cloned from the mouse 129/SvEv lambda genomic library. This genomic fragment contained exons 1 and 2 of the mouse Cst8 gene. The targeting vector was constructed using a 13-kb DNA fragment from NotI to PspOMI as the subclone. The neo cassette replaced 4 kb of the genomic fragment, including part of exon 1 (untranslated region [UTR]) and all of exon 2. The targeting vector was confirmed by restriction analysis after each modification step and by sequencing using primers designed to read from the selection cassette into the short arm and the long arm. Ten micrograms of the targeting vector were linearized by NotI and then transfected by electroporation of iTL1 (129/SvEv) embryonic stem cells. After selection in G418 antibiotic, 200 surviving colonies were expanded for PCR analysis to identify recombinant clones. The correctly targeted embryonic stem cell lines were microinjected into C57BL/6J (B6) blastocysts and implanted into pseudopregnant mice. The resulting chimeric mice were mated to B6 mice to generate germline transmission. Founder male and female heterozygous F1Cst8 mutant mice were shipped to the Texas Tech University Health Sciences Center and bred to B6 mice for two generations to establish the colony and generate a sufficient number of mice for matings before F3 heterozygous mice were intercrossed to produce Cst8−/− homozygous mice.
Genomic DNA from mouse tail snips was isolated and mice were genotyped by PCR using the XNAT kit (Sigma Chemical Co.) and three primers: SA5 (5′-AAGGGAAAGATGGTTCCCAGAACC-3′) and WT2 (5′-GGAAGTGCAGCC AGGCATTCCAGA-3′) to identify the wild-type allele and SA5 and N1 (5′-TGCGAGGCCAGAGGCCACTTGTGTAGC-3′) to identify the targeted allele. PCR conditions were 94° for 3 min, 94° for 30 sec, 62.5° for 30 sec, and 68° for 2 min for 35 cycles, followed by 72° for 7 min. The primer pair to detect the wild-type allele generated a PCR product of 2.2 kb, whereas the primer pair to detect the mutant allele generated a PCR product of 1.55 kb. PCR products were initially confirmed by sequencing.
In Vivo Breeding
The Cst8+/+ or Cst8−/− mice (age, 12–20 wk) were paired with C57BL/6 mice (age, 12 wk) of the opposite sex in a 1:1 ratio at 1700 h. The females were checked every day at 0900 h for a copulation plug as evidence of mating. When a plug was observed, the female was separated from the male into a different cage and euthanized at Day 14 of gestation to count fetuses and implantation sites as a measure of litter size. In some experiments, Cst8+/+ or Cst8−/− mice were housed with littermates of the opposite gender and females checked for copulatory plugs. Litter size was determined by counting both live and dead pups on the day of delivery or the following day.
In Vitro Fertilization
The CD1 female mice (age, 4–8 wk) were induced to superovulate using i.p. injections of 8 IU of equine chronic gonadotropin (Sigma) followed 48 h later by 8 IU of human chorionic gonadotropin (hCG; Sigma). Thirteen hours after the hCG injection, cumulus-oocyte complexes (COCs) were dissected from the oviducts and washed in KRB-Hepes twice and in KRB once. Washing was carried out by transferring the COC by pipette to watch glasses containing the appropriate buffer. One or two COCs (containing 20–25 oocytes) were then transferred to 45-μl drops of KRB in 35-mm Falcon dishes under mineral oil. Cauda epididymides were excised from age-matched Cst8+/+ and Cst8−/− mice into KRB and punctured to allow spermatozoa to disperse. Cauda epididymides were removed, and spermatozoa were capacitated (∼20 × 106 spermatozoa/ml) in KRB for 1.5 h at 37°C in a humidified water-jacketed incubator under 5% CO2. Spermatozoa from retired breeder CD1 males were also included with each experiment as a control. COCs were inseminated with capacitated sperm (5000 sperm in 5 μl [1 × 105 spermatozoa/ml]) and incubated for 3 h. Following fertilization, cumulus cells were removed from the oocytes with hyaluronidase (final concentration, 2 mg/ml; Sigma), and oocytes were washed in KRB-Hepes and then fixed in 5% formalin for 30 min. Next, oocytes were washed in Dulbecco PBS with 0.1% BSA and 0.01% Tween 20, stained with Hoechst 33342 (5 μg/ml) for 20 min, and washed again with Dulbecco PBS with 0.1% BSA and 0.01% Tween 20. Oocytes were mounted on slides and viewed under ultraviolet (UV) light with an Olympus BX60 microscope equipped with epifluorescence. Oocytes were considered to be fertilized by the presence of either two pronuclei or a decondensing sperm head.
For some experiments, 1 mM dbcAMP or 1 mM dbcAMP and 100 μM IBMX were included in the sperm suspension during capacitation. Control spermatozoa received an equivalent volume of the vehicle dimethyl sulfoxide (DMSO) during capacitation. After 90 min of capacitation, the dbcAMP and IBMX were removed from the sperm suspension by serially diluting the spermatozoa 1:1 followed by a 1:10 dilution in capacitating medium that lacked the cAMP components. COCs were inseminated with 1 × 105 spermatozoa/ml and incubated for 3 h. To control for possible direct effects of the small amount of remaining dbcAMP and IBMX on the oocytes, an equivalent volume of dbcAMP and IBMX in medium but without spermatozoa was serially diluted in the same manner as performed for spermatozoa, and this volume was added to the control spermatozoa at the time of addition to the COC. Following fertilization, the cumulus cells were removed and the oocytes washed, fixed, and stained as described above.
Sperm-Zona Pellucida Binding
The CD1 female mice (age, 4–8 wk) were induced to superovulate and COCs isolated as described above. Cumulus cells were removed by incubation in medium containing hyaluronidase (final concentration, 2 mg/ml) and trypsin inhibitor (final concentration, 0.2 mg/ml; Sigma), and oocytes were washed in KRB-Hepes once and KRB twice. Approximately 20 cumulus-free oocytes each were transferred to 45-μl drops of KRB in 35-mm Falcon dishes under mineral oil. Two-cell embryos (generated by standard in vitro fertilization the previous day but allowed to fertilize overnight) were also used as a negative control. Spermatozoa from Cst8+/+ and Cst8−/− mice were isolated and capacitated as described previously and then added to oocytes and embryos (1 × 105 spermatozoa/ml) and incubated for 30 min. Oocytes and embryos were then washed three times in KRB-Hepes using a fire-polished pulled glass pipette to remove loosely bound sperm. Additional washes were performed on all treatment groups if many spermatozoa were still bound to the embryos. Oocytes and embryos were fixed in 5% formalin for 30 min, washed with KRB-Hepes, and mounted on slides. Oocytes and embryos were viewed with an Olympus BX50 microscope, and all bound sperm were counted.
Sperm-Oocyte Fusion
CD1 female mice (age, 4–8 wk) were induced to superovulate and COCs isolated as described previously. COCs were treated with hyaluronidase (final concentration, 2 mg/ml) to remove cumulus cells and were washed once in KRB-Hepes. Oocytes were then treated with acidic Tyrode solution (Sigma) for approximately 15–30 sec to remove the zona pellucida. Oocytes were washed in KRB-Hepes before preloading with 1 μg/ml of Hoechst 33342 for 5 min. Oocytes were subjected to four successive transfers into KRB in watch glasses and six 10-min washes in KRB before being transferred to 45-μl drops of KRB in 35-mm Falcon dishes under mineral oil. Spermatozoa from Cst8+/+ and Cst8−/− mice were isolated and capacitated as described previously. Spermatozoa from CD1 males were also included with each experiment as a control. Oocytes were inseminated with capacitated sperm (1 × 105 spermatozoa/ml) and incubated for 30 min. Following fertilization, oocytes were washed in KRB-Hepes twice and then fixed in 5% formalin for 10 min. Next, the oocytes were washed again in KRB-Hepes, mounted on slides, and viewed under UV light with an Olympus BX60 microscope. Oocytes were considered to be fertilized by the presence of a decondensing sperm head within the ooplasm.
Sperm Motility Analysis
Motility of Cst8+/+ and Cst8−/− spermatozoa capacitated in KRB for 30, 60, 90, and 120 min was assessed using a computer-assisted sperm analysis (CASA; HTM-CEROS Version 12; Hamilton Thorne Research). Sperm concentrations were adjusted to 5–10 × 106 spermatozoa/ml, and 12 μl of sample were loaded on a prewarmed counting chamber and the samples immediately analyzed. Ten fields were examined on duplicate slides for each time point for the following parameters: percentage motile, percentage progressively motile, smoothed path velocity (VAP), track velocity (VCL), straight line velocity (VSL), amplitude of lateral head displacement, beat cross frequency, linearity (VSL:VCL ratio), straightness (VSL:VAP ratio).
Induction of the Sperm Acrosome Reaction
Cauda spermatozoa from Cst8+/+ and Cst8−/− mice were dispersed into KRB lacking CaCl2 and sperm concentrations determined. Next, 1.5 × 106 spermatozoa were aliquoted into 1.5-ml tubes (final concentration, 15 × 106 sperm/ml) and CaCl2 added (final concentration, 1.7 mM) to start capacitation. Capacitation was carried out with the tubes uncapped for 1.5 h at 37°C in a humidified water-jacketed incubator under 5% CO2. At various times, A23187 (final concentration, 10 μM; Sigma) or vehicle DMSO was added and incubations continued for another 10 min. In other experiments, progesterone was used to induce the acrosome reaction. In these studies, 2 × 105 sperm/tube (final concentration, 2 × 106 sperm/ml) were aliquoted and capacitation initiated by the addition of CaCl2 as described. At the various times, progesterone (final concentration, 15 μM; Sigma) or the vehicle DMSO was added and incubation continued for 20 min. For both A23187 and progesterone studies, the experiments were stopped by adding an equal amount of 2× fixative solution (10% formaldehyde in PBS), and 6 × 104 sperm were spread onto duplicate slides to air dry. Slides were stained with 0.22% Coomassie Blue G-250 (Bio-Rad) for 5 min, washed twice with ddH2O, and mounted with coverslips and Fluoromount-G (SouthernBiotech) following a protocol modified from that described by Larson and Miller [16]. Slides were viewed using an Olympus BX50 microscope, and 200 sperm were analyzed per slide for the presence (blue staining) or absence (no staining) of the sperm acrosome.
Protein Tyrosine Phosphorylation
Cauda sperm from Cst8+/+ and Cst8−/− mice were dispersed into KRB medium lacking CaCl2. Sperm were centrifuged at 4000 × g for 5 min and resuspended in 1 ml of KRB medium lacking CaCl2 and sperm concentrations determined. Next, 1.5 × 106 spermatozoa were aliquoted into 1.5-ml tubes (final concentration, 15 × 106 sperm/ml) and CaCl2 added (final concentration, 1.7 mM) to start capacitation. Capacitation was carried out with the tubes uncapped for 1.5 h at 37°C in a humidified water-jacketed incubator under 5% CO2. At 0, 30, 60, 90, and 120 min of capacitation, aliquots were removed from the incubator and placed on ice, and an equal volume of Dulbecco PBS with 4× phosphatase inhibitors was added (1× concentrations: 80 mM β-glycerophosphate, 10 mM sodium fluoride, and 1 mM sodium vanadate [Sigma]). Samples were centrifuged at 6000 × g for 5 min at 4°C. Most of the supernatant fraction was discarded, and an equal volume of 2× Laemmli buffer containing 8% β-mercaptoethanol was added to the remaining pellet for Western blot analysis. In some experiments, immediately after the addition of CaCl2 to initiate capacitation, 1 mM dbcAMP and 100 μM IBMX were added to the sperm samples, whereas control samples received an equivalent volume of DMSO. At various times of capacitation, aliquots were removed and processed as described above for Western blot analysis.
To examine the protein tyrosine phosphorylation on capacitated spermatozoa by immunofluorescence, a procedure modified from that described by Asquith et al. [17] was followed. Briefly, aliquots were removed at 0 or 90 min of capacitation, and an equal volume of Dulbecco PBS containing 4× phosphatase inhibitors was added and samples centrifuged at 500 × g for 5 min to pellet the spermatozoa. The sperm pellet was resuspended in Dulbecco PBS containing phosphatase inhibitors and paraformaldehyde (EMS Sciences) added to a 2% final concentration. The samples were fixed for 30 min at room temperature, and the sperm pellet was washed in PBS and then spread on Superfrost Plus microscope slides (Thermo Scientific) and allowed to air dry. Spermatozoa were permeabilized by incubation in 0.2% Triton X-100/PBS for 20 min at room temperature, rinsed four times with PBS, and blocked in 1% BSA (fatty acid-free) in PBS for 1 h at room temperature in a humidified chamber. The samples were washed four times with PBS and then incubated with an anti-phosphotyrosine antibody (clone 4G10; Millipore) at 1:100 dilution in 0.1% BSA/PBS overnight at 4°C in a humidified chamber. The slides were washed four times in PBS and incubated with a goat anti-mouse Alexa Fluor 594 secondary antibody (Invitrogen) diluted 1:100 in 0.1% BSA/PBS for 1 h at room temperature in the dark. Control slides were incubated with secondary antibody alone. The slides were washed six times in PBS and coverslips mounted with Fluoromount G. The slides were examined using an Olympus BX50 microscope equipped with epifluorescence with excitation at 545–580 nm and emission at greater than 610 nm. To determine the different phosphotyrosine fluorescence patterns, 100–300 spermatozoa/slide were counted and percentages determined. ANOVA followed by Newman-Keuls or Bonferroni posttest was used to determine statistical significance.
Cyclic AMP Assay
The Cst8+/+ and Cst8−/− spermatozoa were dispersed in noncapacitating KRB and concentrations adjusted to 10 × 106 spermatozoa/ml with either capacitating KRB or fresh noncapacitating KRB, both supplemented with 100 μM IBMX to prevent cAMP turnover. Next, 3 × 106 spermatozoa of each genotype were aliquoted into 1.5-ml tubes and incubated for 0, 30, 60, 90, or 120 min. The sperm in capacitating KRB were incubated at 37°C in a humidified water-jacketed incubator under 5% CO2 with open caps. The sperm in noncapacitating KRB were incubated at 37°C with closed caps. At each 30-min interval, samples were removed and centrifuged at 8000 rpm for 5 min at room temperature to pellet spermatozoa. The supernatant was discarded, and 300 μl of 0.1 M HCl were added to the sperm pellet and the sample vortexed for 2 sec and then incubated at room temperature for 20 min to allow the extraction of intracellular cAMP. The sperm sample was then centrifuged at 10 000 × g for 5 min at room temperature and the supernatant collected and stored at −80°C. Cyclic AMP was quantified in extracts isolated from 1 × 106 spermatozoa using the acetylation protocol from the Format A Cyclic AMP “Plus” Enzyme Immunoassay Kit (BIOMOL International). A standard curve was run for each assay, and unknown concentrations of cAMP were determined from these values using the Gen 5 software and the Synergy HT plate reader (BioTek Instruments).
Protein Kinase A Activity Assay
Protein kinase A (PKA) activity was compared between Cst8+/+ and Cst8−/− spermatozoa during capacitation using a method described by Visconti et al. [18] and the SignaTECT PKA Assay kit (Promega). Cauda spermatozoa were dispersed into noncapacitating KRB, counted, and then adjusted to a concentration of 10 × 106 sperm/ml in noncapacitating KRB or capacitating KRB. At each time interval (0, 30, 60, 90, 120, and 180 min) of capacitation, an aliquot of spermatozoa was removed and diluted 1:1 with medium, and 10 μl of diluted spermatozoa (0.5 × 105 sperm) were incubated with 12.28 μl of 2× assay cocktail (1× concentrations: 100 μM biotinylated Kemptide substrate, 100 μM ATP, 0.05 μl of γ32ATP [3000 Ci/mmol, 10 μCi/μl; PerkinElmer], 1% Triton X-100, 80 mM β-glycerophosphate, 10 mM NaF [Sigma], 20 mM Tris, 10 mM MgCl2, 0.05 mg/ml of BSA, and 0.03 μl of protease inhibitor cocktail [Sigma]) and 0.44 μl of DMSO vehicle or 0.22 μl 100 mM dbcAMP and 22 μl of 10 mM IBMX (final concentrations, 1 mM dbcAMP and 100 μM IBMX). The Kemptide, cold ATP, Tris, MgCl2, and BSA were from the SignaTECT PKA Assay kit. The reactions were incubated at 37°C for 15 min and were carried out in triplicate tubes. It was experimentally predetermined that the assay was linear at 15 min by comparing the results from increasing numbers of sperm cells. The blank reaction contained all reagents except spermatozoa. The addition of dbcAMP/IBMX in the reactions served as a positive control for PKA activation, whereas reactions that were supplemented with 10 μM H-89 (Sigma), a PKA inhibitor, served as a negative control.
Reactions were stopped by addition of 11.2 μl of 7.5 M guanidine-HCl (SignaTECT kit), and 10 μl from the mixtures were spotted onto a SAM Biotin Capture Membrane (SignaTECT kit). The membrane was washed with 2 M NaCl once for 1 min, 2 M NaCl three times for 4 min each, 2 M NaCl and 1% H3PO4 four times for 4 min each, and ddH2O two times for 1 min each. The membrane was allowed to dry for 30 min, and then each sample spot was separated by cutting, placed in scintillation vials with 5 ml of Scintiverse (Thermo Scientific), and analyzed by scintillation counting. Three additional, randomly chosen samples were spotted onto the membrane but not washed to calculate total specific activity. The specific activity was calculated by taking the average counts per minute (CPM) from the three random unwashed samples × 3.9 μl (total reaction volume divided by 10-μl volume, which was spotted and counted)/2000 pmol (amount of ATP in the reaction) = CPM/pmol. The PKA activity was then calculated by taking the average of the experimental CPM − blank CPM × 33.9 μl (reaction volume)/10 μl (volume counted) × 15 min × (0.05 × 106 spermatozoa) × specific activity = pmol ATP/min × 106 spermatozoa.
Western Blot Analysis
For protein tyrosine phosphorylation studies, samples were sonicated, and proteins from 1 × 106 spermatozoa were heated at 95°C for 5 min and separated by 10% Criterion Tris-glycine SDS-PAGE (precast gels; Bio-Rad). Separated proteins were transferred to Immobilon-P (Millipore) for 1.5 h at 100 V. Membranes were blocked with 5% milk in TBS with 0.1% Tween 20 (TBST) for 1 h at room temperature, followed by incubation with a mouse anti-phosphotyrosine antibody (clone 4G10; Millipore) at 1:10 000 in TBST overnight at 4°C. Blots were then washed with TBST three times for 10 min each and incubated for 2 h at room temperature with a goat anti-mouse horseradish peroxidase (HRP) secondary antibody (1:30 000) in 3% milk in TBST. Blots were washed with TBST three more times for 10 min each and once with TBS before developing with SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific). To confirm equal loading, the blots were stripped using the Restore Plus Stripping Buffer (ThermoScientific) and incubated with an antibody to α-tubulin (clone B-5-1-2; Sigma) at 1:80 000. Membranes were also stained with Coomassie Blue to examine protein loading. The 116-kDa protein band (also known as p105) corresponds to a constitutively phosphorylated hexokinase and also served as an internal loading control [17].
For the analysis of CRES protein in samples from Cst8+/+, Cst8+/−, and Cst8−/− mice, whole testes and epididymides in antibody lysis buffer containing 20 mM Tris (pH 7.4), 50 mM NaCl, 0.5% NP-40, 0.5% deoxycholate, 0.5% SDS, 1 mM ethylenediaminetetra-acetic acid, and 10 μg/ml of aprotinin were homogenized using a Polytron, centrifuged at 15 000 × g to remove insoluble material, and the protein quantitated by the BCA assay (ThermoScientific). Equal microgram amounts of protein were separated on precast 15% Criterion Tris-glycine gels (Bio-Rad) and proteins transferred to Immobilon-P as described above. The membranes were incubated in 3% milk in TBST for 1 h at room temperature, followed by incubation with an affinity-purified rabbit anti-mouse CRES antibody (1:4000) in milk buffer overnight at 4°C. The membranes were washed in TBST followed by incubation with a goat anti-rabbit HRP secondary antibody (1:40 000; Invitrogen) in milk buffer for 2 h at room temperature. The membranes were washed extensively in TBST and then incubated with SuperSignal West Pico chemiluminescent substrate for 5 min followed by exposure to film.
Northern Blot Analysis
Total RNA was isolated from the testes and epididymides from Cst8+/+, Cst8+/−, and Cst8−/− mice using TRIzol reagent (Invitrogen) following the manufacturer's protocol. Equal microgram amounts of RNA were separated on a 1% agarose gel in borate buffer as described previously [1]. A Cst8 cDNA probe was generated from purified cDNA insert using a random prime labeling method (Prime-It II; Agilent Technologies). After hybridization, the blots were washed twice in 1× SSC (0.15 M sodium chloride and 0.015 M sodium citrate) and 0.1% SDS at room temperature for 15 min and then twice at 65°C for 15 min before exposure to film.
Statistical Analysis
Statistical analysis of the fertilization experiments was performed using one-way ANOVA followed by Bonferroni and Tukey post hoc analysis, which yielded similar P-values. All experiments were included in the analysis except those in which control CD-1 spermatozoa did not achieve a fertilization rate of 50% or higher. Motility studies were analyzed using a t-test. Statistical analyses of the acrosome reaction studies, cAMP, and PKA assays were performed with two-way ANOVA followed by Bonferroni post test.
Results
Generation of Cst8 Mutant Mice
The biological role of CRES in reproduction was examined by generating CRES gene knockout mice by homologous recombination in embryonic stem cells using the targeting vector shown in Figure 1A. The replacement of CRES exons 1 and 2 with the neomycin-resistance gene neo resulted in the loss of 5′ UTR sequences (exon 1) and the initiator methionine and downstream coding sequences in exon 2. The genotypes for the wild-type (Cst8+/+), heterozygous (Cst8+/−) and homozygous (Cst8−/−) mice for the mutation were identified by PCR analysis of genomic DNA (Fig. 1B). DNA from the wild-type mice generated a single PCR product of 2.2 kb, whereas DNA from the homozygous null mice generated a single PCR product of 1.5 kb. The heterozygous mice contained both the wild-type and mutant alleles and, thus, generated both PCR products. Analysis of CRES mRNA in the testis and epididymis from Cst8+/+, Cst8+/−, and Cst8−/− mice showed the expected 700-bp CRES mRNA in the Cst8+/+ mice, a decrease in the amount of CRES mRNA in the Cst8+/− mice relative to that in the Cst8+/+ mice, and a loss of the CRES mRNA from the Cst8−/− mice (Fig. 1C). Further examination of CRES protein in the testis and epididymis by Western blot analysis showed that the 14-kDa and N-glycosylated 19- and 23-kDa CRES proteins were absent from the Cst8−/− mice, with reduced levels of the protein in the Cst8+/− tissues (Fig. 1D). These data show the absence of CRES mRNA and protein from the tissues of the Cst8−/− mice and validate the mouse model for further study.
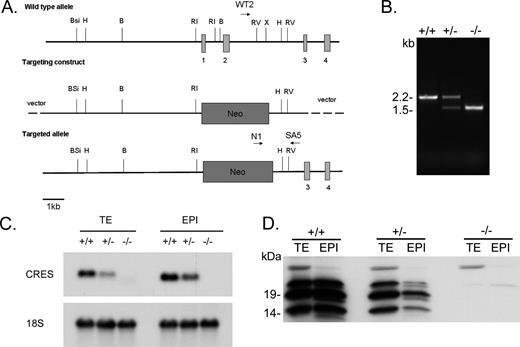
Production of mice lacking Cst8.A) Targeting construct. The first two Cst8 exons (vertical boxes) were replaced by neo. Restriction enzyme sites: B, BamHI; Bsi, BsiWI; H, HindIII; RI, EcoRI; RV, EcoRV; X, XhoI. WT2, N1, and SA5 are primers used for PCR genotyping of the WT (WT2 and SA5) and KO (N1 and SA5) alleles. B) PCR analysis of genomic DNA isolated from tail snips from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mice for the Cst8 mutation. C) Northern blot analysis of CRES mRNA levels in testis (TE) and epididymal (EPI) tissue isolated from Cst8+/+, Cst8+/−, and Cst8−/− mice and probed with a radiolabeled CRES cDNA. The blot was stripped and reprobed with a cDNA generated against 18S rRNA to confirm equal loading. D) Western blot analysis of CRES protein in tissue lysates prepared from testis (TE) and epididymis (EPI) from Cst8+/+, Cst8+/−, and Cst8−/− mice.
Breeding of the Cst8+/− male and female mice resulted in Cst8+/+, Cst8+/−, and Cst8−/− mice at a 1:1.95:0.83 ratio (155 pups from 17 litters), consistent with a mendelian distribution of the Cst8 allele. From the same litters, 58% of the pups were male, and 42% of the pups were female. Both Cst8−/− males and females exhibited normal behavior and body size and appeared to be healthy.
To examine in more detail the effects of the CRES mutation on the male reproductive tract, testis and epididymal weights from Cst8+/+ and Cst8−/− mice of two different age groups were determined and normalized to body weights. As shown in Table 1, the Cst8−/− mice aged 12–16 wk exhibited decreased epididymal weights when normalized to mouse body weight and compared to that of Cst8+/+ mice. However, in the 17- to 21-wk age group, this difference was no longer statistically significant. In contrast, the normalized testis weights in the Cst8−/− exhibited the opposite effect. No difference was found in testis weights from those in the Cst8+/+ mice at 12–16 wk of age, but by 17–21 wk, the normalized testis weights were significantly decreased in the Cst8−/− mice. In both age groups, the number of spermatozoa in the cauda epididymidis was decreased in the Cst8−/− mice compared to the Cst8+/+ mice. Because these results suggest defects in both the testis and epididymis that are not readily apparent on gross examination, an ultrastructural analysis of the Cst8+/+ and Cst8−/− testis and epididymis at both the light-microscopic and electron-microscopic levels is currently being performed. However, preliminary examination suggests that in some Cst8−/− seminiferous tubules, disrupted germ cell-Sertoli cell interactions result in premature sloughing of germ cells, which in turn would result in a slightly reduced number of cauda epididymal spermatozoa (see Note Added in Proof).
Cst8 mouse tissue weights.
| Age and mouse type | Body weight (g) | Testis weight (g) | Testis/body ratio | Epididymis weight (g) | Epididymis/body ratio | Cauda epididymal sperm concentration (x106) |
|---|---|---|---|---|---|---|
| 12–16 Weeks | ||||||
| +/+ (n = 7) | 30.90 ± 4.20 | 0.1130 ± 0.0086 | 0.0037 ± 0.0005 | 0.0386 ± 0.0069 | 0.0013 ± 0.0003 | 29.1 ± 11.5 |
| −/− (n = 12) | 30.53 ± 2.54 | 0.1069 ± 0.0127 | 0.0035 ± 0.0005 | 0.0344 ± 0.0046* | 0.0011 ± 0.0001* | 17.0 ± 6.3* |
| P value | 0.8099 | 0.1190 | 0.2617 | 0.0299 | 0.0402 | 0.008 |
| 17–21 Weeks | ||||||
| +/+ (n = 9) | 29.95 ± 3.63 | 0.1175 ± 0.0073 | 0.0040 ± 0.0005 | 0.0384 ± 0.0025 | 0.0013 ± 0.0001 | 24.2 ± 4.9 |
| −/− (n = 11) | 30.46 ± 4.04 | 0.0998 ± 0.0145* | 0.0033 ± 0.0006* | 0.0365 ± 0.0041 | 0.0012 ± 0.0002 | 19.1 ± 5.6* |
| P value | 0.7719 | < 0.0001 | 0.001 | 0.0852 | 0.1560 | 0.0443 |
| Age and mouse type | Body weight (g) | Testis weight (g) | Testis/body ratio | Epididymis weight (g) | Epididymis/body ratio | Cauda epididymal sperm concentration (x106) |
|---|---|---|---|---|---|---|
| 12–16 Weeks | ||||||
| +/+ (n = 7) | 30.90 ± 4.20 | 0.1130 ± 0.0086 | 0.0037 ± 0.0005 | 0.0386 ± 0.0069 | 0.0013 ± 0.0003 | 29.1 ± 11.5 |
| −/− (n = 12) | 30.53 ± 2.54 | 0.1069 ± 0.0127 | 0.0035 ± 0.0005 | 0.0344 ± 0.0046* | 0.0011 ± 0.0001* | 17.0 ± 6.3* |
| P value | 0.8099 | 0.1190 | 0.2617 | 0.0299 | 0.0402 | 0.008 |
| 17–21 Weeks | ||||||
| +/+ (n = 9) | 29.95 ± 3.63 | 0.1175 ± 0.0073 | 0.0040 ± 0.0005 | 0.0384 ± 0.0025 | 0.0013 ± 0.0001 | 24.2 ± 4.9 |
| −/− (n = 11) | 30.46 ± 4.04 | 0.0998 ± 0.0145* | 0.0033 ± 0.0006* | 0.0365 ± 0.0041 | 0.0012 ± 0.0002 | 19.1 ± 5.6* |
| P value | 0.7719 | < 0.0001 | 0.001 | 0.0852 | 0.1560 | 0.0443 |
Significant difference.
Cst8 mouse tissue weights.
| Age and mouse type | Body weight (g) | Testis weight (g) | Testis/body ratio | Epididymis weight (g) | Epididymis/body ratio | Cauda epididymal sperm concentration (x106) |
|---|---|---|---|---|---|---|
| 12–16 Weeks | ||||||
| +/+ (n = 7) | 30.90 ± 4.20 | 0.1130 ± 0.0086 | 0.0037 ± 0.0005 | 0.0386 ± 0.0069 | 0.0013 ± 0.0003 | 29.1 ± 11.5 |
| −/− (n = 12) | 30.53 ± 2.54 | 0.1069 ± 0.0127 | 0.0035 ± 0.0005 | 0.0344 ± 0.0046* | 0.0011 ± 0.0001* | 17.0 ± 6.3* |
| P value | 0.8099 | 0.1190 | 0.2617 | 0.0299 | 0.0402 | 0.008 |
| 17–21 Weeks | ||||||
| +/+ (n = 9) | 29.95 ± 3.63 | 0.1175 ± 0.0073 | 0.0040 ± 0.0005 | 0.0384 ± 0.0025 | 0.0013 ± 0.0001 | 24.2 ± 4.9 |
| −/− (n = 11) | 30.46 ± 4.04 | 0.0998 ± 0.0145* | 0.0033 ± 0.0006* | 0.0365 ± 0.0041 | 0.0012 ± 0.0002 | 19.1 ± 5.6* |
| P value | 0.7719 | < 0.0001 | 0.001 | 0.0852 | 0.1560 | 0.0443 |
| Age and mouse type | Body weight (g) | Testis weight (g) | Testis/body ratio | Epididymis weight (g) | Epididymis/body ratio | Cauda epididymal sperm concentration (x106) |
|---|---|---|---|---|---|---|
| 12–16 Weeks | ||||||
| +/+ (n = 7) | 30.90 ± 4.20 | 0.1130 ± 0.0086 | 0.0037 ± 0.0005 | 0.0386 ± 0.0069 | 0.0013 ± 0.0003 | 29.1 ± 11.5 |
| −/− (n = 12) | 30.53 ± 2.54 | 0.1069 ± 0.0127 | 0.0035 ± 0.0005 | 0.0344 ± 0.0046* | 0.0011 ± 0.0001* | 17.0 ± 6.3* |
| P value | 0.8099 | 0.1190 | 0.2617 | 0.0299 | 0.0402 | 0.008 |
| 17–21 Weeks | ||||||
| +/+ (n = 9) | 29.95 ± 3.63 | 0.1175 ± 0.0073 | 0.0040 ± 0.0005 | 0.0384 ± 0.0025 | 0.0013 ± 0.0001 | 24.2 ± 4.9 |
| −/− (n = 11) | 30.46 ± 4.04 | 0.0998 ± 0.0145* | 0.0033 ± 0.0006* | 0.0365 ± 0.0041 | 0.0012 ± 0.0002 | 19.1 ± 5.6* |
| P value | 0.7719 | < 0.0001 | 0.001 | 0.0852 | 0.1560 | 0.0443 |
Significant difference.
Analysis of Fertility and Motility in Cst8−/− Mice
To assess the fertility of the Cst8−/− mice, males and females were paired with C57BL6 mice of the opposite gender, and the number of days before plugs were detected as well as the number of implantation sites were determined. The B6 mouse was chosen as the mating partner because of its high fertility, thus removing possible confounding effects of the poorer fertility of the 129 strain on our analysis. The Cst8−/− male and female mice showed no difference in their fertility compared to that of the Cst8+/+ mice in that both phenotypes required a similar number of days before detection of a copulatory plug and generated litters of comparable size (Table 2). Similarly, when intercrosses between Cst8+/+ and Cst8−/− mice were carried out, no differences in fertility were observed with the loss of the CRES gene (Table 2). In contrast to observations in vivo, spermatozoa from Cst8−/− mice exhibited a profound defect in their ability to fertilize COCs in vitro (Fig. 2A). Cst8−/− spermatozoa fertilized only 19% of the oocytes, compared to 66% of oocytes fertilized by Cst8+/+ spermatozoa, resulting in a 72% decrease in fertilizing efficiency of the Cst8−/− spermatozoa (P < 0.01). The ability of Cst8−/− spermatozoa to bind to the egg zona pellucida was reduced by 53% compared to that of Cst8+/+ mice (P < 0.01), and the ability to fuse with the egg plasma membrane was reduced by 68% compared to that of Cst8+/+ spermatozoa (P < 0.01) (Fig. 2, B and C).
Fertility of Cst8−/− mice.*
| F | M | n | Days to plug | n | Average litter size | |
|---|---|---|---|---|---|---|
| +/+ | X | +/+ | - | - | 14 | 7.5 ± 2.3 |
| −/− | X | +/+ | - | - | 13 | 7.2 ± 2.6 |
| +/+ | X | −/− | - | - | 11 | 8.1 ± 1.4 |
| −/− | X | −/− | - | - | 17 | 7.7 ± 1.9 |
| +/+ | X | B6 | 25 | 2.5 ± 1.3 | 9 | 8.9 ± 3.5 |
| −/− | X | B6 | 21 | 2.6 ± 1.6 | 8 | 9.1 ± 2.5 |
| B6 | X | +/+ | 14 | 2.6 ± 1.7 | 2 | 9.5 ± 0.7 |
| B6 | X | −/− | 22 | 2.1 ± 1.9 | 8 | 9.6 ± 1.4 |
| F | M | n | Days to plug | n | Average litter size | |
|---|---|---|---|---|---|---|
| +/+ | X | +/+ | - | - | 14 | 7.5 ± 2.3 |
| −/− | X | +/+ | - | - | 13 | 7.2 ± 2.6 |
| +/+ | X | −/− | - | - | 11 | 8.1 ± 1.4 |
| −/− | X | −/− | - | - | 17 | 7.7 ± 1.9 |
| +/+ | X | B6 | 25 | 2.5 ± 1.3 | 9 | 8.9 ± 3.5 |
| −/− | X | B6 | 21 | 2.6 ± 1.6 | 8 | 9.1 ± 2.5 |
| B6 | X | +/+ | 14 | 2.6 ± 1.7 | 2 | 9.5 ± 0.7 |
| B6 | X | −/− | 22 | 2.1 ± 1.9 | 8 | 9.6 ± 1.4 |
F, female; M, male.
Fertility of Cst8−/− mice.*
| F | M | n | Days to plug | n | Average litter size | |
|---|---|---|---|---|---|---|
| +/+ | X | +/+ | - | - | 14 | 7.5 ± 2.3 |
| −/− | X | +/+ | - | - | 13 | 7.2 ± 2.6 |
| +/+ | X | −/− | - | - | 11 | 8.1 ± 1.4 |
| −/− | X | −/− | - | - | 17 | 7.7 ± 1.9 |
| +/+ | X | B6 | 25 | 2.5 ± 1.3 | 9 | 8.9 ± 3.5 |
| −/− | X | B6 | 21 | 2.6 ± 1.6 | 8 | 9.1 ± 2.5 |
| B6 | X | +/+ | 14 | 2.6 ± 1.7 | 2 | 9.5 ± 0.7 |
| B6 | X | −/− | 22 | 2.1 ± 1.9 | 8 | 9.6 ± 1.4 |
| F | M | n | Days to plug | n | Average litter size | |
|---|---|---|---|---|---|---|
| +/+ | X | +/+ | - | - | 14 | 7.5 ± 2.3 |
| −/− | X | +/+ | - | - | 13 | 7.2 ± 2.6 |
| +/+ | X | −/− | - | - | 11 | 8.1 ± 1.4 |
| −/− | X | −/− | - | - | 17 | 7.7 ± 1.9 |
| +/+ | X | B6 | 25 | 2.5 ± 1.3 | 9 | 8.9 ± 3.5 |
| −/− | X | B6 | 21 | 2.6 ± 1.6 | 8 | 9.1 ± 2.5 |
| B6 | X | +/+ | 14 | 2.6 ± 1.7 | 2 | 9.5 ± 0.7 |
| B6 | X | −/− | 22 | 2.1 ± 1.9 | 8 | 9.6 ± 1.4 |
F, female; M, male.
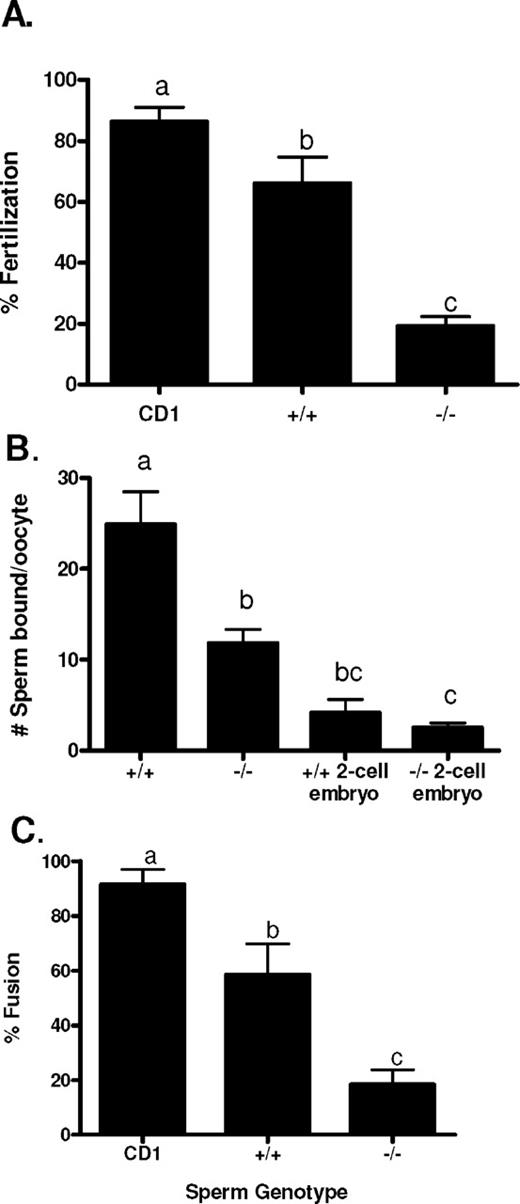
Analysis of Cst8+/+ and Cst8−/− sperm fertilizing ability in vitro. A) Percentage of COCs fertilized after 3 h of incubation with control CD1, Cst8+/+, and Cst8−/− spermatozoa (n = 5; a vs. b, P ≤ 0.05; a vs. c, P ≤ 0.001; b vs. c, P ≤ 0.01). B) Number of Cst8+/+ and Cst8−/− spermatozoa bound to the zona pellucida of cumulus-free oocytes from CD1 female mice. Spermatozoa were also incubated with 2-cell embryos as a control for nonspecific binding (n = 4; a vs. b, P ≤ 0.01; a vs. c, P ≤ 0.001; b vs. c, P ≤ 0.05). C) Percentage sperm-egg fusion of cumulus- and zona pellucida-free oocytes from CD1 females incubated with spermatozoa from control CD1, Cst8+/+, and Cst8−/− mice (n = 5; a vs. b, P ≤ 0.05; a vs. c, P ≤ 0.001; b vs. c, P ≤ 0.01). Values represent the mean ± SEM.
Because actively motile spermatozoa are required for normal fertility, a CASA was carried out on spermatozoa incubated under capacitating conditions for various periods of time. As shown in Figure 3, spermatozoa from the Cst8−/− mice showed a significant decrease from the Cst8+/+ mice in the number of spermatozoa that were motile as well as in the number of spermatozoa that exhibited a progressive motility, but these differences were relatively small (∼10% reduced) and likely do not explain the fertility defect. All other motility parameters, including track speed, path velocity, progressive velocity, lateral displacement, beat cross frequency, straightness, and linearity, were similar between spermatozoa from Cst8+/+ and Cst8−/− mice (data not shown). Because the CASA system available for our motility studies did not allow us to selectively identify spermatozoa that were hyperactivated, we cannot rule out the existence of differences between the Cst8+/+ and Cst8−/− spermatozoa in their ability to undergo hyperactivation. However, a subjective examination of the capacitated spermatozoa did not reveal any obvious differences between the two genotypes.
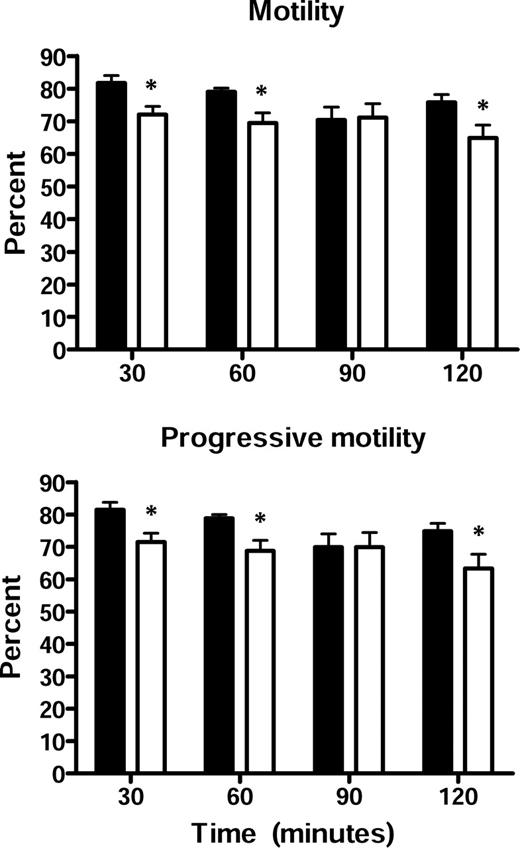
Sperm motility in Cst8+/+ and Cst8−/− mice. Equal concentrations of spermatozoa from Cst8+/+ (black bars) and Cst8−/− (white bars) mice were examined after varying times of capacitation. In each experiment, 10 fields were examined per sample. Values represent the mean ± SEM of four experiments. % motile, percentage of spermatozoa with motility; % progressive motility, percentage of spermatozoa moving forward. An asterisk indicates statistical significance (*P < 0.05).
Sperm Capacitation and Acrosome Reaction in Cst8−/− Mice
Studies were next carried out to determine whether spermatozoa from mice lacking the CRES gene were capable of undergoing the acrosome reaction after increasing incubation times in capacitating medium. Two compounds, including the calcium ionophore A23187 and the more physiological molecule progesterone, were used to induce the acrosome reaction. A23187 induced the acrosome reaction in a time-dependent manner in a similar percentage of capacitated spermatozoa from Cst8+/+ and Cst8−/− mice. Furthermore, the percentage of both Cst8+/+ and Cst8−/− spermatozoa that underwent the induced acrosome reaction was significantly higher than the number of spermatozoa that underwent a spontaneous acrosome reaction in the presence of the vehicle DMSO (Fig. 4A, left). When spermatozoa were induced to acrosome react in the presence of progesterone, Cst8+/+ spermatozoa showed a time-dependent increase in the number of sperm that acrosome reacted, whereas the levels in the Cst8−/− spermatozoa were not different from the spontaneous rates of acrosome reaction in spermatozoa incubated with DMSO (Fig. 4A, right). After 60 and 90 min of capacitation, the percentage of spermatozoa from Cst8−/− mice that underwent a progesterone-stimulated acrosome reaction was significantly lower than that for Cst8+/+ spermatozoa (P < 0.05). The differences in the response of Cst8−/− spermatozoa to A23187 and progesterone suggests that the two agents stimulate the acrosome reaction by different pathways and that the pharmacological induction of the acrosome reaction by A23187 may bypass important biological steps/pathways involving CRES that are required for the more physiological induction of the acrosome reaction by progesterone.
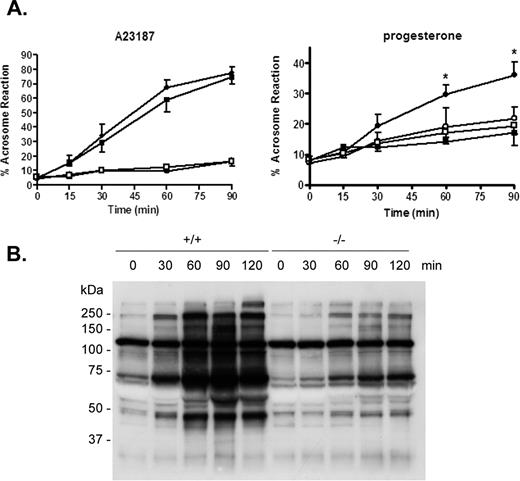
Sperm capacitation and acrosome reaction in Cst8+/+ and Cst8−/− mice. A) Spermatozoa from Cst8+/+ (filled circles) and Cst8−/− (filled squares) mice after various times of capacitation were induced to undergo the acrosome reaction by the calcium ionophore A23187 (left) or progesterone (right). Spermatozoa from Cst8+/+ (open circles) and Cst8−/− mice (open squares) that were incubated in DMSO vehicle only and, thus, represent spontaneous acrosome reactions are also shown. The present or absence of the acrosome was determined by Coomassie Blue staining. Values represent the mean ± SEM (A23187, n = 3; progesterone, n = 4). An asterisk indicates statistical significant (*P ≤ 0.001) comparing Cst8+/+ to Cst8−/− spermatozoa in the presence of progesterone. B) Western blot analysis of protein tyrosine phosphorylation in Cst8+/+ and Cst8−/− spermatozoa after increasing periods of time in capacitation buffer. The blot is representative of that from three different sperm capacitation experiments. Coomassie Blue staining of the blot confirmed equal loading of protein in each lane (not shown).
Because the inability of spermatozoa to acrosome react can reflect disturbances in sperm capacitation, experiments were next carried out to determine whether Cst8−/− sperm proteins become tyrosine phosphorylated during capacitation, an established indicator of the capacitation process [19, 20]. Cst8+/+ and Cst8−/− spermatozoa were incubated in capacitating medium, and at various times, samples were removed and processed for Western blot analysis using an anti-phosphotyrosine antibody. Compared to Cst8+/+ spermatozoa, which showed a time-dependent increase in the number and amount of proteins that became tyrosine phosphorylated during capacitation, Cst8−/− spermatozoa showed a limited ability for protein tyrosine phosphorylation throughout the same capacitation time course (Fig. 4B).
Because protein tyrosine phosphorylation is thought to be a downstream event of earlier cAMP/PKA-mediated signaling pathways critical for capacitation, our observations suggested that spermatozoa from the Cst8−/− mice may have a defect in these signaling pathways, resulting in impaired capacitation and fertilization.
Effects of DbcAMP and IBMX on Cst8−/− Sperm Function
Experiments were next carried out to determine whether supplementing the Cst8−/− spermatozoa with cAMP would circumvent the putative defect in the cAMP-dependent signaling pathways and allow the spermatozoa to undergo normal protein tyrosine phosphorylation associated with capacitation. Spermatozoa from Cst8+/+ and Cst8−/− mice were capacitated in the presence of the vehicle DMSO or the membrane-permeable cAMP agonist, dbcAMP, and the phosphodiesterase inhibitor, IBMX, to prevent cAMP turnover. The presence of dbcAMP and IBMX stimulated sperm protein tyrosine phosphorylation in the Cst8−/− spermatozoa such that the levels were similar to those in Cst8+/+ spermatozoa treated with DMSO (Fig. 5A). Furthermore, the rate of tyrosine phosphorylation was slightly accelerated in the Cst8−/− spermatozoa in the presence of dbcAMP and IBMX. The addition of dbcAMP and IBMX to spermatozoa from Cst8+/+ mice resulted in only a slight increase in the levels of protein tyrosine phosphorylation, suggesting that these spermatozoa were already exhibiting close to maximal levels of tyrosine phosphorylation associated with capacitation. However, similar to the observation with the Cst8−/− spermatozoa, dbcAMP and IBMX accelerated the rate of protein tyrosine phosphorylation in the Cst8+/+ spermatozoa: Almost maximal levels of phosphorylation were observed at 60 min, whereas similar levels were achieved only after 120 min in spermatozoa incubated with DMSO (Fig. 5A).
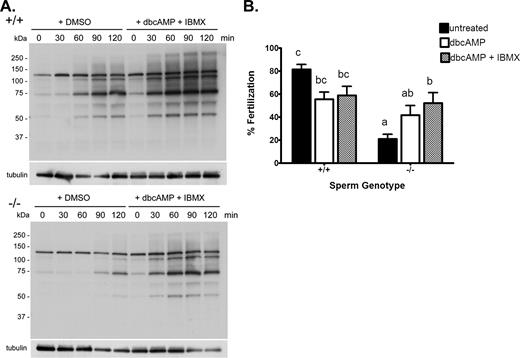
Rescue of sperm protein tyrosine phosphorylation and fertility in spermatozoa from Cst8−/− mice after incubation in dbcAMP and IBMX. A) Western blot analysis of protein tyrosine phosphorylation in Cst8+/+ and Cst8−/− spermatozoa after increasing periods of time in capacitation buffer and in the presence of dbcAMP and IBMX or the vehicle DMSO. Blots were stripped and reprobed with anti-tubulin antibody to verify equal loading of the gel. Blots are representative of those from three different experiments. B) Percentage of COCs fertilized after 3 h of incubation with Cst8+/+ and Cst8−/− spermatozoa that were untreated, incubated with dbcAMP, or incubated with dbcAMP and IBMX before addition to the oocytes. Values represent the mean ± SEM (n = 10 experiments; a vs. b, P ≤ 0.05; c vs. a, P ≤ 0.001; c vs. b, P ≤ 0.05; c vs. ab, P ≤ 0.01; bc vs. a, P ≤ 0.01).
Because the addition of dbcAMP and IBMX to Cst8−/− spermatozoa enhanced the levels of protein tyrosine phosphorylation to that in normal, fertile Cst8+/+ spermatozoa, studies were carried out to determine if the treated Cst8−/− spermatozoa also acquired the ability to fertilize oocytes in vitro. Cst8−/− spermatozoa that were incubated with dbcAMP and IBMX during capacitation fertilized COCs as efficiently as Cst8+/+ spermatozoa treated with dbcAMP and IBMX (Fig. 5B).
The rescue of the fertility defect with dbcAMP and IBMX in mice lacking the CRES gene suggested that the intracellular levels of cAMP and/or PKA may be reduced in spermatozoa from Cst8−/− mice. Assays to measure the total levels of cAMP in Cst8+/+ and Cst8−/− spermatozoa showed no differences between the two sperm populations throughout the capacitation time course (Fig. 6). Both Cst8+/+ and Cst8−/− spermatozoa showed a time-dependent increase in cAMP between 0 and 60 min of capacitation and then a return to levels similar to those of time zero after 90 and 120 min of capacitation. Spermatozoa that were incubated in noncapacitating conditions did not exhibit a capacitation-induced increase in cAMP; rather, they showed only low basal levels of cAMP that did not change throughout the capacitation time course (Fig. 6).
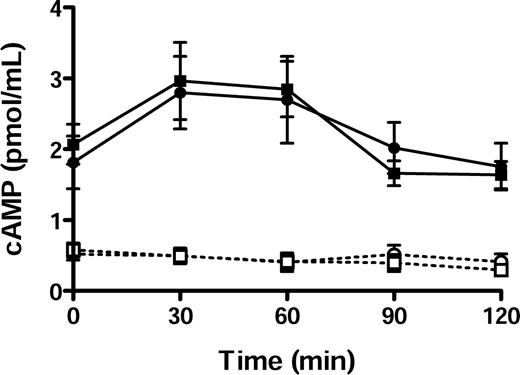
Cyclic AMP levels in Cst8+/+ and Cst8−/− spermatozoa. Cell extracts prepared from Cst8+/+ (circles) and Cst8−/− (squares) spermatozoa incubated in capacitating (filled symbols) or noncapacitating (open symbols) media were analyzed for total levels of cAMP after various times of capacitation. Values represent the mean ± SEM (n = 10 experiments). Cyclic AMP levels in Cst8+/+ and Cst8−/− spermatozoa were significantly increased at all time points in spermatozoa incubated in capacitating medium compared to spermatozoa incubated in noncapacitating medium.
Assays were also carried out to examine the PKA activity in extracts from Cst8+/+ and Cst8−/− spermatozoa. Both sperm populations showed a time-dependent increase in PKA activity throughout the capacitation time course, reaching maximal levels around 90 min, whereas spermatozoa incubated under noncapacitating conditions showed significantly lower levels of PKA activity (P ≤ 0.05) (Fig. 7A). The Cst8−/− spermatozoa exhibited significantly lower levels of PKA activity compared to Cst8+/+ spermatozoa only after 90 min of capacitation, but no differences in PKA activity were observed between the two sperm preparations at any other times (Fig. 7A). The addition of dbcAMP and IBMX to the PKA reaction tubes containing extracts from Cst8+/+ or Cst8−/− spermatozoa resulted in an approximately 3-fold increase in the amount of PKA activity, which remained relatively constant over time and was not different between capacitated and noncapacitated spermatozoa from either genotype (Fig. 7C). These experiments demonstrated that both Cst8+/+ and Cst8−/− spermatozoa can achieve similar maximal levels of PKA activity. Addition of the PKA inhibitor H89 to the reaction tubes resulted in a profound decrease in PKA activity in all sperm samples, indicating that the assay is measuring PKA (Fig. 7B).
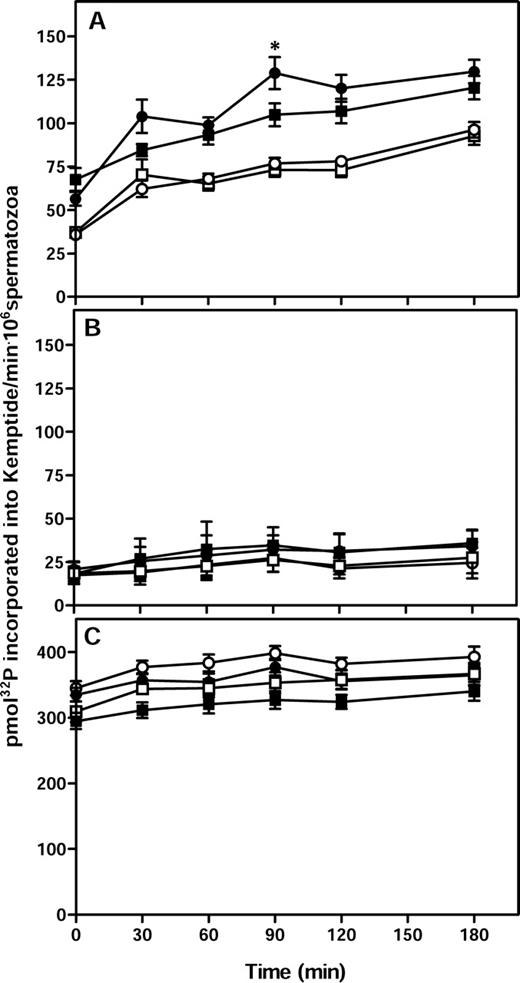
PKA activity in Cst8+/+ and Cst8−/− spermatozoa during capacitation. A) PKA activity in Cst8+/+ (circles) and Cst8−/− (squares) spermatozoa after incubation in capacitating (filled symbols) or noncapacitating (open symbols) medium. Values represent the mean ± SEM (n = 11 experiments). An asterisk indicates statistical significance (*P ≤ 0.05) comparing Cst8+/+ spermatozoa to Cst8−/− spermatozoa. The PKA activity in Cst8+/+ and Cst8−/− spermatozoa incubated under capacitating conditions was increased at all time points compared to PKA activity of spermatozoa incubated under noncapacitating conditions (P ≤ 0.05). PKA activity in capacitated Cst8+/+ spermatozoa was not different from that in capacitated Cst8−/− spermatozoa except at 90 min (P ≤ 0.05). B) PKA activity was measured in similar samples as in A but with 10 μM H89, a PKA inhibitor, included during the assay (n = 2 experiments). C) PKA activity was measured in similar samples as in A but with 1 mM dbcAMP and 100 μM IBMX included during the assay (n = 9 experiments).
Phosphotyrosine Immunofluorescence Analysis
Indirect immunofluorescence analysis was carried out to determine if the decreased protein tyrosine phosphorylation in the Cst8−/− spermatozoa reflected an overall decrease in fluorescence or if protein tyrosine phosphorylation was decreased only in specific regions of the spermatozoa. Studies were also carried out to determine if the addition of dbcAMP and IBMX altered the fluorescence staining pattern. As shown in Figure 8A, and as expected, both Cst8+/+ and Cst8−/− spermatozoa demonstrated an increase in protein tyrosine phosphorylation immunofluorescence after 90 min of capacitation compared to that observed at time zero. However, at both time zero and 90 min, the levels of fluorescence in the Cst8−/− spermatozoa were reduced overall compared to levels in the Cst8+/+ spermatozoa. Control spermatozoa that were incubated with secondary antibody alone showed only a faint midpiece staining (data not shown).
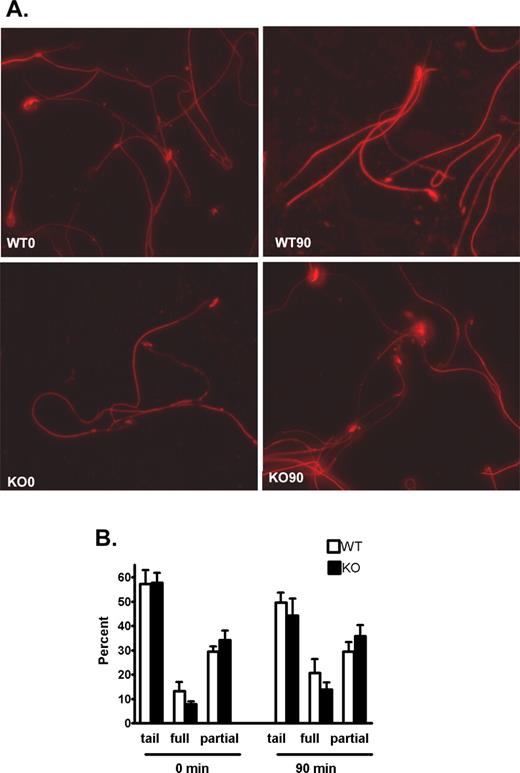
Phosphotyrosine fluorescence in capacitated Cst8+/+ and Cst8−/− spermatozoa. A) At 0 and 90 min of capacitation, Cst8+/+ (WT) and Cst8−/− (KO) spermatozoa were air-dried onto slides and incubated with the monoclonal anti-phosphotyrosine antibody followed by an Alexa Fluor 594-labeled secondary antibody. All images were captured using the same exposure times and were set to that of the strongest fluorescent labeling in WT 90-min sperm samples. Fluorescent images are superimposed on those obtained under phase contrast. Original magnification ×40. B) Percentage of spermatozoa that exhibited phosphotyrosine fluorescence on the midpiece and principle piece only (tail), tail plus full head fluorescence (full), or tail and partial head fluorescence (partial) at 0 and 90 min of capacitation. Value represent the mean ± SEM (n = 3 experiments).
Because different sperm fluorescence patterns were noted in both sperm populations, a more detailed evaluation was carried out to assess for differences between the Cst8+/+ and Cst8−/− spermatozoa. All spermatozoa exhibited protein tyrosine phosphorylation fluorescence in the midpiece and principle piece (tail), and this group of spermatozoa seemed to predominate at both 0 and 90 min of capacitation. Some spermatozoa also had fluorescent heads, which included those with the majority of the head fluorescent (full) and those with only some of the head fluorescent (partial) (Fig. 9A). No significant differences were found between Cst8+/+ and Cst8−/− mice in the percentage of spermatozoa that were distributed among these categories at 0 and 90 min, although there did appear to be a trend for more spermatozoa to acquire full head fluorescence with increased capacitation time and for Cst8−/− spermatozoa to haves less full and more partial head fluorescence (Fig. 8B).
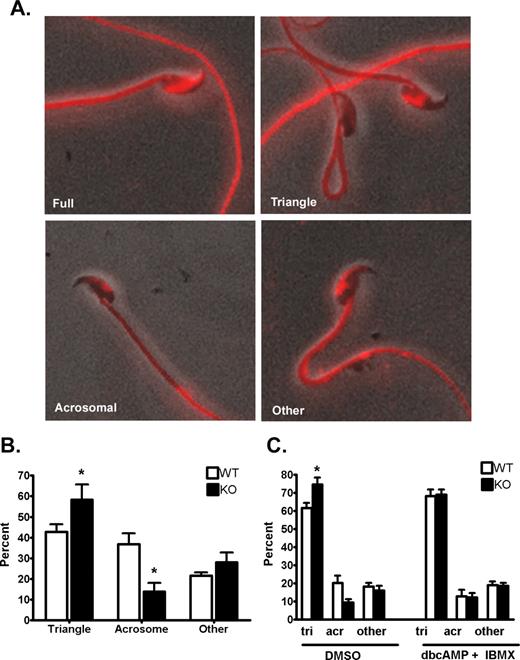
Phosphotyrosine fluorescence patterns observed on the sperm heads of Cst8+/+ and Cst8−/− spermatozoa. Images are to show pattern only and are not relative fluorescence levels between the varying patterns. A) Full fluorescence over most of the sperm head; partial head staining included that in the triangular, acrosomal, or other patterns. Fluorescent images are superimposed on those obtained under phase contrast. Original magnification ×40. B) Percentage of Cst8+/+ (WT) and Cst8−/− (KO) sperm heads exhibiting the triangular, acrosomal, or other patterns of phosphotyrosine fluorescence after 90 min of capacitation. Value represent the mean ± SEM (n = 3 experiments). C) Percentage of WT and KO sperm heads exhibiting triangular (tri), acrosomal (acr), or other patterns of phosphotyrosine fluorescence after 90 min of capacitation in the presence of DMSO or 1 mM dbcAMP plus 100 μM IBMX. Value represent the mean ± SEM (n = 4 experiments). An asterisk indicates statistical significance (*P < 0.05).
Within the partial head fluorescence group, further distinctions could be made based on the fluorescence pattern; these patterns included triangular, acrosomal, and other, which usually represented sperm that possessed both an acrosomal and small triangular pattern of phosphotyrosine fluorescence (Fig. 9A). The Cst8−/− spermatozoa after 90 min of capacitation had significantly more sperm heads exhibiting the triangular phosphotyrosine fluorescence and fewer sperm heads with the acrosomal staining pattern compared to Cst8+/+ spermatozoa (Fig. 9B). Cst8−/− spermatozoa that were capacitated for 90 min in the presence of dbcAMP and IBMX showed phosphotyrosine fluorescence patterns similar to those in the Cst8+/+ spermatozoa (Fig. 9C).
Discussion
The present study demonstrates that despite exhibiting normal fertility in vivo, spermatozoa from mice lacking the CRES gene (Cst8−/−) have severe defects in their ability to fertilize in vitro. Cst8−/− spermatozoa show a profound decrease in their ability to fertilize COCs in vitro, to bind to the egg zona pellucida, and to fuse with the egg plasma membrane when compared to Cst8+/+ spermatozoa. More detailed analyses of the defect in the Cst8−/− spermatozoa indicated these spermatozoa exhibited an impaired ability to undergo capacitation, as evidenced by their inability to acrosome react in the presence of progesterone and the limited tyrosine phosphorylation of sperm proteins during capacitation. We cannot rule out that the presence of the neo cassette in the Cst8−/− mouse genome could contribute to the phenotype, but that other mouse gene knockout models generated using the neo cassette, such as the sperm hyaluronidase Hyal5 knockout, exhibit normal fertility in vitro suggests this is not the case [21].
The differences between the Cst8−/− phenotype in vitro and in vivo could reflect heterogeneity in the sperm population that is not detected in the breeding experiments, because the lower-quality spermatozoa are filtered out during their transit in the female reproductive tract. Indeed, a small proportion of the Cst8−/− spermatozoa are still capable of fertilizing in vitro, and it may be this population of cells that also successfully fertilizes in vivo. Alternatively, it may be that during ejaculation or transit in the female reproductive tract, Cst8−/− spermatozoa are exposed to molecules that restore their fertility. This would not be unlike that recently observed in the mouse model lacking the sperm serine protease Prss21(also known as Tesp5). Exposure of Prss21−/− spermatozoa to the uterine microenvironment or in vitro exposure to isolated uterine fluids rescued the fertility defect [22]. The relevant molecule (or molecules) that restored PRSS21 fertility is not known.
Our studies show that Cst8−/− spermatozoa recover the same level of protein tyrosine phosphorylation and fertility as Cst8+/+ spermatozoa after exposure to dbcAMP and IBMX. These studies would imply that the endogenous levels of cAMP or PKA may be decreased in the Cst8−/− spermatozoa. However, repeated analyses of Cst8−/− spermatozoa revealed no differences in the level or timing of the capacitation-induced rise in cAMP from that in Cst8+/+ spermatozoa. Furthermore, examination of PKA activity in the Cst8−/− spermatozoa revealed no differences from that in Cst8+/+ spermatozoa during capacitation except at one time point. We are being conservative in our interpretation of this result, because throughout the 11 assays that were performed, many experiments showed no difference at any time of capacitation in the levels of PKA activity between Cst8+/+ and Cst8−/− spermatozoa. Thus, inherent variation, likely within the sperm populations but also in the assays themselves, makes these data difficult to interpret. We cannot rule out that the reduced level of PKA activity in Cst8−/− spermatozoa after 90 min of capacitation is biologically relevant and contributes to the fertility defect, but overall PKA activity in Cst8+/+ and Cst8−/− spermatozoa does not seem to be greatly different during capacitation. Furthermore, there does not appear to be a difference in the maximal levels of PKA activity that can be achieved in the two sperm populations, because PKA activity increased similarly in both Cst8+/+ and Cst8−/− spermatozoa that were exposed to dbcAMP and IBMX. The levels of PKA activity observed in our studies were similar to that previously measured in mouse sperm undergoing capacitation [23].
The similarity in the capacitation- induced increase in cAMP and PKA levels in the Cst8+/+ and Cst8−/− mice would suggest that the fertility defect in mice lacking the CRES gene does not directly involve cAMP/PKA; however, these assays measured total levels of activity throughout the sperm cell and not local levels of activity. Indeed, cAMP is distributed between different cellular compartments [24, 25], and PKA is compartmentalized in spermatozoa, with both the catalytic and regulatory subunits I and II as well as PKA activity present in both the Triton X-100-soluble and -insoluble fractions [18]. The increase in PKA activity associated with capacitation has been shown to be localized to only the Triton X-100-soluble fraction, with a population of PKA remaining present in the insoluble fraction, possibly tethered to the sperm cytoskeleton by A kinase anchoring proteins [18]. However, addition of dbcAMP and IBMX resulted in release of the catalytic subunit from the Triton X-100-insoluble material and maximal levels of PKA activity [18]. In mice lacking the CRES gene, the partitioning/tethering of cAMP and/or PKA in the spermatozoa may be abnormal such that local levels are not sufficient to stimulate early and later events of capacitation, including protein tyrosine phosphorylation, thus resulting in impaired fertility. However, when the Cst8−/− spermatozoa are incubated with dbcAMP and IBMX, these reagents permit maximal stimulation of cAMP and activation of PKA that overrides the defect, allowing normal capacitation and fertility. This would suggest that the CRES defect is upstream of these initial signaling events. It is also possible that the loss of CRES results in a defect that does not directly involve the cAMP/PKA-mediated signaling pathways but other pathways that can mediate protein tyrosine phosphorylation and capacitation. This could include other cyclic nucleotide receptors or regulation of PKA by other mechanisms. Studies are currently ongoing to examine PKA activity in the Triton X-100-soluble and -insoluble fractions from Cst8+/+ and Cst8−/− spermatozoa.
At this time, we cannot determine whether the fertility defect in mice lacking the CRES gene is caused by the absence of CRES that is synthesized in germ cells during spermatogenesis and/or CRES that is synthesized and secreted by the proximal caput epididymal epithelium and that may interact with spermatozoa during epididymal maturation. However, in normal CD1 strain mice, we have not been able to detect CRES associated with the epididymal sperm surface, and our previous studies have indicated that CRES localized to within the sperm acrosome, supporting a role for this population of CRES in fertilization [26]. Thus, taken together, these results would favor the idea that the loss of CRES from within spermatozoa, not that secreted by the epididymis, is causing the fertility defect.
The biological role of CRES in the sperm acrosome and how its absence causes the fertility defect remains to be elucidated. One possibility is that CRES functions as a protease inhibitor necessary for the regulation of critical proteases involved in early signaling events during fertilization. Alternatively, CRES function may be as a structural component of the acrosome. We previously demonstrated that CRES self-aggregates and forms amyloid, a highly organized and stable structure that typically is present in protein deposits associated with neurodegenerative diseases, such as Alzheimer disease [27]. It is possible that within spermatozoa, the CRES amyloid structure is functional and may act as a scaffold that allows key signaling molecules to be in the correct proximity to one another, thus allowing appropriate signaling critical for capacitation and fertilization. In support, during capacitation, reorganization of the sperm membrane does occur, and trafficking of signaling complexes is thought to be an integral part of this process [28, 29]. Recently, it was shown that the PMEL17 protein forms a functional amyloid structure that directs melanin synthesis within melanosomes [30], whereas other studies have demonstrated that pituitary hormones are stored as stable amyloid structures before secretion [31]. Thus, functional amyloids in other cell systems, including the reproductive tract, are quite likely. Studies are currently ongoing to examine CRES amyloid in spermatozoa.
The different phosphotyrosine fluorescence labeling that was observed on the sperm heads of the Cst8+/+ and Cst8−/− spermatozoa would also support the idea that cell signaling is altered in the Cst8−/− spermatozoa. In addition to the narrow band of phosphotyrosine fluorescence that appeared to be in the region of the acrosome of some spermatozoa, very strong fluorescence was detected in the equatorial segment (full pattern) in other spermatozoa, whereas in still other sperm populations, phosphotyrosine fluorescence (triangular or other pattern) was in a region that has been termed the equatorial subsegment, which is rich in tyrosine phosphorylated proteins, and has been proposed to be an organizational center for assembly of signaling complexes [32]. The precise phosphotyrosine labeling that is necessary for normal capacitation and fertilization is not known, but the difference between the Cst8+/+ and Cst8−/− spermatozoa in the acrosomal and triangular patterns of phosphotyrosine fluorescence and the loss of these differences with a corresponding gain of fertility with dbcAMP and IBMX treatment would support the idea of these regions as being important.
Taken together, the results presented herein demonstrate a role for CRES in fertilization and suggest an important function during cAMP-mediated cell signaling in sperm capacitation.
Note Added in Proof
Additional information regarding CRES and its affects on the male reproductive tract can be found in an article by Parent et al., [33] which was recently accepted for publication.
Acknowledgments
The authors thank Steve Tardif, Ph.D., for helpful discussions concerning the capacitation and acrosome reaction studies; Pablo Visconti, Ph.D., for helpful discussions with the cAMP and PKA assays; and Stephen Cox, Ph.D., for assistance with the statistical analyses. The authors would also like to thank Seethal Johnson for her excellent technical assistance and help with the mouse colony.
References
Author notes
Supported by NIH HD33903, HD44669, and HD56182 (G.A.C.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}