





Abstract
The herbicide paraquat is an environmental factor that may be involved in the etiology of Parkinson's disease (PD). Systemic exposure of mice to paraquat causes a selective loss of dopaminergic neurons in the substantia nigra pars compacta, although paraquat is not selectively incorporated in dopaminergic neurons. Here, we report a contribution of endogenous dopamine to paraquat-induced dopaminergic cell death. Exposure of PC12 cells to paraquat (50μM) caused delayed toxicity from 36 h onward. A decline in intracellular dopamine content achieved by inhibiting tyrosine hydroxylase (TH), an enzyme for dopamine synthesis, conferred resistance to paraquat toxicity on dopaminergic cells. Paraquat increased the levels of cytosolic and vesicular dopamine, accompanied by transiently increased TH activity. Quinone derived from cytosolic dopamine conjugates with cysteine residues in functional proteins to form quinoproteins. Formation of quinoprotein was transiently increased early during exposure to paraquat. Furthermore, pretreatment with ascorbic acid, which suppressed the elevations of intracellular dopamine and quinoprotein, almost completely prevented paraquat toxicity. These results suggest that the elevation of cytosolic dopamine induced by paraquat participates in the vulnerability of dopaminergic cells to delayed toxicity through the formation of quinoproteins.
Human epidemiological studies indicate an association between an exposure to pesticides and an increased risk for Parkinson's disease (PD) (Gorell et al., 1998; Seidler et al., 1996) . The herbicide paraquat has been particularly implicated as a potential risk factor for the development of PD (Hertzman et al., 1990; Liou et al., 1997; Tanner et al., 2011). Systemic exposure of mice to paraquat caused a loss of dopaminergic cells in the substantia nigra pars compacta, although GABAergic neurons in the substantia nigra pars reticulata and hippocampal neurons did not display any change (McCormack et al., 2002). This report suggested selective toxicity of the herbicide in dopaminergic neurons. The details of the mechanism of paraquat-induced dopaminergic cell damage are unknown; however, this possibly involves increased production of oxygen-free radicals via a redox cycling mechanism. Thus, it means that diaphorase and/or Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase could catalyze the NADPH-dependent one-electron reduction of paraquat. The radical product of paraquat transfers its electron onto molecular oxygen, ultimately leading to the formation of the superoxide anion, and regenerates the parent compound (Bus and Gibson, 1984).
A previous study demonstrated that paraquat was taken up into the brain by the neutral amino acid system (Shimizu et al., 2001). On the basis of the structural similarity between paraquat and the 1-methyl-4-phenylpyridinium ion (MPP+), it has been speculated that paraquat may enter dopaminergic neurons via the dopamine transporter. Paraquat toxicity was independent of the dopamine transporter (Richardson et al., 2005), whereas an inhibitor of the dopamine transporter suppressed the dopaminergic toxicity of paraquat (Shimizu et al., 2003a). Although the involvement of this transporter in the uptake of paraquat has not been demonstrated sufficiently over the past decade (Ossowska et al., 2005a; Richardson et al., 2005). Rappold et al. (2011) recently reported that the monovalent cation paraquat, rather than the compound in its native divalent cation state, was a substrate for the dopamine transporter. Moreover, they showed that the monovalent cation paraquat was more effectively transported by the organic cation transporter 3, which is abundantly expressed in nondopaminergic neurons (Cui et al., 2009). Therefore, the reason for more vulnerability of dopaminergic neurons to paraquat, in animal models, remains to be clarified.
It was hypothesized that nigrostriatal dopaminergic neurons may be particularly vulnerable to oxidative damage because they are rich in dopamine, which can undergo both enzymatic oxidation and autoxidation. Thus, oxygen radical production triggered by paraquat could lead to more pronounced injury to dopaminergic neurons. In fact, a number of studies have suggested that dopamine mediates the vulnerability of dopaminergic neurons to neurotoxins and PD-related genes (Izumi et al., 2009; Nakaso et al., 2013; Sakka et al., 2003; Xu et al.2002). Here, we examined whether endogenous dopamine was involved in the vulnerability of dopaminergic cells to paraquat toxicity. In addition, we investigated the mechanism underlying the paraquat-induced dopaminergic cell death mediated by endogenous dopamine.
MATERIALS AND METHODS
Materials
Paraquat dichloride and reserpine were purchased from Nacalai Tesque (Kyoto, Japan). α-Methyl-dl-p-tyrosine methyl ester hydrochloride (α-MT), digitonin, and ascorbic acid were obtained from Sigma (St Louis, MO). Trolox was obtained from Cayman Chemical (Ann Arbor, MI). GBR12909 was purchased from Tocris Cookson (Bristol, UK).
Cell cultures
Rat adrenal pheochromocytoma PC12 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% fetal calf serum (FCS) and 10% horse serum (HS). PC12 cells were seeded in culture plates at a density of 5.3 × 104 cells/cm2 and incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were grown for at least 24 h before exposure to drugs.
Cultures of the rat mesencephalon were established according to methods described previously (Izumi et al., 2009). In brief, the ventral two-thirds of the mesencephalon was dissected from rat embryos on the 16th day of gestation. The specimens were then chemically and mechanically dissociated into single-cell suspensions. Cells were plated onto 0.1% polyethyleneimine-coated plastic coverslips at a density of 1.3 × 105 cells/cm2. Cultures were maintained in Eagle's minimum essential medium (EMEM) containing 10% FCS [1–4 days in vitro (DIV)] or HS (5–12 DIV). Cultures were incubated at 37°C in a humidified atmosphere of 5% CO2 in air. Primary cultures were exposed to drugs at 8 DIV. The animals were treated in accordance with the guidelines of the Kyoto University Animal Experimentation Committee and Japanese Pharmacological Society.
Evaluation of viability of PC12 cells
Cell viability was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay and the lactate dehydrogenase (LDH) release assay. The activity of LDH released into the medium during exposure to drugs was measured using an LDH assay kit (Kyokuto Pharmaceutical Industrial, Tokyo, Japan). Accordingly, 10 μl of culture supernatant was mixed with 90 μl of the LDH substrate mixture contained in the assay kit. After incubation for 30 min at room temperature, absorbance was measured at 570 nm. For the MTT assay, the culture medium was replaced with serum-free DMEM containing 0.5 mg/ml MTT tetrazolium salt (Nacalai Tesque, Kyoto, Japan), and the cells were incubated at 37°C for 30 min. The medium was removed and the cells were solubilized into a 2-propanol solution. Then, absorbance was measured at 570 nm. The viability of the cells was expressed as a percentage of the absorbance measured in control cells.
Immunocytochemistry
Following fixation with 4% paraformaldehyde for 30 min, cells were incubated with 0.2% Triton X-100 for 15 min, with primary antibodies for 2 h, and with secondary antibodies for 1 h. For diaminobenzidine staining, cells were subsequently incubated with avidin-biotinylated horseradish peroxidase complex (Vector Laboratories Inc., Burlingame, CA) for 1 h and reacted with diaminobenzidine solution (Dojindo Laboratories, Kumamoto, Japan) for 8 min. For nuclear staining, cells were incubated with Hoechst 33258 (100 μg/ml; Life Technologies, Carlsbad, CA) for 15 min. The following antibodies were obtained commercially: rabbit antityrosine hydroxylase (TH) IgG from Millipore (Billerica, MA); mouse anti-TH IgG from Sigma; biotinylated antirabbit IgG (H + L) from Vector Laboratories Inc., and Alexa Fluor 488 goat antimouse IgG (H + L) from Life Technologies.
Evaluation of viability in dopaminergic neurons
The number of TH-immunoreactive cells was counted in 20 randomly selected fields (858 × 1287 μm2/field). Counts were performed by an observer blind to the experimental treatments. The viability of dopaminergic neurons was calculated as a percentage of the number of the neurons in sham-treated cultures.
Western blotting
Western blot analysis using cell lysates was performed as previously described (Izumi et al., 2009). In brief, whole-cell lysates were centrifuged at 17,000 × g for 30 min at 4°C. The protein contents of the supernatants were normalized, and proteins were separated on SDS-polyacrylamide gel, followed by transfer to a polyvinylidene fluoride membrane (Millipore). The membranes were probed with primary antibody [anti-TH (Sigma), anti-phospho-Ser TH (PhosphoSolutions, Aurora, CO), antiglyceraldehyde-3-phosphate dehydrogenase (GAPDH; Ambion, Austin, TX)] and with horseradish peroxidase-conjugated secondary antibody (GE Healthcare, Little Chalfont, UK) for 1 h. The membrane-bound secondary antibody was detected with an enhanced chemiluminescence detection system (GE Healthcare). The band intensities were analyzed with computer software, ImageJ 1.33u (National Institute for Health, Bethesda, MD).
Evaluation of cellular uptake and release of [3H]dopamine
Cells were incubated with Krebs-Ringer-HEPES buffer (125mM NaCl, 4.8mM KCl, 25mM HEPES, 1.2mM MgSO4, 1.2mM KH2PO4, 5.6mM glucose, 2.2mM CaCl2, 10μM pargyline, and 1mM ascorbate, pH7.4) containing 50nM [7,8-3H]dopamine (GE Healthcare) for 30 min. After the loading of [3H]dopamine, cells were rinsed four times with Krebs-Ringer-HEPES buffer to remove unbound radioactivity. To measure the amount of dopamine incorporated, cells were lysed with 0.4% Tween X-100. To measure the amount of dopamine released, cells were further incubated with Krebs-Ringer-HEPES buffer in the presence or absence of drug for 30 min at 37°C. The amount of radioactivity in the Tween X-100 solution or the Krebs-Ringer-HEPES buffer was measured with a liquid scintillation counter.
Measurement of vesicular transport of [3H]dopamine
Vesicular dopamine uptake was determined as described previously (Izumi et al., 2008). PC12 cells were washed twice with 10mM phosphate-buffered saline (pH 7.5) and harvested with potassium glutamate buffer (KG buffer; 150mM potassium glutamate, 20mM HEPES, 4mM EGTA, 1mM MgCl2, 10μM pargyline, and 1mM ascorbate, adjusted to pH 7.0 with KOH). This cell suspension was pelleted by centrifugation and resuspended in KG buffer supplemented with 10μM digitonin and incubated for 5 min at 37°C to selectively permeabilize the plasma membrane. Permeabilized cells were distributed into individual tubes and washed by adding 500 μl of ice-cold KG buffer and spun down (1000 g, 2 min, 4°C). Vesicular transport of dopamine was started by adding 100 μl of KG buffer containing 2mM Mg-ATP and 50nM [7,8-3H]dopamine in the presence or absence of drug. Incubation was performed for 30 min at 37°C and stopped by adding 500 μl of ice-cold KG buffer followed by a rapid centrifugation. The cell pellet was lysed in 0.4% Triton X-100 to measure radioactivity by scintillation counting. A nonspecific value was determined using 10μM of reserpine and subtracted from the experimentally determined values.
Measurement of dopamine content by high-performance liquid chromatography with electrochemical detection
Dopamine content was measured by high-performance liquid chromatography (HPLC) with electrochemical detection as described previously (Izumi et al., 2009) with a slight modification. For measurement of intracellular dopamine, cells were sonicated in extraction solution containing 0.1N perchloric acid, 10mM sodium disulfite, and 1mM EDTA. For measurement of cytosolic dopamine, cells were incubated with Krebs-Ringer-HEPES buffer containing digitonin for 15 min at 37°C, and the supernatant was collected for measurement. To measure vesicular dopamine, the digitonin-permeabilized cells were sonicated in the extraction solution. After centrifugation at 17,000 × g for 30 min, the supernatants were analyzed with the Prominence HPLC system (Shimazu Corporation, Kyoto, Japan). The HPLC system was equipped with an Eicompak SC-5ODS reverse phase column (Eicom, Kyoto, Japan; inside diameter 3.0 mm, length 150 mm) and a Coulochem III electrochemical detector (ESA Inc., Chelmsford, MA). The guard cell (ESA model 5020) was set at +500 mV, and the analytical cell (ESA model 5011) was set at +400 mV. The mobile phase consisted of 0.1M citrate/0.1M sodium acetate buffer (pH 2.6) containing 110 mg/l sodium octylsulfate, 5 mg/l EDTA-2Na, and 13% methanol (vol/vol), and the flow rate was set at 0.5 ml/min.
Measurement of TH activity
TH activity was determined by measuring the amount of l-dihydroxyphenylalanine (l-DOPA) accumulated by the presence of carbidopa, an inhibitor of aromatic l-amino acid decarboxylase, according to methods described previously (Lindgren et al., 2000) with a slight modification. Treated cells were incubated at 37°C for additional 1 h in serum-containing DMEM supplemented with or without carbidopa (200μM). The cells were sonicated in extraction solution containing 0.1N perchloric acid, 10mM sodium disulfite, and 1mM EDTA. After centrifugation at 17,000 × g for 30 min, supernatants were collected for measurement. The amount of l-DOPA was determined in the same way used for intracellular dopamine described above. The amount of l-DOPA accumulated by the presence of carbidopa was calculated by subtraction of the amount of l-DOPA in the presence and absence of carbidopa.
Measurement of aromatic l-amino acid decarboxylase activity
Aromatic l-amino acid decarboxylase (AADC) activity was measured by the method described by Ishikawa et al. (2009). AADC activity was determined by using l-DOPA as substrate and measuring the dopamine formed. Treated cells were lysed with a buffer containing 150mM NaCl, 1mM EDTA, 20mM Tris (pH 8.0), and 0.5% Nonidet P-40. After normalization of the protein contents, lysates were reacted with 10mM phosphate-buffered saline (PBS; pH 7.0) containing 250μM l-DOPA, 125μM EDTA, 1.25μM 2-mercaptoethanol, 1.25μM DTT, 12.5μM pyridoxal-5-phosphate, and 125μM pargyline (to prevent dopamine breakdown) at 37°C for 15 min, and the reaction was stopped by adding a solution containing 1N perchloric acid, 100mM sodium disulfite, and 10mM EDTA in one-tenth volume to the reaction mixture. After centrifugation at 17,000 × g for 30 min, supernatants were collected for measurement. Newly synthesized dopamine was analyzed by HPLC as described above.
Measurement of monoamine oxidase activity
Monoamine oxidase (MAO) activity was measured fluorimetrically using the Amplex Red Monoamine Oxidase Assay Kit (Life Technologies). MAO activity was determined by using p-tyramine as substrate and detecting hydrogen peroxide in a horseradish peroxidase-coupled reaction. In brief, treated cells were harvested into 50mM Tris-buffered saline (pH 7.5) and homogenized. After centrifugation at 1000 × g for 10 min to remove cellular debris, the protein contents of the supernatants were normalized. The supernatants were mixed in equal amounts with a working solution composed of amplex red (100μM), horseradish peroxidase (0.5 U/ml), and p-tyramine (2mM), and incubated at 37°C for 1 h. Fluorescence intensity was measured using a Wallac ARVO SXFL 1420 Multilabel Counter (Perkin Elmer Life Sciences, Boston, MA) at an excitation wavelength of 544 nm and an emission wavelength of 580 nm. Background fluorescence derived from the control without p-tyramine was subtracted from the experimentally determined values.
Measurement of catechol-O-methyltransferase activity
As described previously (Vieira-Coelho and Soares-da-Silva, 1996), catechol-O-methyltransferase (COMT) activity was determined by using adrenaline as substrate and measuring the metanephrine formed. Treated cells were harvested into 10mM PBS (pH 7.2) containing 500μM DTT, and homogenized. After centrifugation at 1000 × g for 10 min to remove cellular debris, the protein contents of the supernatants were normalized. The supernatants were mixed in equal amounts with PBS containing 200μM adrenaline, 200μM magnesium chloride, 200μM S-adenosylmethionine, and 200μM pargyline, and incubated at 37°C for 1 h. The reaction was stopped by adding a solution containing 1N perchloric acid, 100mM sodium disulfite, and 10mM EDTA in one-tenth volume to the reaction mixture. After centrifugation at 17,000 × g for 30 min, the supernatants were collected for measurement. Newly synthesized metanephrine was analyzed by HPLC with a mobile phase consisted of 0.1M citrate/0.1M sodium acetate buffer (pH 2.6) containing 110 mg/l sodium octylsulfate, 5 mg/l EDTA-2Na, and 3% methanol (vol/vol), as described above.
Measurement of quinoprotein content
Cells were lysed with PBS (pH 7.4) containing 10μM phenylmethylsulfonyl fluoride, 1% NP-40, and 0.5% sodium deoxycholate. After centrifugation at 17,000 × g for 15 min, the protein contents of the supernatants were normalized. For detection of quinoprotein, the nitroblue tetrazolium (NBT)/glycinate colorimetric assay was performed as described previously (Paz et al., 1991). In brief, the protein samples were added to NBT reagent (0.24mM NBT in 2M potassium glycinate, pH 10) followed by incubation in the dark for 30 min on a shaker. Absorbance was measured at 540 nm.
Statistics
The statistical significance of the difference between three or more groups of individual data was analyzed by one-way analysis of variance and post hoc multiple comparisons using Turkey's test, unless otherwise stated. Statistical significance was defined as p < 0.05. Data are expressed as mean ± SEM.
RESULTS
Delayed Cytotoxicity Induced by Paraquat in PC12 Cells
TH is a rate-limiting enzyme for catecholamine synthesis. TH was abundantly expressed in PC12 pheochromocytoma cells (Fig. 1A) . When PC12 cells were exposed to paraquat (10–100μM) for 48 h, a decrease in cell viability was observed at concentrations ≥30μM (Fig. 1B). In agreement with a previous report (Shimizu et al., 2003a), simultaneous treatment with GBR12909, an inhibitor of the dopamine transporter, suppressed paraquat-induced cytotoxicity (Fig. 1C). An intermediate concentration of paraquat (50μM) was selected, and time course of cytotoxicity was quantified with the MTT reduction assay. Reduction of cell viability was observed in a time-dependent manner following 36 h of exposure with paraquat (50μM). In other words, treatment for >24 h was required for paraquat at a concentration of 50μM to cause toxicity in PC12 cells (Fig. 1D). However, treatment with paraquat (50μM) for 6 or 12 h followed by incubation in drug-free medium for 42 or 36 h resulted in delayed cytotoxicity in PC12 cells (Fig. 1E). It is suggested that the alteration of cell function within 12 h of exposure with paraquat (50μM) is enough to cause the delayed-onset toxicity.
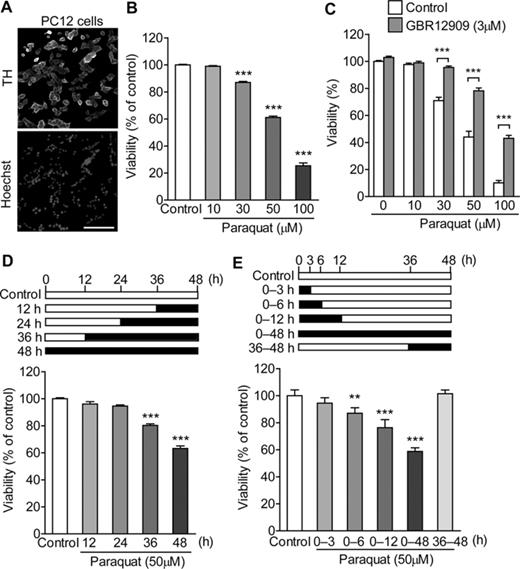
Effect of paraquat on cell viability in PC12 cells. (A) Representative micrographs of immunostained PC12 cells. PC12 cells were stained with an antibody to TH and all nuclei were stained with Hoechst 33258. Scale bar = 100 μm. (B) Concentration-dependency of paraquat-induced cytotoxicity in PC12 cells. PC12 cells were treated with paraquat (10–100μM) for 48 h. Cell viability was determined by the LDH release assay. (C) Effect of GBR12909 on paraquat-induced cytotoxicity in PC12 cells. PC12 cells were exposed to paraquat (10–100μM) in the presence or absence of GBR12909 (3μM) for 48 h. Cell viability was determined by the LDH release assay. Statistical analyses were performed using two-way ANOVA and post hoc multiple comparison using Bonferroni's test. ***p < 0.001. (D) Time-dependency of paraquat-induced cytotoxicity. PC12 cells were treated with 50μM paraquat for 12–48 h. The horizontal closed bar represents the period of treatment with paraquat. The horizontal open bar represents the period of incubation in drug-free medium. Cell viability was determined by the MTT assay at the 48-h time point. (E) Delayed cytotoxicity induced by paraquat. PC12 cells were treated with 50μM paraquat. In some cases, the cells were further incubated in drug-free medium. The horizontal closed bar represents the period of treatment with paraquat. The horizontal open bar represents the period of incubation in drug-free medium. Cell viability was determined by the MTT assay at the 48-h time point. **p < 0.01, ***p < 0.001 compared with control.
Involvement of Intracellular Dopamine in Paraquat-Induced Cytotoxicity
To examine whether endogenous dopamine participates in the vulnerability of dopaminergic cells to paraquat toxicity, α-MT, an inhibitor of TH, was used to decrease the intracellular dopamine content. Exposure of PC12 cells to α-MT (0.1–1mM) for 24 h reduced the level of intracellular dopamine in a concentration-dependent manner (Table 1). When α-MT (1mM) was applied from 24 h before and simultaneously with paraquat (10–100μM) for 48 h, paraquat-induced cytotoxicity was significantly abrogated (Fig. 2A). Pretreatment and simultaneous treatment with α-MT (0.1–1mM) concentration-dependently suppressed paraquat-induced toxicity, consistent with the reduction in intracellular dopamine content (Fig. 2B). Similar results were obtained with the MTT assay (data not shown). To confirm the involvement of endogenous dopamine in dopaminergic cell death, primary-cultured dopaminergic neurons were prepared from the rat fetal mesencephalon. As previously reported (Izumi et al., 2009), exposure of mesencephalic cultures to α-MT (0.1–1mM) for 24 h reduced the level of intracellular dopamine in a concentration-dependent manner. Paraquat-induced dopaminergic neuronal death was significantly attenuated by 24 h of pretreatment and coadministration of α-MT (0.1–1mM) with paraquat (Figs. 2C and D).
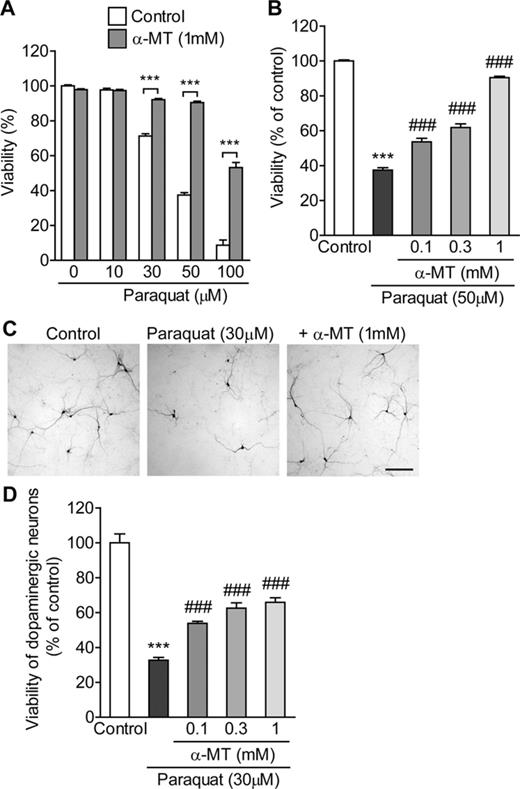
Involvement of intracellular dopamine in paraquat-induced cytotoxicity. (A) Effect of α-MT on paraquat-induced cytotoxicity in PC12 cells. PC12 cells were pretreated with α-MT (1mM) for 24 h and exposed to paraquat (10–100μM) with α-MT (1mM) for 48 h. Cell viability was determined by the LDH release assay. Statistical analyses were performed using two-way ANOVA and post hoc multiple comparison using Bonferroni's test. ***p < 0.001. (B) Concentration-dependency of α-MT-induced cytoprotection against paraquat toxicity in PC12 cells. PC12 cells were pretreated with α-MT (0.1–1mM) for 24 h and exposed to 50μM paraquat with α-MT (0.1–1mM) for 48 h. Cell viability was determined by the LDH release assay. (C and D) Effect of α-MT on paraquat-induced dopaminergic neurotoxicity in primary mesencephalic cultures. (C) Representative micrographs of dopaminergic neurons immunostained with anti-TH antibody. Scale bar = 200 μm. (D) α-MT (0.1–1mM) was added to mesencephalic cultures for 24 h before and 24 h during exposure to paraquat (30μM). The viability of dopaminergic neurons was determined by counting the number of TH-immunoreactive cells. ***p < 0.001 compared with control. ###p < 0.001 compared with paraquat alone.
Effect of α-MT on Dopamine Content in PC12 Cells
| Treatment | Dopamine content (%) |
|---|---|
| Control | 100.0 ± 13.4 |
| α-MT (0.1mM) | 51.6 ± 5.5** |
| α-MT (0.3mM) | 25.8 ± 1.4*** |
| α-MT (1mM) | 9.5 ± 1.0*** |
| Treatment | Dopamine content (%) |
|---|---|
| Control | 100.0 ± 13.4 |
| α-MT (0.1mM) | 51.6 ± 5.5** |
| α-MT (0.3mM) | 25.8 ± 1.4*** |
| α-MT (1mM) | 9.5 ± 1.0*** |
Note. PC12 cells were exposed to α-MT (0.1–1mM) for 24 h.
**p < 0.01 compared with control.
***p < 0.001 compared with control.
| Treatment | Dopamine content (%) |
|---|---|
| Control | 100.0 ± 13.4 |
| α-MT (0.1mM) | 51.6 ± 5.5** |
| α-MT (0.3mM) | 25.8 ± 1.4*** |
| α-MT (1mM) | 9.5 ± 1.0*** |
| Treatment | Dopamine content (%) |
|---|---|
| Control | 100.0 ± 13.4 |
| α-MT (0.1mM) | 51.6 ± 5.5** |
| α-MT (0.3mM) | 25.8 ± 1.4*** |
| α-MT (1mM) | 9.5 ± 1.0*** |
Note. PC12 cells were exposed to α-MT (0.1–1mM) for 24 h.
**p < 0.01 compared with control.
***p < 0.001 compared with control.
Elevation of Dopamine Content by Paraquat
Next, the influence of exposure to paraquat on intracellular dopamine content was examined in PC12 cells. The amount of intracellular dopamine was time-dependently increased by treatment with paraquat (50μM). The level of dopamine peaked at 9 h, then gradually decreased (Fig. 3A). Meanwhile, short-term exposure to paraquat (50μM) for 60 min or less had no effect on the amount of intracellular dopamine (Fig. 3B). Dopamine, when left unsequestered in the pH-neutral cytosolic compartment, is highly susceptible to oxidation to toxic dopamine-quinones and reactive oxygen species (Izumi et al., 2005). Therefore, distinguishing between cytosolic and vesicular dopamine is crucial for understanding the role of elevated dopamine content. For this purpose, cytosolic dopamine was extracted by mild, selective permeabilization of the plasma membrane with the steroid glycoside digitonin (Fiskum et al., 1980; Vergo et al., 2007). The amount of dopamine extracted from the extracellular milieu was increased by digitonin in a concentration-dependent manner (Fig. 3C). To find the optimal concentration of digitonin that would permeabilize the plasma membrane sufficiently and preclude the potential rupture of intracellular membranes, i.e., vesicles, we measured the activity of the cytosolic protein LDH in the extracellular milieu in response to different concentrations of digitonin (Fig. 3D). Complete disruption of the plasma membrane was obtained after application of 100 μg/ml digitonin. Because the release of LDH reached a plateau at 30 μg/ml, this concentration of digitonin was deemed optimal for extracting the cytosolic dopamine pool into the extracellular milieu. Eisenhofer et al. (2004) reported that most of the dopamine in dopaminergic neurons (∼90%) is packaged into synaptic vesicles by vesicular monoamine transporter 2, whereas ∼10% escapes sequestration and remains in the cytosol. Using this digitonin-based permeabilization technique, we found that exposure to paraquat (50μM) increased the levels of both cytosolic and vesicular dopamine (Fig. 3E). Sequestration of dopamine into the vesicles via the vesicular monoamine transporters (VMAT) could have protective effect by decreasing cytosolic dopamine levels. To block dopamine storage into the vesicles, reserpine, an inhibitor of VMAT, was applied simultaneously with paraquat. Simultaneous treatment with reserpine enhanced paraquat-induced cytotoxicity (Fig. 3F).
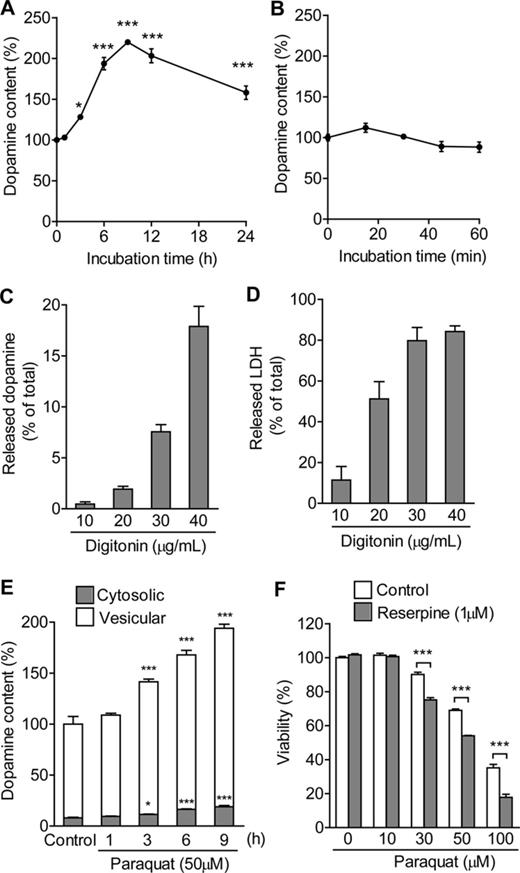
Effect of paraquat on intracellular dopamine. (A and B) Temporal changes in intracellular dopamine content in response to paraquat. PC12 cells were treated with 50μM paraquat for 1–24 h (A) or 15–60 min (B). (C) Release of dopamine into the extracellular milieu in response to digitonin. (D) Release of LDH into the extracellular milieu in response to digitonin. PC12 cells were incubated with 10–40 μg/ml digitonin for 15 min. After centrifugation, the supernatant was collected for measurement. (E) Time-dependent changes in cytosolic and vesicular dopamine content in response to paraquat. PC12 cells were treated with 50μM paraquat for 1–9 h. *p < 0.05, ***p < 0.001 compared with control. (F) Effect of reserpine on paraquat-induced cytotoxicity in PC12 cells. PC12 cells were exposed to paraquat (10–100μM) in the presence or absence of reserpine (1μM) for 48 h. Cell viability was determined by the LDH release assay. Statistical analyses were performed using two-way ANOVA and post hoc multiple comparison using Bonferroni's test. ***p < 0.001.
Elevation of TH Activity by Paraquat
One possible mechanism for the increase in dopamine content is elevation of dopamine synthesis. TH activity was increased early during exposure to paraquat, and then gradually decreased (Fig. 4A). Phosphorylation of TH at serine (Ser) residues, Ser19, Ser31 and Ser40, directly or indirectly increases its activity (Dunkley et al., 2004). However, exposure to paraquat did not change phosphorylation of TH at any time point during the 12 h of the experiment (Fig. 4B). AADC is another enzyme responsible for the synthesis of dopamine, but exposure to paraquat also failed to increase AADC activity (Fig. 4C). Enhancement of the uptake of extracellular dopamine and inhibition of the release of intracellular dopamine could contribute to the increase in dopamine content. Using 3H-labeled dopamine, we showed that paraquat had no effect on cellular uptake and release of dopamine (Figs. 4D and E). Besides, intracellular dopamine is predominantly stored in the vesicles, but paraquat did not affect vesicular dopamine transport in digitonin-permeabilized cells (Fig. 4F). Although inhibition of dopamine turnover may participate in the increase in dopamine content, the activity levels of MAO and COMT, which are predominant metabolic enzymes for intracellular dopamine degradation, were not altered by exposure to paraquat (Figs. 4G and H).
![Influence of paraquat on dopamine synthesis, metabolism, release, and uptake. (A) Effect of paraquat on TH activity. PC12 cells were treated with 50μM paraquat for 1–9 h. (B) Effect of paraquat on phosphorylation of TH. PC12 cells were treated with 50μM paraquat for 1–12 h. Levels of phosphorylated THs (p-THs) were normalized to those of GAPDH. n = 4. (C) Effect of paraquat on AADC activity. PC12 cells were treated with 50μM paraquat for 6 h. (D) Effect of paraquat on dopamine uptake. PC12 cells were incubated with Krebs-Ringer-HEPES buffer containing [3H]dopamine in the presence or absence of 50μM paraquat for 30 min. (E) Effect of paraquat on dopamine release. PC12 cells loaded with [3H]dopamine were treated with 50μM paraquat for 30 min. (F) Effect of paraquat on vesicular dopamine transport. Digitonin-permeabilized PC12 cells were loaded with [3H]dopamine in the presence or absence of 50μM paraquat for 30 min. (F) Effect of paraquat on MAO activity. PC12 cells were treated with 50μM paraquat for 1–12 h. (G) Effect of paraquat on COMT activity. PC12 cells were treated with 50μM paraquat for 6 h. **p < 0.01, ***p < 0.001 compared with control.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/toxsci/139/2/10.1093_toxsci_kfu054/1/m_kfu054fig4.jpeg?Expires=1716598714&Signature=uIgtvKBvaqOh0gLuaLDAxMH3bWhduwR-Hm8whethBZO4f0RR9pkmZlFgYVw97N4XZHjJi7u7KfWL~T4gV1IEDr3P0~Xnh0wZ480CnTOALjjEJIldoqAsGef9FYdygF6oxjvc6JeVr3aLuXUy0DSPNk3qrMZOMJ56-RbziiboL2bvmyy2QYReWZ3jEsNXOPeKbMvOfVwQ2NkrC3EptmN-Oy~WE4gAu87hH7JwdXL79t1clqPwEmQvHwB16w82-GRpuFG001Ca2rrHmzF6kRpmo7nH-BJSxAtUOKkE9xft0JkG2HIG1lCuSR5dlPFC6I9C35b0YKyrABDzlzypyeoj8Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Influence of paraquat on dopamine synthesis, metabolism, release, and uptake. (A) Effect of paraquat on TH activity. PC12 cells were treated with 50μM paraquat for 1–9 h. (B) Effect of paraquat on phosphorylation of TH. PC12 cells were treated with 50μM paraquat for 1–12 h. Levels of phosphorylated THs (p-THs) were normalized to those of GAPDH. n = 4. (C) Effect of paraquat on AADC activity. PC12 cells were treated with 50μM paraquat for 6 h. (D) Effect of paraquat on dopamine uptake. PC12 cells were incubated with Krebs-Ringer-HEPES buffer containing [3H]dopamine in the presence or absence of 50μM paraquat for 30 min. (E) Effect of paraquat on dopamine release. PC12 cells loaded with [3H]dopamine were treated with 50μM paraquat for 30 min. (F) Effect of paraquat on vesicular dopamine transport. Digitonin-permeabilized PC12 cells were loaded with [3H]dopamine in the presence or absence of 50μM paraquat for 30 min. (F) Effect of paraquat on MAO activity. PC12 cells were treated with 50μM paraquat for 1–12 h. (G) Effect of paraquat on COMT activity. PC12 cells were treated with 50μM paraquat for 6 h. **p < 0.01, ***p < 0.001 compared with control.
Enhancement of Quinoprotein Formation by Paraquat
Dopamine-quinones react with cellular nucleophiles such as cysteinyl residues of proteins to form covalently linked quinoprotein adducts (Asanuma et al., 2003). Formation of quinoprotein was increased early during exposure to paraquat, and then gradually decreased (Fig. 5A). Pretreatment with α-MT (1mM) not only decreased the basal level of quinoprotein, but also completely inhibited the elevation of quinoprotein formation caused by paraquat (Fig. 5B).
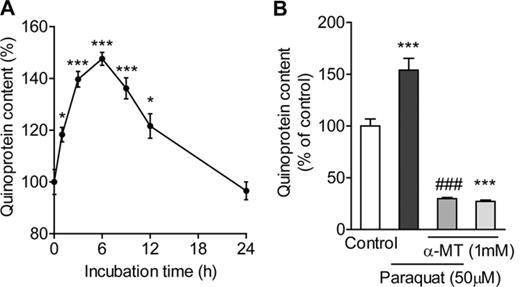
(A) Effect of paraquat on quinoprotein formation. PC12 cells were treated with 50μM paraquat for 1–24 h. (B) Effect of α-MT on paraquat-induced quinoprotein formation. PC12 cells were pretreated with 1mM α-MT for 24 h and exposed to 50μM paraquat with α-MT for 6 h. *p < 0.05, ***p < 0.001 compared with control. ###p < 0.001 compared with paraquat alone.
Suppression of Paraquat-Induced Alteration of Dopamine Systems by Ascorbic Acid
In order to modulate the cellular redox state, ascorbic acid was applied from 24 h before and simultaneously with paraquat. Relatively low concentrations (1–10μM) of ascorbic acid attenuated paraquat-induced cytotoxicity in a concentration-dependent manner (Fig. 6A). Under this condition, ascorbic acid also inhibited paraquat-induced elevation of intracellular dopamine and quinoprotein contents (Figs. 6C and E). On the other hand, trolox, a water-soluble analogue of α-tocopherol, provided limited protection against paraquat toxicity, and it had no effect on the paraquat-induced elevation of intracellular dopamine (Figs. 6B and D).
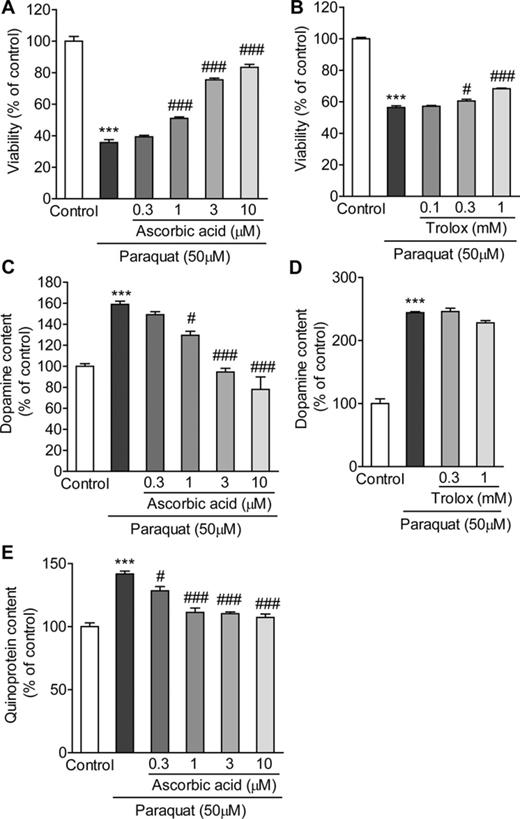
The effects of ascorbic acid and trolox on paraquat-induced alteration of dopamine systems. (A and B) The effects of ascorbic acid and trolox on paraquat-induced toxicity. PC12 cells were pretreated with 0.3–10μM ascorbic acid (A) or 0.1–1mM trolox (B) for 24 h, and exposed to 50μM paraquat with ascorbic acid or trolox for 48 h. Cell viability was determined by the LDH release assay. (C and D) The effects of ascorbic acid and trolox on the elevation of dopamine content induced by paraquat. PC12 cells were pretreated with 0.3–10μM ascorbic acid (C) or 0.3–1mM trolox (D) for 24 h, and exposed to 50μM paraquat with ascorbic acid or trolox for 9 h. (E) Effect of ascorbic acid on paraquat-induced quinoprotein formation. PC12 cells were pretreated with 0.3–10μM ascorbic acid for 24 h, and exposed to 50μM paraquat with ascorbic acid for 6 h. ***p < 0.001 compared with control. #p < 0.05, ###p < 0.001 compared with paraquat alone.
DISCUSSION
The results of this study indicate that endogenous dopamine plays an important role in paraquat-induced dopaminergic cell death. We found that paraquat increased the level of cytosolic dopamine accompanying the activation of TH, resulting in the formation of quinoprotein.
When intraperitoneally injected into mice, paraquat was reported to preferentially reduce the number of dopaminergic neurons in the substantia nigra pars compacta and noradrenergic neurons in the locus ceruleus (Fernagut et al., 2007; McCormack et al., 2002). In addition, although our previous study reported that the half-maximal cytotoxic concentration of paraquat in primary-cultured striatal neurons was not <100μM (Osakada et al., 2003), this study showed that 30μM paraquat was sufficient to induce neuronal death in primary-cultured dopaminergic neurons. Moreover, this study demonstrated that a decrease in intracellular dopamine content conferred resistance to paraquat toxicity on both PC12 cells and primary-cultured dopaminergic neurons. These findings suggest that endogenous catecholamine is involved in the vulnerability of catecholaminergic cells to paraquat toxicity.
Exposure to paraquat may affect dopamine systems. It was reported that extracellular dopamine efflux in the striatum was elevated 1h after administration of paraquat to animal models (Shimizu et al., 2003b). Elevated level of dopamine metabolite in the striatum was observed 24 h after the paraquat treatment (Ossowska et al., 2005a). In addition to these reports, we observed an elevation of intracellular dopamine content by paraquat in PC12 cells. We propose that this influence of paraquat on dopamine systems may be crucial for its toxic effect on dopaminergic cells. Our data suggest that increased TH activity is one of the cellular mechanisms underlying the increased intracellular dopamine content associated with paraquat. However, phosphorylation of TH was not related to the increased TH activity. For catalysis of TH, the redox state of iron in the active site must be ferrous, but molecular oxygen can oxidize ferrous iron to the inactive ferric form (Ramsey et al., 1996). The superoxide anion radical, as a one-electron reductant, is capable of donating one electron to reduce ferric iron to the ferrous state (Bus and Gibson, 1984). Therefore, it can be assumed that the superoxide anion radical via a redox cycling of paraquat reduce the iron in TH to the ferrous form, regenerating the active enzyme. In fact, tetrahydropterins, cofactors for TH, are capable of donating up to four electrons to reduce the ferric form to the ferrous form of the enzyme (Ramsey et al., 1996). In support of the idea that the superoxide anion radical derived from paraquat affected TH activity, ascorbic acid abrogated the paraquat-induced elevation of intracellular dopamine content. Interestingly, trolox, in contrast to ascorbic acid, failed to suppress the elevation of dopamine content. This apparent discrepancy may be explained by a lower efficiency of scavenging of the superoxide anion by trolox than by ascorbic acid (Taubert et al., 2003). Although further investigations will be necessary to confirm this hypothesis, the present study showed that exposure to paraquat increased TH activity. The elevation of TH activity gradually disappeared after 6–9 h of exposure to paraquat, followed by a return to the basal level of intracellular dopamine. The decline in TH activation may be explained by feedback inhibition by the elevated level of intracellular dopamine (Gordon et al., 2008).
It was been reported that subchronic administration of paraquat increased TH activity in the striatum (McCormack et al., 2002). Although a loss of nigral dopaminergic neurons was detected, paraquat did not have an effect on the levels of dopamine and TH protein in the commonly used regimens (McCormack et al., 2002; Ossowska et al., 2005b ). Up-regulation of TH activity in the striatum after paraquat injection is therefore thought to be a compensatory change to restore tissue levels of the neurotransmitter. In addition, we propose that up-regulation of TH activity itself after paraquat treatment may contribute to its toxic effect. This is because exposure of PC12 cells to paraquat increased TH activity before cell death. Besides, as described above, elevations of striatal dopamine efflux also occurred just after administration of paraquat (Shimizu et al., 2003b). However, one must keep in mind that PC12 cells have their limitations, although these cells have the machinery to synthesize, release, take up, and store catecholamines. Any result obtained with PC12 cells does not assure that the same finding will hold in in vivo models or in the human diseases. Therefore, any hypotheses arising from PC12 cell studies should be verified in primary neuronal cultures and in vivo models.
Cytosolic dopamine is highly susceptible to oxidation into toxic dopamine-quinones. Dopamine-quinones conjugate with cysteine residues of various functional proteins, including several key molecules involved in the pathogenesis of PD (e.g., TH, dopamine transporter, and parkin) to form quinoproteins, resulting in dysfunction of these proteins (Kuhn et al., 1999; LaVoie et al., 2005; Whitehead et al., 2001). Furthermore, dopamine oxidation inhibits mitochondrial respiration and induces mitochondrial permeability transition (Ben-Shachar et al., 1995; Berman and Hastings, 1999). Whereas quinoprotein formation returned to the basal level at 24 h after treatment, paraquat significantly induced cell death in PC12 cells from 36 h onward. Considering that treatment with paraquat for 6 or 12 h caused delayed cytotoxicity (Fig. 1E), it is suggested that quinoprotein formation induced by paraquat contributes to its toxic effect, even though quinoprotein formation was transient. Although the level of intracellular dopamine peaked at 9 h after exposure to paraquat, the level of quinoprotein peaked at 6 h. Because quinoprotein may be generated by a variety of quinone products, the amount of quinoprotein does not necessarily correlate with the level of dopamine-quinones. However, considering that α-MT almost completely deprived intracellular quinoprotein (Fig. 5B), the majority of quinoprotein in PC12 cells may be derived from dopamine-quinones. It is known that quinoprotein formation is suppressed by glutathione (Kuhn et al., 1999). Because our unpublished data showed that exposure of PC12 cells to paraquat caused a compensatory increase in intracellular glutathione in a time-dependent manner, the time courses of the levels of intracellular dopamine did not correspond with those of quinoprotein.
The results of this study suggest that the vulnerability of dopaminergic neurons to paraquat toxicity is attributable to endogenous dopamine-mediated stress. In previous reports (Izumi et al., 2009; Sakka et al., 2003; Xu et al.2002), dopaminergic neurons exhibited enhanced vulnerability to cell damage induced by PD-relevant toxicants and genes, such as rotenone, glutamate, and α-synuclein, which do not specifically act on dopaminergic neurons. Similarly to this study, these reports indicate that dopamine itself could serve as a susceptibility factor for dopaminergic neurons. Taken together, these findings imply that endogenous dopamine may be a key molecule for the selective vulnerability of dopaminergic neurons in the pathogenesis of PD. In addition, our findings demonstrate that the increase in TH activity caused by paraquat induced an elevation of cytosolic dopamine and subsequent quinoprotein formation. A better understanding of the quinoproteins, which are formed by paraquat, that are involved in dopaminergic neuronal death may provide insight into the pathogenic process of PD and provide targets for therapeutic intervention aimed at slowing the progression of PD.
FUNDING
This work was supported by JSPS KAKENHI (21790070, 23790083).
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments