





Abstract
Nucleotide polymorphism in Scots pine (Pinus sylvestris) was studied in the gene encoding phenylalanine ammonia-lyase (Pal, EC 4.3.1.5). Scots pine, like many other pine species, has a large current population size. The observed levels of inbreeding depression suggest that Scots pine may have a high mutation rate to deleterious alleles. Many Scots pine markers such as isozymes, RFLPs, and microsatellites are highly variable. These observations suggest that the levels of nucleotide variation should be higher than those in other plant species. A 2,045-bp fragment of the pal1 locus was sequenced from five megagametophytes each from a different individual from each of four populations, from northern and southern Finland, central Russia, and northern Spain. There were 12 segregating sites in the locus. The synonymous site overall nucleotide diversity was only 0.0049. In order to compare pal1 with other pine genes, sequence was obtained from two alleles of 11 other loci (total length 4,606 bp). For these, the synonymous nucleotide diversity was 0.0056. These estimates are lower than those from other plants. This is most likely because of a low mutation rate, as estimated from between–pine species synonymous site divergence. In other respects, Scots pine has the characteristics of a species with a large effective population. There was no linkage disequilibrium even between closely linked sites. This resulted in high haplotype diversity (14 different haplotypes among 20 sequences). This could also give rise to high per locus diversity at the protein level. Divergence between populations in the main range was low, whereas an isolated Spanish population had slightly lower diversity and higher divergence than the remaining populations.
Introduction
Mating system and life history have major influences on the patterns of genetic variation (Hamrick and Godt 1996 ). Such patterns are well known from studies of isozymes. Most data on nucleotide variation in plants concern annuals and short-lived perennials, partly cereals or other domesticated crops, or predominantly selfing species, such as Arabidopsis thaliana (Gaut and Clegg 1993 ; Innan et al. 1996 ; Huttley et al. 1997 ; Kawabe, Miyashita, and Terauchi 1997 ; Kawabe et al. 1997 ; Stephan and Langley 1998 ; Kawabe and Miyashita 1999 ; Small, Ryburn, and Wendel 1999 ). Only a few studies compare patterns of nucleotide diversity in plant species with different mating systems (Liu, Zhang, and Charlesworth 1998 ; Liu, Charlesworth, and Kreitman 1999 ; Savolainen et al. 2000 ). So far, outcrossing long-lived plants have not been studied for nucleotide variation.
In this study, we examine nucleotide variation in Scots pine (Pinus sylvestris L.). It is a long-lived predominantly outcrossing perennial with continuous distribution that ranges from Scotland to eastern Siberia and from northern Finland to Turkey (fig. 1 ). It has high variability at enzyme loci, RFLPs, and microsatellites (Gullberg et al. 1985 ; Muona and Harju 1989 ; Müller-Stark, Baradat, and Bergmann 1992; Karvonen and Savolainen 1993 ; Goncharenko, Silin, and Padutov 1994 ; Karhu et al. 1996 ). In 10 allozyme studies, the average heterozygosity values per population ranged from 0.25 to 0.39 for Scots pine (Müller-Stark, Baradat, and Bergmann 1992), among the highest for the European tree species compared. These values are high compared with many other groups, such as Drosophila allozyme heterozygosity with an average of 0.14. The differentiation between populations at marker loci is low within Scandinavia (FST = 0.02) (Karhu et al. 1996 ) and even between Scandinavia and the eastern part of the range (Wang, Szmidt, and Lindgren 1991 ). There is much recombination in the genome of pines: the genetic genome size of Pinus taeda is 1,700–1,800 cM, likely to be similar to that in many pines (Remington et al. 1999 ). No linkage disequilibrium was found between enzyme loci either in pollen grains or female gametes (Muona and Szmidt 1985 ). Scots pine has very high early and late inbreeding depression (Koski 1971 ; Kärkkäinen, Koski, and Savolainen 1996). In a partially selfing species, most deleterious alleles should be rapidly eliminated. The most reasonable way to account for high inbreeding depression in a partially selfing species is a high genome-wide mutation rate to deleterious alleles (Lande, Schemske, and Schultz 1994 ), and Scots pine seems to conform to this model (Koelewijn, Koski, and Savolainen 1999 ).
This background generates predictions with respect to nucleotide diversity. First, as the effective population size (Ne) and mutation rate (μ) (to deleterious genes) are known to be high, we could predict high levels of neutral nucleotide diversity, 𝛉 = 4Neμ (Kimura 1983, pp. 194–252 ). The earlier findings of enzyme gene variation also predict high diversity. Second, as the populations at molecular markers have only low differentiation, the differentiation at the nucleotide level could be low as well, if both are predominantly governed by gene flow and drift.
However, earlier data do not allow us to make a well-justified prediction on the range of linkage disequilibrium in the genome. The level of disequilibrium between neutral genes is governed by the product 4Nec, where c is the recombination rate between loci (Hill and Robertson 1968 ). Close linkage or inbreeding will restrict effective recombination. The larger this product, the less disequilibrium we expect between neutral loci. The current population size is very large and there is much recombination between genes, but we might still observe effects of historical bottlenecks between closely linked nucleotide sites. The current population structure of Scots pine in Europe appears to have been formed mainly by the postglacial migration of the species from refugia located in southwestern, southern, and southeastern parts of its present range. This expansion took place about 7,000–9,000 years ago (Hyvärinen 1987 ). Scots pine mitochondrial DNA variants provide further evidence for multiple origins after the last glaciation from the south: through western Europe via France, Germany, and Denmark and from the northeast via Finland (Sinclair, Morman, and Ennos 1999 ; Soranzo et al. 2000 ). Are we able to detect traces of the glacial history at the nucleotide level, or has ample gene flow because of pollen migration (Koski 1970 ) and abundant recombination already eliminated these traces? Are the populations at mutation drift equilibrium, or do they still show traces of the possible glacial bottlenecks, in disequilibria between closely linked nucleotide sites or in frequency spectra that are predicted in expanding populations (Harpending 1994 ).
We studied nucleotide polymorphism in the gene encoding phenylalanine ammonia-lyase (Pal, EC 4.3.1.5), a key enzyme in the secondary metabolism of higher plants catalyzing the nonoxidative conversion of l-phenylalanine into transcinnamate, the initial substrate of the phenylpropanoid pathway in wood formation. In Scots pine, Pal is believed to be related to ozone tolerance (Rosemann, Heller, and Sandermann Jr. 1991 ), defense against pathogens (Lange et al. 1994 ), and metabolism of exogenous compounds (Laukkanen and Sarjala 1997 ). cDNA sequences of pal have been reported for a number of angiosperms (e.g., Yamada et al. 1992 ). In angiosperms this gene usually consists of two exons and one intron. In gymnosperms, pal was first reported from loblolly pine, P. taeda, as a single-copy gene with no introns (Whetten and Sederoff 1992 ). Later, Butland, Chow, and Ellis (1998) sequenced a 366-bp–long conserved region of pal from Pinus banksiana and discovered that pal is actually a multigene family of at least 8–10 loci.
Our results first show that Scots pine has rather low nucleotide diversity at synonymous sites. Following this finding, we examined whether other aspects of the nucleotide data are consistent with the predictions for a species with a large effective population size.
Materials and Methods
Plant Materials
Seeds of Scots pine, P. sylvestris, were obtained from four natural populations: two Finnish (Bromarv and Kolari), one Russian (Kirov), and one Spanish (Puebla de Lillo in the province of Leon) (fig. 1 ). Seeds were stored at 4°C. For DNA extraction, the seeds were kept on paper towels moistened with distilled water for 4–5 h at room temperature to make tissue preparation easier. Only haploid tissue (megagametophyte) of the seed was used for DNA isolation. Five megagametophytes from different trees from each population were analyzed. We also obtained two sequences from 11 other loci. The two alleles were from two widely separated trees in southern Finland.
DNA Isolation, Amplification, and Sequencing
Total genomic DNA from the megagametophytes was purified either by CTAB method (Doyle and Doyle 1990 ) or by FastDNA® Kit (BIO101). On the basis of the P. taedapal-gene nucleotide sequence (one exon of 2,265 bp, GenBank U39792), we designed PCR-primers that would amplify the pal1 gene in two overlapping parts in P. sylvestris. To amplify the other P. sylvestris genes used in this study, we designed primers on the basis of P. sylvestris and P. taeda gene sequences from GenBank (table 1 ). Short fragments were directly sequenced, first using AmpliTaq Gold (PE Applied Biosystems) for the PCR. The PCR products were separated in 1% agarose gels (0.5 TBE), from which they were purified by centrifugation through aquarium filter fibers. Direct sequencing was done from both strands with BigDye-kit (PE Applied Biosystems). For cloning of longer parts of the genes, PCR was done with Dynazyme EXT (with proofreading property, Finnzyme). The amplification products were first cloned with the TOPO TA cloning kit (version J, Invitrogen) and then sequenced (BigDye, PE Applied Biosystems). The PCR-program for amplification of the pal1 gene reactions with AmpliTaq Gold-enzyme was: 94°C 12 min; 94°C 1.10 min, 53°C 1 min, 72°C 1.50 min, 30 cycles; 72°C 10 min, and for Dynazyme-enzyme: 94°C 4 min; 94°C 1.10 min, 53°C 1 min, 72°C 1 min, 30 cycles; 72°C 10 min. Three cloned products were sequenced in order to avoid errors made by the DNA polymerase. If nucleotide differences between the three clones were detected, then the majority nucleotide was taken to be the correct one in that position. In both sequencing methods, we used several sequencing primers designed on the basis of the P. taeda sequence and located every 300–400 bp. Newly determined P. sylvestris sequences are deposited in the GenBank database and the accession numbers are listed in table 1 .
Data Analyses
We obtained estimates of the scaled mutation parameter, 𝛉, based on nucleotide diversity, π (Nei and Li 1979 ) and on the proportion of segregating sites, S (Watterson 1975 ). Differences between populations were measured by FST, estimated with the analysis of molecular variance (AMOVA) (Excoffier, Smouse, and Quattro 1992 ) as implemented in the Arlequin 2.000 software (Schneider, Roessli, and Excoffier 2000 ). We also estimated the number of recombinations (Hudson 1987 ) and the linkage disequilibrium. The degree of codon bias was measured by the effective number of codons (ENC; Wright 1990 ). The ENC values can range from 20 (strong codon bias) to 61 (no codon bias). Unless otherwise stated, we used DnaSP (Rozas and Rozas 1999 ) for all analyses.
Results
Structure and Divergence of the pal1 Locus of P. sylvestris
The pal1 locus in Scots pine is similar to the pal1 locus of jack pine, P. banksiana (Butland, Chow, and Ellis 1998 ), and to the pal reported in loblolly pine, P. taeda (Whetten and Sederoff 1992 ). In all these species, the pal1 locus is represented by a single exon without introns. Within the region studied, there were no insertions or deletions. There were, on an average, only 12.2 nonsynonymous substitutions between the species (Ka nonsynonymous = 0.0079).
Among the sequenced 2,045 sites in 20 alleles, there were 12 segregating sites (table 2 ). Nine of the polymorphisms were synonymous and three were nonsynonymous. A transition of C to T at site 243 resulted in a change of alanine to valine in two individuals of the Kolari population. In the Kirov population, a transversion of T to G at site 762 resulted in a change of valine into glycine in one individual, and a transversion of A to T at site 915 alters histidine to leucine. These amino acid changes are considered to be conservative for protein function (Graur and Li 2000 , p. 20).
The species-wide estimate of synonymous diversity was 0.0049. For comparison we obtained nucleotide sequences from 11 other pine genes (table 1 ). The average synonymous nucleotide diversity at these loci was 0.0056, with a range of 0.0000–0.0232. The comparable nucleotide diversity at the pal1 locus in the Bromarv population in southern Finland was 0.0056 (table 3 ). Thus, based on these very preliminary comparisons, pal1 seems representative of other genes as well.
Within-population Nucleotide Diversity
Scots pine has a low level of within-population nucleotide diversity in the pal1 locus (table 3 ). For synonymous sites, the mean within-population nucleotide diversity πs was 0.0045, with the highest value found in the populations from Bromarv and Kirov, 0.0056, and the lowest one in the Spanish population from Puebla de Lillo, 0.0024. At nonsynonymous sites, polymorphism was only detected in two of the populations, Kolari and Kirov, at a very low level (πa = 0.0004 and 0.0005, respectively) with an overall average of populations of 0.0002. The lowest total nucleotide polymorphism value was observed in the Spanish population (π = 0.0006), and the highest was detected in the Russian population (π = 0.0018). The three populations from the main range of Scots pine distribution (Bromarv, Kolari, and Kirov) had significantly higher levels of total nucleotide variation than the geographically isolated population of Puebla de Lillo from Spain (P < 0.05, t-test).
All polymorphic sites had only two alternative nucleotides, and four of the sites were singletons. As the populations showed so little divergence, we combined them into one set for Tajima's test. A significant negative Tajima's test could suggest either purifying selection or recent population expansion. However, the value of −0.56 was not statistically significantly different from zero.
Differentiation Between Populations
The genetic differentiation between the populations as estimated with AMOVA (table 4 ) was very low. If the population from Spain was excluded, the value of FST was only 0.017, but if it was included in the calculations, the FST increased to 0.110. Statistically significant pairwise differentiation was found only between the most distant populations from Puebla de Lillo and Kirov (FST = 0.310).
Linkage Disequilibrium and Recombination
As the level of differentiation was so low, we analyzed the total set of 20 sequences jointly for linkage disequilibrium and for recombination parameter R (Hudson 1987 ). There was significant pairwise correlation by chi-square test between two pairs of sites (112 and 915, 139 and 762), but this is what is to be expected by chance, and after Bonferroni correction there was no evidence for linkage disequilibrium. Because of the low sample sizes the power of test is low, but table 2 shows no haplotype structure. The high recombination observed in the locus results in high haplotype diversity. A total of 14 haplotypes were detected among the 20 sequences analyzed. The overall haplotype diversity, H (Nei 1987, pp. 259–260 ), was 0.95 ± 0.03. The smallest number of the haplotypes, two, occurred in the Spanish population from Lillo. The other populations had five haplotypes each.
Discussion
Low Level of Within-population DNA Variability in Scots Pine
Our data do not support the predictions of high nucleotide polymorphism in Scots pine. Species-wide silent nucleotide diversity, πs, was 0.0049. Table 5 shows that this is lower than the diversity in the short-lived plants, e.g., in A. thaliana, Leavenworthia stylosa, or Zea mays. Some plants do have lower estimates of diversity, such as Arabidopsis lyrata at Adh, or Dioscorea tokoro (Terauchi, Terachi, and Miyashita 1997 ). It is higher than the value in humans (Cargill et al. 1999 ) but lower than that in Drosophila melanogaster and Drosophila simulans (Moriyama and Powell 1996 ). The organisms with the longest generation times, pines and humans, have the lowest nucleotide diversity. Our estimate of synonymous nucleotide diversity in P. sylvestris at 11 other loci, with just two sequences for each locus was πs = 0.0056 (table 5 ). Thus, the pal1 gene is not exceptional among genomic areas of P. sylvestris.
However, the level of replacement nucleotide polymorphism in the pal1 locus (πa = 0.0003) was about seven times lower than in the other genes of Scots pine (πa = 0.0022) and equal to the πa in humans (table 5 ). This suggests a very high functional constraint on the protein.
Our results on rather low diversity could be because of several factors that we can examine. First, the effective population size (at least for this locus) could be lower than that assumed. Second, the mutation rate could be lower than that suggested by the inbreeding depression data. Third, the synonymous sites may in fact not be fully neutral, but under directional selection for major codons, e.g., for reasons of translational efficiency (see Akashi 1997 ). Further, the low variability could be due to selective forces at other loci that reduce levels of variation, such as hitchhiking (Maynard Smith and Haigh 1974 ; Kaplan, Hudson, and Langley 1989 ), background selection (Charlesworth, Morgan, and Charlesworth 1993 ), or to pseudohitchhiking due to alternating direction of selection in large populations (Gillespie 2000 ). The high generation time and short time after glaciation bottlenecks may also mean that nucleotide polymorphism is not at equilibrium, even if our small data set did not show evidence of this (see below).
Large Effective Population Size and Mutation Drift Equilibrium
Many aspects of the data are consistent with a large effective population size. The differentiation between populations separated by thousands of kilometers was very low (e.g., Kolari in northern Finland and Kirov in Russia). Even the currently isolated Lillo was not much diverged. Further, there was no evidence of linkage disequilibrium in the data set, even between the very closely linked polymorphic sites. Linkage disequilibrium at neutral sites is governed by 1/(4Nec), and as c between closely linked sites is very small, Ne must be large to account for the lack of disequilibrium.
We also examined the data for signs of departure from demographic equilibrium, i.e., of postglacial expansion from several refugia, as explained earlier. A recent expansion could show up as an excess of singletons, which can be measured by Tajima's D. The value of −0.56 for the nuclear loci did not provide evidence for recent expansion. However, there are still local traces of possible refugia. The small Spanish populations may be remnants of a larger distribution. The observed lower level of DNA polymorphism in the Spanish population than in the Russian and Finnish populations agree with the previous data obtained with allozymes. The Spanish populations of Scots pine are slightly less variable than the populations from the continuous range of the distribution, with He of 0.325 and 0.363, respectively (Prus-Glowacki and Stephan 1994 ). In all, the data on the Scots pine main range are consistent with a large effective population size and mutation drift equilibrium.
High Mutation Rate in Pines?
Because P. sylvestris does not have higher nucleotide diversity compared with other species, we need to examine again the hypothesis of a high mutation rate in pines. As the mutation rate governs the rate of neutral substitution between species, the synonymous divergence between species (Ks) provides an estimate of the mutation rate. Thus, μ = Ks/2T, where T is the time since divergence.
The best estimate of divergence time, based on fossil evidence, for P. sylvestris and P. taeda is 120 Myr (Millar 1998 ), and the rate of synonymous substitutions is 0.15 × 10−9 per year for pal1 and about 0.05 × 10−9 per year for Adh (table 6 ). The pal1 and the Adh genes have similar rates of neutral nucleotide substitutions, and both appear to evolve much more slowly in pines than in the monocots or dicots. Lu, Szmidt, and Wang (1998) have observed a low number of substitutions between Gingko biloba and Larix sibirica at the nuclear coxI locus (based on their data, Ks = 0.016). This Ks estimate suggests an even lower mutation rate in those lineages than in pines because Gingko and Larix have diverged before the pines (about 200 MYA). A similar low rate (0.24 × 10−9) has also been found in the chloroplast gene rbcL of the chestnut tree (family Fagaceae), another group of long-lived woody perennials (Frascaria et al. 1993 ). As shown in table 6 , the estimates of angiosperm mutation rates are higher by an order of magnitude. Note that even if we use a generation time of 25 years for pine, the per generation mutation rate estimates are not higher than in other plants in either pal or Adh.Lande, Schemske, and Schultz (1994) have suggested that mutation rates in pines must be much higher than those measured in annual plants to account for the levels of inbreeding depression that pines maintain. It seems that the observed high inbreeding depression in Scots pine fits the model by Lande, Schemske, and Schultz (1994) quite well (Koelewijn, Koski, and Savolainen 1999 ). Mutation rates per generation at nuclear genes leading to chlorophyll deficiency have also been estimated to be about 10 times as high in pines and other woody plants as in annuals (Kärkkäinen, Koski, and Savolainen 1996; Klekowski 1998 ). Thus, the genome-wide per generation mutation rate to deleterious alleles gives a very different picture from the neutral mutation rate per site per year. This difference merits further study.
High Diversity at Many Marker Loci in Pines
Several measures of population diversity show pines to be quite variable. Scots pine has high levels of morphological, isozyme, RFLP, and microsatellite polymorphism (e.g., Karhu et al. 1996 ). Also, finding variable markers for mapping has been easy in many pine species (e.g., Sewell, Sherman, and Neale 1999 ). Enzyme gene markers have been used in several studies to quantify genetic variation at the species level in Scots pine (see review by Müller-Stark, Baradat, and Bergmann 1992). For example, in a study of 10 enzyme loci (2–5 alleles per locus) heterozygosity was between 0.075 and 0.482, with mean heterozygosity of 0.322 (Muona, Harju, and Kärkkäinen 1988 ). In 10 studies, the average heterozygosity values per population ranged from 0.25 to 0.39 for Scots pine (Müller-Stark, Baradat, and Bergmann 1992). How can the low nucleotide polymorphism at pal1 be reconciled with the previous data? First, RFLPs measure both nucleotide polymorphism and length variation. Large direct repeats seem to be widespread in introns of conifer genes, at least at the Adh gene (Perry and Furnier 1996 ). We have also observed that pine genes often have indel variation, sometimes involving these repeats. These could lead to the high RFLP diversity. For microsatellites and random amplified polymorphic DNA (RAPDs), high mutation rates at repetitive areas of the genome occur in many organisms. Allozyme diversity is based on the number of alleles that give rise to electrophoretically different proteins. If we examine the pal1 data, we find that the low per nucleotide diversity results in a high haplotype diversity of 0.95. Much of this diversity is because of silent sites. If we just take into account the three nonsynonymous sites (243, 762, and 915), we observe five different haplotypes, with frequencies 0.8, 0.1, 0.05, and 0.05, with results in expected per locus heterozygosity of 0.35. Thus, it is possible that at least a part of the high locus-level diversity in pines is because of the lack of within-locus disequilibrium. This suggestion, of course, needs to be further studied at a larger number of loci.
Table 5 shows that Dr. melanogaster, like Scots pine, also has high enzyme variation compared with other species, but low nucleotide diversity compared with Dr. simulans. Enough comparisons are not yet available to account for these differences. We cannot exclude the possibility that the high variability at allozyme loci is partly maintained by selection, and thus not directly comparable with the patterns at presumably neutral sites.
Are Synonymous Sites Neutral: Codon Usage in P. sylvestris
Mutations occurring in the third positions of codons should be selectively neutral because they do not cause amino acid changes (Kimura 1968, 1983 ). However, unequal use of synonymous codons points to selection at these sites also. Selection may favor some synonymous codons over the others, especially in highly expressed genes. There is a negative correlation between synonymous substitution rate and codon usage bias in many species (e.g., Sharp and Li 1986, 1989 ). Selection for alternative codons is expected to be rather weak, and thus efficient only in large populations. Drosophila simulans has high nucleotide diversity and codon bias, and Dr. melanogaster has lower diversity and less codon bias (see table 5 ). This is presumably because of the larger effective population size of Dr. simulans (Akashi 1997 ). A further point of evidence for the large effective population size of Scots pine could be found by examining codon bias. As Scots pine has very large populations, even weak selection could be efficient. Table 5 shows a comparison of codon bias in Scots pine and P. radiata, a species with very limited population size and less genetic diversity than Scots pine (Moran, Bell, and Elridge 1988 ). There is no evidence for increased codon bias in Scots pine relative to P. radiata, and thus at least not for the kind of selection at third positions because of major codon bias in large populations.
Selection at Linked Loci
Selection at linked loci has proved to have a major influence on patterns of diversity, especially in areas of low recombination (Aguadé, Miyashita, and Langley 1989 ). This could be because of selective sweeps (Kaplan, Hudson, and Langley 1989 ), background selection (Charlesworth, Morgan, and Charlesworth 1993 ), or alternating selection pressures (pseudohitchhiking) (Gillespie 2000 ). These selective explanations require that there be linkage disequilibrium between the neutral site and the target of selection, and then make additional assumptions. We do not yet know anything about the distribution of the rate of recombination in the P. sylvestris genome. Scots pine has high levels of deleterious mutations (as required by background selection), selection at many loci is efficient in large populations (as required by hitchhiking), and the direction of selection at these many loci can vary (as assumed in the pseudohitchhiking model). However, the similarity of the nucleotide diversity at pal1 gene to the 11 other loci and the low level of disequilibrium within the gene suggest that disequilibrium will also be low between the locus itself and adjacent areas. Thus, selection at linked loci is not the main cause for the low diversity. More genomic areas need to be studied.
Wolfgang Stephan, Reviewing Editor
Present address: Osteoporosis Research Center, Omaha, Nebraska, USA.
Keywords: nucleotide polymorphism SNP Pinus sylvestris codon bias linkage disequilibrium
Address for correspondence and reprints: Outi Savolainen, Department of Biology, University of Oulu, P.O. Box 3000, FIN-90014 Oulu, Finland. outi.savolainen@oulu.fi .
Table 1 The Genes and Primers Used and the GenBank Accession Numbers of the Sequences Determined in this Study
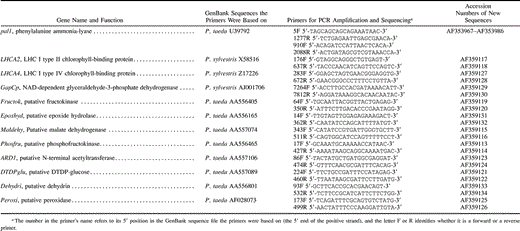
Table 1 The Genes and Primers Used and the GenBank Accession Numbers of the Sequences Determined in this Study
Table 2 Summary of Nucleotide Polymorphism in the Phenylalanine Ammonia-lyase (pal1) Locus in P. sylvestris
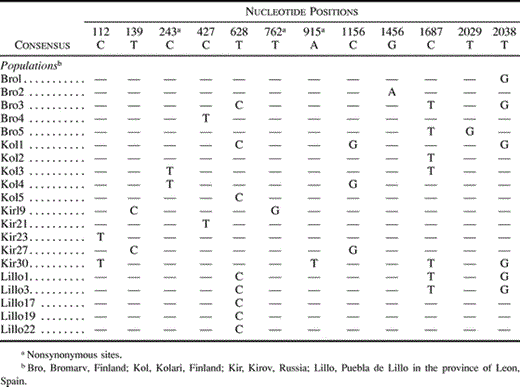
Table 2 Summary of Nucleotide Polymorphism in the Phenylalanine Ammonia-lyase (pal1) Locus in P. sylvestris
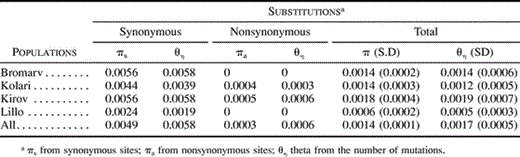

Table 5 Allozyme Heterozygosity, Nucleotide Polymorphism, and Codon Bias (measured as effective number of codons, ENC)
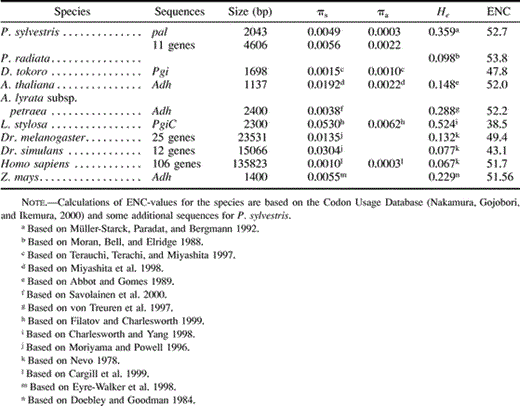
Table 5 Allozyme Heterozygosity, Nucleotide Polymorphism, and Codon Bias (measured as effective number of codons, ENC)
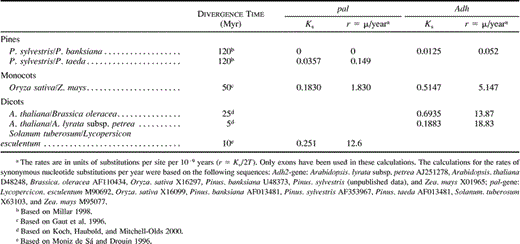
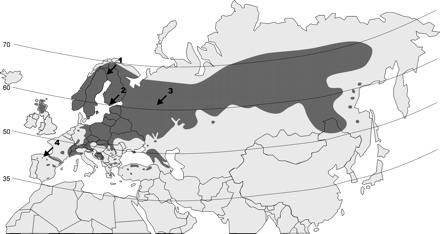
Fig. 1.—The distribution of P. sylvestris natural populations has been marked in gray. The numbers indicate the locations sampled in this study: 1, Kolari (Finland); 2, Bromarv (Finland); 3, Kirov (Russia); 4, Puebla de Lillo (Spain)
We gratefully acknowledge the financial support from the Research Council for Environment and Natural resources and the Center for International Mobility (CIMO). The Finnish Forest Research Institute, Dr. Igor Yakovlev, Mari-El State Technical University, Yoshkar-Ola, Russia, and Dr. Ricardo Alia, INIA (Instituto National de Investigación Agropecuaria) kindly provided the seed samples. We thank Dr. Helmi Kuittinen and Prof. Pekka Pamilo for comments on the manuscript.
References
Abbot R. J., M. F. Gomes,
Aguadé M., N. T. Miyashita, C. H. Langley,
Akashi H.,
Butland S. L., M. L. Chow, B. E. Ellis,
Cargill M., D. Altshuler, J. Ireland, et al. (17 co-authors)
Charlesworth B., M. T. Morgan, D. Charlesworth,
Charlesworth D., Z. Yang,
Excoffier L., P. E. Smouse, J. M. Quattro,
Eyre-Walker A., R. B. Gaut, H. Hilton, D. L. Feldman, B. S. Gaut,
Filatov D. A., D. Charlesworth,
Frascaria N., L. Maggia, M. Michaud, J. Bousquet,
Gaut B. S., M. T. Clegg,
Gaut B. S., B. R. Morton, B. C. McCaig, M. T. Clegg,
Gillespie J. H.,
Goncharenko G. G., A. E. Silin, V. E. Padutov,
Graur D., W.-H. Li,
Gullberg U., R. Yazdani, D. Rudin, N. Ryman,
Hamrick J. L., M. J. W. Godt,
Harpending H. C.,
Hill W. G., A. Robertson,
Hudson R. R.,
Huttley G. A., M. L. Durbin, D. E. Glover, M. T. Clegg,
Hyvärinen H.,
Innan H., F. Tajima, R. Terauchi, N. T. Miyashita,
Kaplan N. L., R. R. Hudson, C. H. Langley,
Karhu A., P. Hurme, M. Karjalainen, P. Karvonen, K. Kärkkäinen, D. Neale, O. Savolainen,
Kärkkäinen K., V. Koski, D. Savolainen,
Karvonen P., O. Savolainen,
Kawabe A., H. Innan, R. Terauchi, N. T. Miyashita,
Kawabe A., N. T. Miyashita,
Kawabe A., N. T. Miyashita, R. Terauchi,
Kimura M.,
Kimura M.,
Koch M. A., B. Haubold, T. Mitchell-Olds,
Koelewijn H. P., V. Koski, O. Savolainen,
Koski V.,
Lande R., D. W. Schemske, S. T. Schultz,
Lange B. M., M. Trost, W. Heller, C. Langebartels, H. Sandermann Jr.,
Laukkanen H., T. Sarjala,
Liu F., D. Charlesworth, M. Kreitman,
Liu F., L. Zhang, D. Charlesworth,
Lu M. Z., A. E. Szmidt, X.-R. Wang,
Millar C. I.,
Miyashita N. T., A. Kawabe, H. Innan, R. Terauchi,
Moniz de Sá M., G. Drouin,
Moran G. F., J. C. Bell, K. Elridge,
Moriyama E. N., J. R. Powell,
Müller-Starck G., P. H. Baradat, F. Bergmann,
Muona O., A. Harju,
Muona O., A. Harju, K. Kärkkäinen,
Muona O., A. E. Szmidt,
Nakamura Y., T. Gojobori, T. Ikemura,
Nei M., W.-H. Li,
Nevo E.,
Perry D. J., G. R. Furnier,
Prus-Glowacki W., B. R. Stephan,
Remington D. L., R. W. Whetten, B. H. Liu, D. M. O' Malley,
Rosemann D., W. Heller, H. Sandermann Jr.,
Rozas J., Rozas R.,
Savolainen O., C. H. Langley, B. P. Lazzaro, H. Freville,
Schneider S., D. Roessli, L. Excoffier,
Sewell M. M., B. K. Sherman, D. B. Neale,
Sharp P. M., W.-H. Li,
Sharp P. M., W.-H. Li,
Sinclair W. T., J. D. Morman, R. A. Ennos,
Small R. L., J. A. Ryburn, J. F. Wendel,
Soranzo N., R. Alia, J. Provan, W. Powell,
Stephan W., C. H. Langley,
Terauchi R., T. Terachi, N. T. Miyashita,
von Treuren R., H. Kuittinen, K. Kärkkäinen, E. Baena-Gonzalez, O. Savolainen,
Wang X. R., A. E. Szmidt, D. Lindgren,
Watterson G. A.,
Whetten R. W., R. R. Sederoff,

{kind=link}