





Abstract
A known phylogeny was generated using a four-step serial bifurcate PCR method. The ancestor sequence (SSU rDNA) evolved in vitro for 280 nested PCR cycles, and the resulting 15 ancestor and 16 terminal sequences (2,238 bp each) were determined. Parsimony, distance, and maximum likelihood analysis of the terminal sequences reconstructed the topology of the real phylogeny and branch lengths accurately. Divergence dates and ancestor sequences were estimated with very small error, particularly at the base of the phylogeny, mostly due to insertion and deletion changes. The substitution patterns along the known phylogeny are not described by reversible models, and accordingly, the probability substitution matrix, based on the observed substitutions from ancestor to terminal nodes along the known phylogeny, was calculated. This approach is an extension of previous studies using bacteriophage serial propagation, because here mutations were allowed to occur neutrally rather than by addition of a mutagenic agent, which produced biased mutational changes. These results provide for the first time biochemical experimental support for phylogenies, divergence date estimates, and an irreversible substitution model based on neutrally evolving DNA sequences. The substitution preferences observed here (A to G and T to C) are consistent with the high G+C content of the Thermus aquaticus genome. This suggests, at least in part, that the method here described, which explores the high Taq DNA polymerase error rate, simulates the evolution of a DNA segment in a thermophilic organism. These organisms include the bacterial rod T. aquaticus and several Archaea, and thus, the method and data set described here may well contribute new insights about the genome evolution of these organisms.
Introduction
Experimental phylogenetics is a convincing means of understanding basic processes of nucleotide change in phylogenies (Hillis, Mable, and Moritz 1996 ). Hillis and collaborators evolved the bacteriophage T7 by sequential propagation and generated a known phylogeny which provided for the first time experimental support for phylogeny inference methods (Hillis et al. 1992 ; Bull et al. 1993 ). Gene phylogenies can be inferred from sequence data using several algorithms under three basic optimality criteria, namely, parsimony (Fitch 1977 ), pairwise distances (Saitou and Nei 1987 ), and maximum likelihood (Felsenstein 1981 ). Among these, the use of explicit models of sequence evolution, or maximum likelihood, which is computationally very intensive, has been increasing recently because of improvements in computer hardware speed and software optimization (Olsen et al. 1994 ; Strimmer and von Haeseler 1996 ; Korber et al. 2000 ). Gene phylogenies may represent species phylogeny if the substitutions in a particular gene represent orthologous steps (Li and Graur 1991 ). Divergence dates of genes and species can also be estimated from phylogenetic distances (Rambaut and Bromham 1998 ; Yoder and Yang 2000 ). These estimates are based on the concept of a molecular clock (Zuckerkandl and Pauling 1962 ), either global or local, which can be tested for a set of sequences, models, and trees, using relative rate tests (Sarich and Wilson 1973 ), triplets rate test (Tajima 1993 ), and the likelihood ratio test (Felsenstein 1988 ).
In the maximum likelihood method for phylogeny inference, the explicit model of nucleotide substitution (the state transition matrix) is of primary importance (Felsenstein 1981 ; Posada and Crandall 1998 ). This varies from the single parameter Jukes and Cantor model to the general time reversible model (Jukes and Cantor 1969 ; Rodriguez et al. 1990 ). However, these models assume reversible matrices; in other words, they assume that the probability of the forward change over time (e.g., A to G) is equal to the probability of the reverse event (G to A). Other models, based on irreversible matrices, were proposed, but require the position of the phylogeny root to be inferred by maximizing the likelihood (Yang and Roberts 1995 ; Galtier and Gouy 1998 ; Schadt, Sinsheimer, and Lange 1998 ; Galtier, Tourasse, and Gouy 1999 ).
Neutral substitutions are very interesting for phylogenetic purposes, although their use for divergence date estimates and phylogeny inference has not yet been tested explicitly by experimental phylogenetics. In the experimental phylogeny of bacteriophage T7, the accelerated rate of change was induced by the presence of a mutagenic agent which changed not only the tempo, but also the mode of evolution, because the mutagenic agent likely biased substitutions from G to A and C to T (Bull et al. 1993 ). Also, in the T7 sequential propagation, lineages that replicate faster or more effectively tend to be overrepresented compared with slower replicating lineages, and therefore the system is not neutral (Hillis et al. 1992 ). Neutral substitutions, with proper corrections for superimposed mutations and stochastic variations, likely reflect the elapsed time, and therefore history, since the divergence of two sequences (Li and Graur 1991 ). Substitutions in sites with selective pressure mostly reflect the selection regime and thus may cause convergent substitutions, under negative selection, or anomalously long phylogeny branches, under positive selection. Accordingly, divergence dates could be underestimated or overestimated, respectively.
Here we present a method, based on serial PCR, that extends previous studies (Hillis et al. 1992 ; Bull et al. 1993 ) by generation of a data set of neutrally evolving sequences. Analysis of this data set provided experimental support for maximum likelihood, with modelfit analysis, for finding the correct topology, reconstruction of ancestor sequences, and divergence date estimates. This data set was also used to calculate a probability transition matrix which describes an irreversible dynamics, based on Taq DNA polymerase error rate, and should contribute to further research on coalescence theory (Kingman 1982 ), phylogeny inference methods, and evolution of thermophilic organisms (Galtier, Tourasse, and Gouy 1999 ).
Materials and Methods
Gene Amplification and Sequencing
The ancestor sequence (Trypanosoma cruzi SSU rDNA, GenBank AF288660) was used as template in a 35 cycle PCR using primers RIBA (5′-CCGAATTCGTCGACAACCTGGTTGATCCTGCCA GT-3′) and RIBB (5′-CCCGGGATCCAAGCTTGATCCTTCTGCAGGTTCACCTAC-3′) which enables the amplification of the complete SSU rDNA sequence. The amplification started with amplification of 0.35 μg of cloned ancestor sequence (0.102 pmol, 6.14 × 1010 molecules) in a 100-μl solution containing 5 units of Taq DNA pol (GIBCO), 7 mM MgCl2, 40 nmol each deoxynucleotide triphosphates (dNTP), 20 mM tris(hydroxymethyl)aminomethane (Tris)–HCl pH 8.4, 50 mM KCl, and 10 pmol of each primer. After 35 cycles amplicons were purified from gel (GFX, Amersham-Pharmacia) serially diluted 1:1,000, and 4 μl of the diluted solution were used as template for an additional 35 cycles in 100-μl solutions containing 5 units of Taq DNA pol (GIBCO), 7 mM MgCl2, 40 nmol each dNTP, 20 mM Tris–HCl p H8.4, 50 mM KCl, and 10 pmol of each primer (fig. 1 ). Cycling conditions were 94°C for 1 min, 41°C for 1 min, and 72°C for 2 min, and final extension 72°C for 7 min. After each 70 nested cycles the amplicons were cloned into pBluescript, and two randomly selected clones were picked to be the ancestor of another 70 cycles nested for a total of four rounds of 70 nested cycles. This procedure was repeated four times, resulting in four rounds of 70 nested PCR cycles. Amplification of cloned products was done using M13 forward and reverse primers. The nested reaction has 280 cycles, and all 16 terminal sequences coalesce to ancestor sequence 1.1 (fig. 2 ). The 70 cycles from each round were divided into two 35 nested cycles, because at 35 cycles the reaction is close to its linear stage. All 31 sequences derived from the process (16 terminal nodes plus 15 ancestors), along with six additional clones of the first 70 cycles, were determined completely, in a total of 37 complete SSU rDNA sequences (total of 82,806 assembled bases). Sequencing was done using BigDye Terminators (Applied Biosystems) in an ABI377/96 automated sequencer, by primer walking using internal primers of the 18S rRNA gene. Sequences were assembled using PHRED+PHRAP+CONSED (Gordon, Abajian, and Green 1998 ) from ABI chromatograms, available upon request. Quality of assembled sequences ranged from phred scores 30 to 40 as estimated by CONSED (Gordon, Abajian, and Green 1998 ). Sequences presented here have been deposited in GenBank under accession numbers AF288660 (ancestor 1.1) and AF359461 to AF359496 (from 2.1 to T16). Supplementary data, such as alignments and phylogenies, are available on the World Wide Web site (http://compbio.epm.br/ievol) of one of us.
Phylogenetic Analysis
Terminal sequences were aligned by eye using SEAVIEW sequence editor (Galtier, Gouy, and Gautier 1996 ). Trees were constructed using PAUP 4.0b6 (Swofford 1998 ) and TREE-PUZZLE (Strimmer and von Haeseler 1996 ), and modelfit tests were performed using the hierarchical likelihood ratio test implemented in MODELTEST 3.04 (Posada and Crandall 1998 ). Maximum likelihood trees were constructed using the model selected by MODELTEST (TVMef [Posada and Crandall 1998] with equal base frequencies; the rate matrix A–C = 0.4397, A–G = 13.2362, A–T = 4.9778, C–G = 0.2123, C–T = 13.2362, and G–T = 1.0; proportion of invariable sites = 0; and equal rates for all sites) with heuristic tree-bisection–reconnection (TBR) search. Parsimony (Fitch 1977 ) trees were built using accelerated transformation (ACCTRAN) and TBR searching with collapse option. Distance trees were constructed using Neighbor-Joining (Saitou and Nei 1987 ) with a maximum likelihood distance matrix using the model selected by MODELTEST. The standard errors of branch lengths were estimated using PAUP 4.0b6 (Swofford 1998 ). The number of molecules after 280 cycles was calculated by quantitation of templates and PCR products by absorbance at 260 nm, applying corresponding corrections for dilutions, and conversion to number of molecules using Avogadro's number. Divergence dates with low and high confidence intervals and associated evolutionary rates were estimated using the quartet analysis implemented in QDATE version 1.11 (Bromham et al. 1998 ; Rambaut and Bromham 1998 ). The number of substitutions of each rate category was directly quantitated from polymorphic sites along the real phylogeny.
Model Construction
The instantaneous rate matrix (Q-matrix) was built from the observed number of changes of each of 16 categories, in the real topology. Each element Qij was calculated by dividing the observed number of changes from nucleotide i to nucleotide j by the total number of changes. The diagonal elements Qii were calculated by using the relation Qii = −Σj≠i Qij. The Q-matrix was then written as in table 1 .
The substitution probability matrix (P-matrix) was calculated from the relation P(t) = eQt (Swofford et al. 1996 ). In order to find the elements of the P-matrix we had to decompose the Q-matrix into its eigenvalues and eigenvectors. The eigenvalues were obtained by calculating the λ values (vn) that satisfied the equation det(Q − λI) = 0 (where I is the identity matrix), namely, v1 = −0.5187, v2 = − 0.4218, v3 = − 0.0595, and v4 = −5.7958 × 10−6. For each λ value the respective eigenvector (Vλ) was determined as that satisfying the equation (Q − λI)Vλ = 0. These vectors are: V1 = (−0.6060,−0.1701, 0.1669, 0.7589)T; V2 = (0.5025, −0.2510, −0.2801, 0.7785)T; V3 = (0.5243, −0.5155, 0.6207,−0.2722)T; and V4 = (−0.4999, −0.5001, −0.4998, −0.5000)T. The Pij(t) elements in the P-matrix were calculated using standard transformation procedures of the Q-matrix, using the previously calculated eigenvalues and eigenvectors. All mathematical calculations were confirmed using MATHEMATICA software (Wolfram Research).
Results and Discussion
Here we wanted to simulate neutral evolution and therefore we present a model which explores the high error rate of Taq DNA polymerase (Saiki et al. 1988 ) (fig. 1 ). A known SSU rRNA gene sequence was used as the ancestor to start the process of sequential PCR evolution (fig. 1 ). Every 70 cycles the PCR products were cloned, and two clones were completely sequenced and used as templates (ancestors) for the next round of 70 cycles (fig. 1 ). Accordingly, the ancestor 1.1 originated 16 terminal sequences or terminal nodes, T1 to T16, after 280 PCR nested cycles (generations) (fig. 1 ). The full-length sequences of the 16 terminal nodes and the 15 internal nodes (2.1 to 4.8, fig. 3A ), or ancestors, were determined. The alignment of all 37 sequences used here revealed 196 polymorphic sites, including gaps, and the alignment of the 16 terminal sequences had 169 polymorphic sites (fig. 2 ). After PCR evolution of 280 generations (fig. 1 ), the total number of molecules, 9.92 × 1011, was estimated from PCR product quantitation. Sequences 2.3 to 2.8 were used only to calculate the mutation rate of Taq DNA polymerase after 70 PCR cycles, as described later.
The real phylogeny obtained (fig. 3A ) has a series of 15 dichotomies from the initial ancestor to the 16 terminal sequences, and substitutions that occurred along the phylogeny involved 7.6% of the total number of positions (table 1 ). We observed that the number of substitutions per time interval (along a branch length) varies significantly. This might be due to stochastic effects once lineages are sampled and propagated randomly. The 16 terminal sequences (T1 to T16) were used to reconstruct the real phylogeny by maximum likelihood, parsimony, and neighbor-joining (Fitch 1977 ; Felsenstein 1981 ; Saitou and Nei 1987 ). Topologies obtained by the three methods were identical and found the real topology. In fig. 3B we show the reconstructed maximum likelihood phylogeny which has a topology identical to the true tree (fig. 3A ). This phylogeny has ln(Likelihood) = −4259.7384 and was inferred using a submodel of the General Time Reversible Model (TVMef) with equal rates for all sites, with parameter determination by hierarchical likelihood ratio tests (Rodriguez et al. 1990 ; Posada and Crandall 1998 ). The best parsimony tree found had 82 informative sites, 157 steps, rescaled consistency index of 0.962, homoplasy index of 0.038, and ln(Likelihood) = −4259.7384, but was not statistically different from the six other parsimony trees with up to 160 steps, as tested by the Kishino–Hasegawa topology test (Kishino and Hasegawa 1989 ). The maximum likelihood tree differs from the real tree by 30% (9 out of 30) with respect to branch lengths, particularly in clusters where the number of substitutions where higher (fig. 3 ). However, the differences in branch lengths between the known phylogeny (fig. 3A ) and the inferred phylogeny (fig. 3B ) are within the range of the maximum likelihood standard error for branch lengths, as estimated in the inferred tree (fig. 3B ), and therefore, these differences were expected by the inference method. In addition to MODELTEST, optimization of model parameters was also done with simultaneous estimation of tree searching. For this, an initial topology was inferred by neighbor-joining with simultaneous estimation of nucleotide frequencies and the rate matrix (R-matrix). This initial topology was then rearranged by TBR with simultaneous estimation of the gamma distribution rate parameter and the proportion of invariant sites, and reestimation of nucleotide frequencies and the R-matrix. This approach is as close as possible, in the scope of the present study, to escape from point estimates of model parameters, as done by MODELTEST, and to letting parameters behave freely during tree searching. The tree obtained by this heuristic full optimization (ln[Likelihood] = −4254.74116) found that the real topology (fig. 3A ) and the parameters were very similar to those obtained by MODELTEST, except in the proportion of invariant sites, which in the full optimization was 0.43. At least in the case of the sequences presented here, MODELTEST gave results similar to those of the heuristic full optimization. A complete full optimization would have to be done using either an exhaustive search of all trees (more than 2 × 1014 trees for 16 taxa) or an exact algorithm, such as branch-and-bound, with simultaneous parameter optimization, and this would take an impractical computational time.
To verify the evolutionary rate of the real phylogeny (Taq DNA polymerase error) we used eight sequences, 2.1 to 2.8 (fig. 2 ), of the first 70 nested cycles. Among 2,238 positions, we observed 44 polymorphic sites, being 2 insertions, 4 deletions, and 48 base substitutions. The average change from the ancestor 1.1 to sequences 2.1 to 2.8 (fig. 2 ) is six misincorporations (excluding indels), which gives 0.0027 errors per position and an evolutionary rate of 0.38 × 10−4 substitutions per site per generation. This is higher than other estimates for Taq DNA polymerase (0.27 × 10−4) (Bracho, Moya, and Barrio 1998 ) and might be because of the concentration of MgCl2 (7 mM) used in our study, in order to accelerate the substitution rate. The substitutions at terminal sequences are evenly distributed along the SSU rRNA sequence length (table 1 ) which demonstrates that we generated a neutral evolution simulation. Therefore, selection is not responsible for the branch length variation observed in the real tree (fig. 3A ).
We also tested if divergence times could be estimated from inferred phylogenies. The 16 terminal sequences were analyzed by a maximum likelihood quartet method (Rambaut and Bromham 1998 ), using the same substitution model and parameters used to infer the maximum likelihood phylogeny (fig. 3B ). All terminal pairs diverged 70 cycles earlier and were clustered into quartets to infer the divergence of internal nodes. Divergence times of ancestors 1.1, 2.1, 2.2, 3.2, and 3.4 were estimated correctly, and ancestors 3.1 and 3.3 had dates outside the 95% confidence interval. The inferred tree, however, passes the likelihood ratio test for molecular clocks (Felsenstein 1988 ) as the difference between the likelihoods of the molecular clock constrained tree and the unconstrained tree is not statistically significant at 95% confidence level (2 × ΔLnL = 21.2818, and the Chi-square critical value for 14 degrees of freedom at P < 0.05 is 23.685). The maximum likelihood quartet analysis also estimated the evolutionary rate between 0.24 × 10−4 and 0.42 × 10−4 substitutions per site per generation.
Ancestor sequences 1.1 to 4.8 were also predicted from the maximum likelihood tree (hypothetical ancestors, HA) and compared with ancestors from the real phylogeny (fig. 4 ). Ancestors of the real phylogeny were reconstructed with accuracy from 99.46% to 99.87% (3 to 12 differences) by maximum likelihood. Most of the inaccurately assigned ancestor states were in regions with insertions and deletions and in two positions with two substitutions at the same site, positions 101 and 637 (fig. 4 and table 1 ). The reconstruction with the most errors (12) was ancestor 1.1, and the most accurate reconstructions were from ancestors 3.4, 4.7, and 4.8 (fig. 4 ). This suggests that as we move deeper in the tree, ancestor sequence reconstructions might be more sensitive to insertion and deletion events and multiple substitutions at the same site. Insertions and deletions occurred much more frequently after runs of homopolymeric regions, as observed elsewhere, and might be caused by the slippage of Taq DNA polymerase (Bracho, Moya, and Barrio 1998 ).
The substitution model selected by the hierarchical likelihood ratio test was compared with the actual changes (table 2 ). It shows that the changes in the real tree follow an irreversible model, particularly when A > G-G > A, A > T-T > A, and C > T-T > C changes are compared. Base frequencies in the data set (16 terminal sequences) are f(A) = 0.24938; f(C) = 0.22548; f(G) = 0.26574; and f(T) = 0.25941; among constant sites they are (0.24374, 0.23225, 0.27171, 0.25230) and among variable sites (0.32895, 0.12972, 0.18143, 0.35990). This suggests that the direction of change is not biased toward more abundant residues in the nucleotide pool.

To construct the substitution probability matrix, the P-matrix (table 3 ; table 2 , in italics), the Q-matrix was decomposed into its eigenvalues and eigenvectors and used in calculations as described in Materials and Methods. The resulting P-matrix (table 3 ) is a stochastic Markov model (Swofford et al. 1996 ) which is also irreversible, as are other models proposed (Yang and Roberts 1995 ; Galtier and Gouy 1998 ; Schadt, Sinsheimer, and Lange 1998 ; Galtier, Tourasse, and Gouy 1999 ). Another variation in the models is regarding their homogeneity along the phylogeny (Galtier and Gouy 1998 ; Galtier, Tourasse, and Gouy 1999 ). To verify whether the model is homogeneous or nonhomogeneous along the known phylogeny (fig. 3A ), we compared the rate matrices (R-matrix) after 280 cycles with the rate matrix after 70 cycles (based on sequences 2.1 to 2.8). The R-matrix at 70 cycles (not shown) is nearly identical to the R-matrix after 280 generations (table 2 ) which suggests that the model of sequence evolution is homogeneous along the know phylogeny in fig. 3A.
The experimental approach to phylogeny inference by a biochemical assay, as presented here, is advantageous over plain computer simulations, because the introduction of variations in sequences along the phylogeny is driven by errors of an enzymatic reaction, which is a better approximation of the real event. The process of substitution along a phylogeny is not understood in its full extent, and therefore, it is very unlikely that a computer simulation will include all the variations and nuances underlying this process. Consequently, computer simulations will be dependent on the particular parameters and bias that the programmer chooses to include.
The results presented here, using a biochemical assay, show that the phylogeny topology was reconstructed correctly, even with differences between the observed model and the maximum likelihood modelfit selected model (table 2 ). However, inference of branch lengths, divergence dates, and hypothetical ancestors are more sensitive to stochastic rate variations which should be corrected by using a more accurate model. Also, the variation in branch lengths, meaning variation in evolutionary rates, observed in the true tree (fig. 3A ) occurred in the absence of selection. Interestingly, the molecular clock is not rejected by the likelihood ratio test of the inferred phylogeny (fig. 3B ) (Felsenstein 1988 ), which suggests that even with considerable variation of rates among lineages, at least to the extent shown here, divergence date estimates from phylogenies are accurate (Bromham et al. 1998 ; Rambaut and Bromham 1998 ). We employed a strategy analogous to the one used by Hillis et al. (1992) . However, in our study the number of molecules at each stage and the exact number of generations (PCR cycles) are known. Because we simulated neutral evolution of an SSU rDNA, changes in the secondary structure of the gene product did not interfere with our in vitro evolutionary process. The SSU rRNA in vivo has a functionally conserved secondary structure, and most substitutions tend to occur within regions of unpaired loops (Hillis and Dixon 1991 ).
The serial PCR method described here could be used in studies of evolution of thermophilic organisms. The substitution bias observed here (A > G and T > C, table 1 ) is consistent with the high G + C content of the Thermus aquaticus genome, and might be a consequence of specific properties of Taq DNA polymerase. However, the polymerase domains of Taq DNA polymerase and the Klenow fragment of Escherichia coli DNA polymerase I are nearly identical. In Taq DNA polymerase, two of the catalytically critical carboxylate residues on the 3′–5′ exonuclease activity are missing (Kim et al. 1995 ). The simulation described here might well reflect the neutral evolution of a DNA segment in a thermophilic organism, such as in the bacterial rod T. aquaticus and in several Archaea, which implies that the method presented here could be developed to address questions about the genome evolution of these organisms (Woese 1987 ; Pace 1997 ).
As a perspective, the serial PCR evolution method and the data set presented here can contribute to future studies on coalescence theory (Kingman 1982 ) and to divergence date estimate methodology (Rambaut and Bromham 1998 ; Yoder and Yang 2000 ), because the number of generations, the phylogeny, and the mutation rate per generation are known. The in vitro generation of a phylogeny with no selection and no migration should be particularly useful for estimating the 𝛉 parameter (Kuhner, Yamato, and Felsenstein 1995 ). Nevertheless, this study provides for the first time biochemical experimental support for phylogeny inference from neutral substitutions using maximum likelihood with modelfit optimization.
Wolfgang Stephan, Reviewing Editor
Keywords: molecular evolution experimental phylogenetics SSU rRNA gene maximum likelihood
Address for correspondence and reprints: Marcelo R. S. Briones, Departamento de Microbiologia, Imunologia e Parasitologia, Escola Paulista de Medicina, Universidade Federal de São Paulo, Rua Botucatu, 862, 3° andar. CEP 04023–062, São Paulo, S.P. Brazil. marcelo@ecb.epm.br .
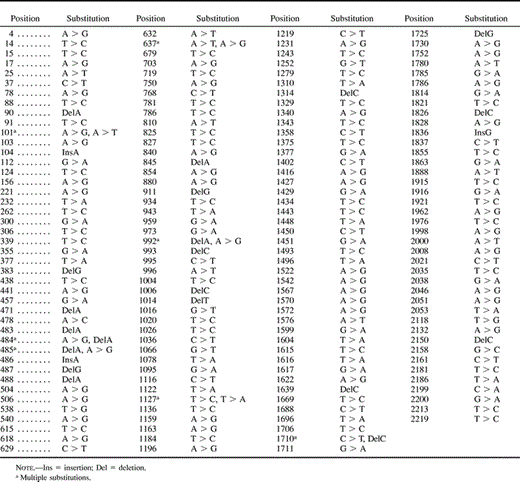
Table 2 Rate-Matrix Selected by Modelfit Analysis (reversible) Compared with the Rate-Matrix Observed from Real Data (in parenthesis)
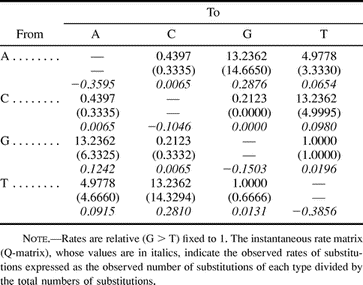
Table 2 Rate-Matrix Selected by Modelfit Analysis (reversible) Compared with the Rate-Matrix Observed from Real Data (in parenthesis)
Table 3 Substitution Probability Matrix Elements of the P-matrix, Derived from Observed Substitutions Along the Real Phylogeny (fig. 3A)
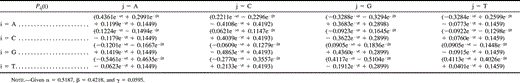
Table 3 Substitution Probability Matrix Elements of the P-matrix, Derived from Observed Substitutions Along the Real Phylogeny (fig. 3A)
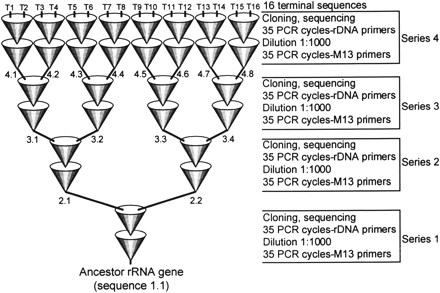
Fig. 1.—Evolution of DNA sequences by a series of bifurcate PCRs. An ancestor SSU rDNA cloned in pBluescript was used as template for series 1 of 70 nested PCR cycles with M13 primers. After the initial 35 cycles, reaction products were diluted 1:1,000 and used as templates for the subsequent 35 cycles, with rDNA primers RIBA and RIBB. After 70 cycles amplicons were cloned, and two clones were picked randomly and used as templates for the next series of nested PCR cycles. Lineages are propagated at random, and therefore the evolution is neutral and behaves as a stochastic process. Tree nodes T1 to T16 indicate terminal sequences, and 1.1 to 4.8, internal ancestors

Fig. 2.—Polymorphic sites of sequences generated by serial PCR neutral evolution. Sequences 1.1 to 4.8 represent the internal ancestors, and T1 to T16, the terminal sequences. Sequences 2.3 to 2.8 were not used in subsequent propagation, except to estimate the Taq DNA polymerase error rate at 70 cycles. Numbers above sequences indicate the position number in the alignment (total number of positions considered is 2,238). Dots indicate residues that are identical to sequence 1.1
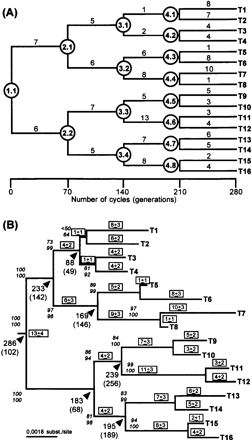
Fig. 3.—Comparison of real phylogeny with inferred maximum likelihood phylogeny (Felsenstein 1981 ; Posada and Crandall 1998 ; Swofford 1998 ). The serial PCR in vitro evolution resulted in the topology depicted (A) with varying branch lengths whose ancestors (1.1 to 4.8, circled) and terminal sequences (T1 to T16) were sequenced in full length. Scale bar indicates the number of cycles between tree internodes and nodes. The inferred phylogeny (B) has a topology identical to the real tree (A) and 9 out of 30 branch lengths were estimated correctly. Boxed numbers indicate branch lengths (number of substitutions), numbers in italics represent the percentage of a given cluster in 100 bootstrap replicates, with reestimation of parameters at each bootstrap replicate (top), and without reestimation at each replicate (bottom). Numbers below arrows indicate the estimated divergence (cycles ago), with the low–high confidence interval range (in parenthesis) as calculated by maximum likelihood quartet analysis (Rambaut and Bromham 1998 ). Numbers of substitutions, with corresponding standard errors, in the inferred tree (B) were calculated by multiplying the branch lengths (in substitutions per site) by the total number of positions (2,238 bp)
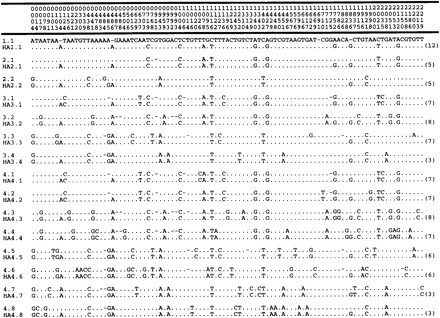
Fig. 4.—Comparison between ancestor sequences in the real phylogeny (fig. 3A ) and maximum likelihood reconstruction of ancestral states, HA. Only polymorphic positions are shown. Dots indicate residues identical to sequence 1.1. Numbers above the alignment indicate the position numbers. Numbers in parenthesis indicate the differences between the HA and their corresponding real ancestor sequences
We thank J. F. Perez, Scientific Director of Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for sequencing equipment made available to us through the ONSA Brazilian sequencing network, and the excellent suggestions from the two anonymous referees who reviewed this manuscript. G.F.O.S. and S.Y.K. received graduate and undergraduate fellowships, respectively, from CNPq (Brazil). This work was supported by grants to M.R.S.B. from FAPESP and CNPq (Brazil), and the International Research Scholars Program of the Howard Hughes Medical Institute (United States).
References
Bracho M. A., A. Moya, E. Barrio,
Bromham L., A. Rambaut, R. Fortey, A. Cooper, D. Penny,
Bull J. J., C. W. Cunningham, I. J. Molineux, M. R. Badgett, D. M. Hillis,
Felsenstein J.,
———.
Galtier N., M. Gouy,
Galtier N., M. Gouy, C. Gautier,
Galtier N., N. Tourasse, M. Gouy,
Gordon D., C. Abajian, P. Green,
Hillis D. M., J. J. Bull, M. E. White, M. R. Badget, I. J. Molineux,
Hillis D. M., M. T. Dixon,
Hillis D. M., B. K. Mable, C. Moritz,
Jukes T. H., C. R. Cantor,
Kim Y., S. H. Eom, J. Wang, D.-S. Lee, S. W. Suh, T. A. Steitz,
Kishino H., M. Hasegawa,
Korber B., M. Muldoon, J. Theiler, F. Gao, R. Gupta, A. Lapedes, B. H. Hahn, S. Wolinksy, T. Bhattacharya,
Kuhner M. K., J. Yamato, J. Felsenstein,
Olsen G. J., H. Matsuda, R. Hagstrom, R. Overbeek,
Posada D., K. A. Crandall,
Rambaut A., L. Bromham,
Rodriguez F., J. F. Oliver, A. Marin, J. R. Medina,
Saiki R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis, H. A. Erlich,
Saitou N., M. Nei,
Sarich V. M., A. C. Wilson,
Schadt E. E., J. S. Sinsheimer, K. Lange,
Strimmer K., A. von Haeseler,
Swofford D. L.,
Swofford D. L., G. J. Olsen, P. J. Waddell, D. M. Hillis,
Tajima F.,
Yang Z., D. Roberts,
Yoder A. D., Z. Yang,

{kind=link}
{kind=link}
{kind=link}
{kind=link}