





Abstract
The cell-specific information of transcriptional regulation on microRNAs (miRNAs) is crucial to the precise understanding of gene regulations in various physiological and pathological processes existed in different tissues and cell types. The database, mirTrans, provides comprehensive information about cell-specific transcription of miRNAs including the transcriptional start sites (TSSs) of miRNAs, transcription factor (TF) to miRNA regulations and miRNA promoter sequences. mirTrans also maps the experimental H3K4me3 and DHS (DNase-I hypersensitive site) marks within miRNA promoters and expressed sequence tags (ESTs) within transcribed regions. The current version of database covers 35 259 TSSs and over 2.3 million TF-miRNA regulations for 1513 miRNAs in a total of 54 human cell lines. These cell lines span most of the biological systems, including circulatory system, digestive system and nervous system. Information for both the intragenic miRNAs and intergenic miRNAs is offered. Particularly, the quality of miRNA TSSs and TF-miRNA regulations is evaluated by literature curation. 23 447 TSS records and 2148 TF-miRNA regulations are supported by special experiments as a result of literature curation. EST coverage is also used to evaluate the accuracy of miRNA TSSs. Interface of mirTrans is friendly designed and convenient to make downloads (http://mcube.nju.edu.cn/jwang/lab/soft/mirtrans/ or http://120.27.239.192/mirtrans/).
INTRODUCTION
The microRNA (miRNA) is a subset of non-coding RNAs regarded as ‘small RNAs of big roles’ (1). It prevents or suppresses protein translation by binding to the 3’UTR of mRNA. The target mRNA of miRNA is usually in vast numbers, which enables miRNA to participate in almost all the biological processes. The essential roles of miRNA in gene regulation attract huge research interests, which in turn drives the rapid development of miRNA study. In recent years, miRNAs are also widely recognized as the potential biomarker for clinical diagnoses, treatments and prognoses of various kinds of diseases, especially cancers (2,3). Till 2014, about twenty years after the first miRNA was found (4), 28645 hairpin precursor miRNAs (pre-miRNAs) and 35828 mature miRNAs from 223 species have been recorded in the most comprehensive repository of miRNAs, miRBase (version 21) (5). For human, 1881 pre-miRNAs and 2588 mature miRNAs were revealed.
A full understanding of miRNA function depends on the knowledge of miRNA regulation network, including the upstream transcriptional regulation and their downstream targets. Compared to the abundance of miRNA expression databases and target databases, the resource for regulating miRNA expression is far behind. The main reason is that the 5’ end of primary miRNA transcript is rapidly sliced by Drosha after transcription so that it is very hard to capture the full-length transcript of miRNA (6,7). This eventually lead to the difficulties in the gain of accurate miRNA transcriptional start sites (TSSs), miRNA promoters and the regulations of transcription factors (TFs) on the miRNA transcription (i.e. TF-miRNA regulations). Several approaches have been conducted by combining high throughput data with appropriate algorithms or literature mining. A few databases about miRNA transcription were built to provide information of either TF-miRNA regulation or miRNA TSSs. ChIPBase (8) provides abundant resources of TF-miRNA regulations and other TF-target regulations by parsing ChIP data. CircuitsDB (9) discovered TF-miRNA regulation circuits for human and mouse. TransmiR (10) offers highly reliable yet somewhat limited number (735 in total) of TF-miRNA regulations with literature curation. However, without accurate annotation of miRNA TSSs and promoters, it's hard to tell whether the parsed TF truly functions in the transcription of the miRNA that is downstream to the binding site of the TF (i.e. TFBS). While miRT (11) provides miRNA TSSs that are collected from a couple of experiments, no TF-miRNA regulations are offered. All of these databases developed earlier did not tackle the problem of cell-specificity of miRNA transcription.
Recent studies have shown that miRNAs expression is distinctly tissue-specific or cell-type specific, which are fundamental to tissue development and function maintenance (12). Increasing number of tissue specific miRNAs are reported to be disease associated (13–16). These miRNAs could be of clinical potentials as biomarkers for the diagnoses, treatments and prognoses of diseases (2,3). So, the knowledge about cell-specific transcription of miRNAs is eagerly demanded to facilitate the investigation of miRNAs in fundamental biological aspects as well as in clinical applications. TSmiR (17) offers the regulations of TFs on tissue-specific miRNAs for 12 tissues based on ChIP-seq data. DIANA-miRGen (18) provides information of both miRNA TSSs and TF-miRNA regulations for 428 intergenic miRNAs in nine cell lines including human and mouse.
Here, we introduce mirTrans database to provide comprehensive information about cell-specific transcription of miRNA. mirTrans offers abundant cell-specific data which includes both miRNA TSSs and TF-miRNA regulations, together with some supportive data derived from other public resources [e.g. H3K4me3 peaks and DHS peaks]. Up to now, it contains 35 259 TSSs and 2 340 406 TF-miRNA regulations for 1513 miRNAs in 54 human cell lines. The 54 cell lines belong to 17 tissues which cover most of biological systems, including circulatory system, digestive system, nervous system, reproductive system and respiratory system. So, the database could be applied in quite a variety of biological or medical fields. Information of both intragenic miRNAs and intergenic miRNAs is reported. Specifically, quality evaluation on every miRNA TSS and TF-miRNA regulation is presented by literature evidence and EST evidence. Literature evidence gives the documentary support to miRNA TSS and TF-miRNA regulations, and EST evidence evaluates the accuracy of miRNA TSS by EST coverage. mirTrans also provides a convenient and friendly interface for customers. Flexible query is allowed by either one of the three types of keywords (i.e. miRNA, cell line and TF) or any combinations among them. Genomic distribution graph illustrates the intuitive view of miRNA cell-specific transcription. All the data in mirTrans are offered for free downloads.
RESULTS
Database content
mirTrans generally covers two types of data, i.e. the core data and the supportive data. The core data were produced on our own, which mainly include cell-specific miRNA TSSs and TF-miRNA regulations; the supportive data are derived from other resources, which are the primary materials for either the production or the quality evaluation of core data. Figure 1 illustrates how these data were produced or applied.
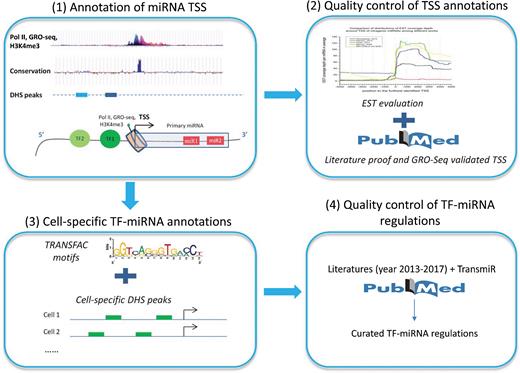
Workflow for the production of core data.
Core data
Cell-specific miRNA TSSs
TSS data are produced through a python implementation that is developed on the bases of a method previously published by the authors (19). This implementation allows the input in the form of ‘.bed’, where the TSS-related segments are recorded. The ‘.bed’ file could be obtained when peak-calling tools [e.g. MACS2 (20) and Homer (21)] are used to catch the peak areas in TSS-related sequencing data (e.g. H3K4me3, Pol II and GRO/PRO-seq). With the help of the implementation, it is convenient to integrate multiple types of high-throughput data for the expansion of TSS data. In the future, TSS data in mirTrans will be updated once a certain amount of additional TSS-related data are collected. Up to now, the TSSs in mirTrans are predicted based on H3K4me3 and DHSs (19).
Usually, predictions on miRNA TSSs are focused on intergenic miRNAs, because intragenic miRNAs are considered to be co-transcribed with its host gene. However, more and more evidences have shown that intragenic miRNA is also capable of transcription independent to its host gene (22). Our implementation has a special module to tell whether the transcription is dependent or independent on its host gene, and also returned the right TSSs for every intragenic miRNA in certain cell lines.
In particular, mirTrans provides quality evaluations for all the TSSs, which is quite helpful for customers to make choice. The quality of TSSs is annotated in two ways. One is so called ‘ESTs (expressed sequence tags) evidence’, which evaluates TSS quality with the significance of EST coverage difference around TSS. EST is the expressed segment of transcripts. Theoretically, significant differences of EST coverage between the upstream and the downstream of TSS should be observed. The less the P value of EST evidence is, the higher the TSS quality is. The additional evaluation is termed as ‘literature evidence’, which annotates TSS quality by literature curation. ‘Strong evidence’ means the supports from literatures of traditional molecular experiments, while ‘evidence’ refers to the supports from GRO-Seq, CAGE and other high-throughput transcriptional signals (23–25).
Cell-specific TF-miRNA regulations
TF-miRNA regulation is produced based on the data of miRNA TSS. In general, the promoter that is 1kb upstream to the cell-specific miRNA TSSs is scanned with geneXplain platform (http://platform.genexplain.com) in order to obtain the cell-specific transcription factor binding sites (TFBSs). In particular, for the cell lines with DHSs data, TFBSs are scanned on the DHS segments within the promoter, since DHSs data gives the marks for TFBS regions (26). TF-miRNA regulations are obtained when the related TFs are retrieved from TRANSFAC (27) by TFBSs. Since ChIP-seq data for TF binding is able to provide the experiment-proved TFBSs for certain TFs, it will be employed for the refinement of cell-specific TF-miRNA regulations in the future, as the updating of mirTrans.
Two parameters i.e. the affinity score and the conservation scores, are additionally offered for TFBSs. The affinity score which scales the affinity of TF binding to TFBS is calculated with MATCH (28) provided by TRANSFAC, while the conservation score which measures the conservation of TFBS sequence across species is calculated based on data of UCSC track ‘phastCons100way’ (29,30). Till now, >2.3 million cell specific TF-miRNA regulations are available in mirTrans.
The quality of cell-specific TF-miRNA regulations is also verified with literature evidences. The literature pool was constructed on the basis of TransmiR database (http://www.cuilab.cn/transmir) combined with new papers from PubMed during the year 2013–2017. The TF-miRNA regulations of ‘strong evidence’ are supported by promoter-related experiments, e.g. ChIP and luciferase report assays detecting the activity of promoters (31); while the regulations of ‘evidence’ are supported by gene expression experiments or the computational analysis based on expression data (32). The other TF-miRNA regulations recorded in this database were only based on DHS evidence.
Supportive data
So far, H3K4me3 peaks, DHSs peaks, ESTs and pre-miRNAs are the supportive data in mirTrans. H3K4me3 and DHSs data were collected from ENCODE (33), ESTs data were derived from UCSC (34), and pre-miRNAs were derived from miRBase (miRBase version 21). H3K4me3, DHSs, and pre-miRNA data are the source data for the production of cell-specific miRNA TSSs and cell-specific TF-miRNA regulations. ESTs are used to evaluate the quality of TSSs.
Most of the supportive data are graphically illustrated together with miRNA TSSs and TFBSs when miRNA TSSs are retrieved. Theoretically, H3K4me3 peak represents the location of miRNA TSS, DHSs peak represent the region of TFBSs, and EST the position of transcripts. Therefore, the genomic distributions of all these data gives the intuitive illustration of cell-specific transcription of miRNAs.
Database statistics
Up to now, 35 259 TSSs and 2 340 406 TF-miRNA regulations for 1513 pre-miRNAs (546 intergenic miRNA and 967 intragenic miRNA) in 54 human cell lines are recorded in mirTrans. Comparing to the previous comparable databases, mirTrans offers the most abundant repository for the cell-specific transcription of miRNAs (see Table 1). Two recent researches reported a large amount of miRNA transcriptional regulation data by using GRO/PRO-seq and CAGE techniques respectively (24,25). The statistics on the results of these two papers are also shown in Table 1. In addition, literature evidence is provided to indicate the data quality. So far, there are all together 23 447 TSS records and 2148 TF-miRNA regulations supported by the specified experimental evidences as described above and which are regarded as the most reliable data. Note that literature evidence will be increasingly accumulated as the update of mirTrans.
Summary of the contents in mirTrans and other comparable databases/data sets
| Database | Cell lines | miRNAs | miRNA TSSs | miRNA TSSs with literature evidence | TF-miRNA regulations | TF-miRNA regulations with literature evidence |
|---|---|---|---|---|---|---|
| mirTrans | 54 | 1513 (546 intergenic / 967 intragenic) | 35259 | 23447 | 2340406 | 2148 |
| Liu Q et al. [24] (2017) | 27 | 480 (intergenic) | 5839 | N/A | 1893 | 9 |
| de Rie D et al. [25] (2017) | 84 | 1357 (371 intergenic / 986 intragenic) | 1118 | N/A | N/A | N/A |
| ChIPBase (2016) | N/A | unknown | N/A | N/A | 273761 | None |
| DIANA-miRGen (2016) | 9 | 428 (intergenic) | 276 | None | unknown | None |
| TSmiR (2014) | 12 tissues | 116 | N/A | N/A | 2347 | None |
| CircuitsDB (2013) | N/A | 180 | N/A | N/A | 115 TFs to 180 miRNAs | None |
| miRT (2012) | N/A | 588 (206 intergenic / 382 intragenic) | 670 | None | N/A | N/A |
| TransmiR (2009) | N/A | 100 | N/A | N/A | 735 | 735 |
| Database | Cell lines | miRNAs | miRNA TSSs | miRNA TSSs with literature evidence | TF-miRNA regulations | TF-miRNA regulations with literature evidence |
|---|---|---|---|---|---|---|
| mirTrans | 54 | 1513 (546 intergenic / 967 intragenic) | 35259 | 23447 | 2340406 | 2148 |
| Liu Q et al. [24] (2017) | 27 | 480 (intergenic) | 5839 | N/A | 1893 | 9 |
| de Rie D et al. [25] (2017) | 84 | 1357 (371 intergenic / 986 intragenic) | 1118 | N/A | N/A | N/A |
| ChIPBase (2016) | N/A | unknown | N/A | N/A | 273761 | None |
| DIANA-miRGen (2016) | 9 | 428 (intergenic) | 276 | None | unknown | None |
| TSmiR (2014) | 12 tissues | 116 | N/A | N/A | 2347 | None |
| CircuitsDB (2013) | N/A | 180 | N/A | N/A | 115 TFs to 180 miRNAs | None |
| miRT (2012) | N/A | 588 (206 intergenic / 382 intragenic) | 670 | None | N/A | N/A |
| TransmiR (2009) | N/A | 100 | N/A | N/A | 735 | 735 |
| Database | Cell lines | miRNAs | miRNA TSSs | miRNA TSSs with literature evidence | TF-miRNA regulations | TF-miRNA regulations with literature evidence |
|---|---|---|---|---|---|---|
| mirTrans | 54 | 1513 (546 intergenic / 967 intragenic) | 35259 | 23447 | 2340406 | 2148 |
| Liu Q et al. [24] (2017) | 27 | 480 (intergenic) | 5839 | N/A | 1893 | 9 |
| de Rie D et al. [25] (2017) | 84 | 1357 (371 intergenic / 986 intragenic) | 1118 | N/A | N/A | N/A |
| ChIPBase (2016) | N/A | unknown | N/A | N/A | 273761 | None |
| DIANA-miRGen (2016) | 9 | 428 (intergenic) | 276 | None | unknown | None |
| TSmiR (2014) | 12 tissues | 116 | N/A | N/A | 2347 | None |
| CircuitsDB (2013) | N/A | 180 | N/A | N/A | 115 TFs to 180 miRNAs | None |
| miRT (2012) | N/A | 588 (206 intergenic / 382 intragenic) | 670 | None | N/A | N/A |
| TransmiR (2009) | N/A | 100 | N/A | N/A | 735 | 735 |
| Database | Cell lines | miRNAs | miRNA TSSs | miRNA TSSs with literature evidence | TF-miRNA regulations | TF-miRNA regulations with literature evidence |
|---|---|---|---|---|---|---|
| mirTrans | 54 | 1513 (546 intergenic / 967 intragenic) | 35259 | 23447 | 2340406 | 2148 |
| Liu Q et al. [24] (2017) | 27 | 480 (intergenic) | 5839 | N/A | 1893 | 9 |
| de Rie D et al. [25] (2017) | 84 | 1357 (371 intergenic / 986 intragenic) | 1118 | N/A | N/A | N/A |
| ChIPBase (2016) | N/A | unknown | N/A | N/A | 273761 | None |
| DIANA-miRGen (2016) | 9 | 428 (intergenic) | 276 | None | unknown | None |
| TSmiR (2014) | 12 tissues | 116 | N/A | N/A | 2347 | None |
| CircuitsDB (2013) | N/A | 180 | N/A | N/A | 115 TFs to 180 miRNAs | None |
| miRT (2012) | N/A | 588 (206 intergenic / 382 intragenic) | 670 | None | N/A | N/A |
| TransmiR (2009) | N/A | 100 | N/A | N/A | 735 | 735 |
The 54 cell lines in mirTrans come from 17 tissues, which cover most of the biological systems including circulatory system, digestive system, nervous system, reproductive system and respiratory system (see Figure 2). About 30% of cell lines are cancer cells. This enables mirTrans a broad range of application in cancer study.
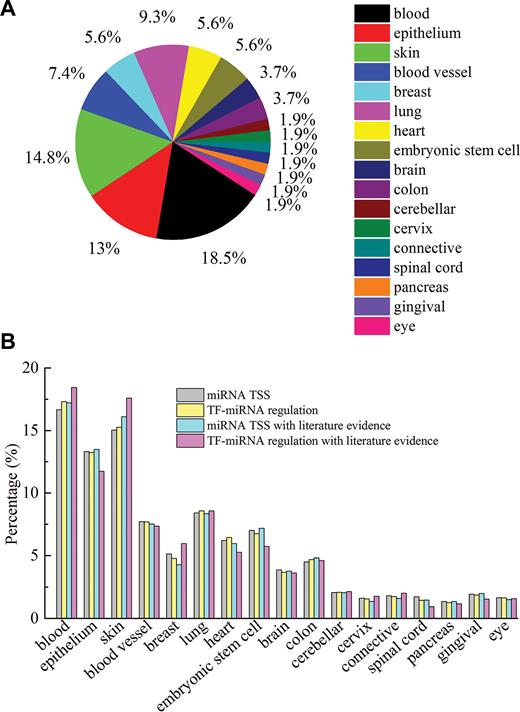
Statistics on the data in mirTrans. (A) Distribution of cell lines in 17 tissues; (B) distribution of miRNA TSSs, TF-miRNA regulations, miRNA TSSs with literature evidence, TF-miRNA regulations with literature evidence in 17 tissues.
The segment that starts from miRNA TSS to the end of pre-miRNA is annotated as the confident transcript of miRNA. Statistics on EST distribution show that 96.5% (10627/11016) of the confident transcripts are covered by at least one EST. Moreover, the coverage difference of EST between the upstream and the downstream of miRNA TSS is calculated for quality evaluation of TSSs (i.e. EST evidence mentioned above). 78.4% (8640/11016) of TSSs are with significant difference of EST coverage (P < 0.05, by t-test), which indicates the high-quality of miRNA TSSs in mirTrans.
Database implementation
System facility
All the data in mirTrans are well organized with MySQL. The website is implemented with PHP, JavaScript and HTML. Genomic graphs are illustrated using ‘Gviz’ package of Bioconductor.
Data query
Data retrieving starts from data query. mirTrans offers two ways of queries, i.e. simple search and advanced search (Figure 3A). Simple search allows the query with a single keyword, while advanced search allows the query with multiple keywords. Fuzzy words are supported in both simple and advanced search.

Interface for data query and download. (A–D) Data query; (E) data download. Red square in (B–D) marks the button for customized download or filter option.
For simple search, there are three entries which respectively relate to three types of keywords, i.e. miRNA, cell line and TF. Different entries go to different forms of return.
Simple search by miRNA returns a graph followed by a table. The graph shows the genomic distribution of pre-miRNA, miRNA TSS miRNA TFBSs, H3K4me3 peak, DHS peak and ESTs (Figure 3B). The table lists the following information that is associated with the input pre-miRNA: genomic location, TSS location, distance from TSS to pre-miRNA, cell line where the TSS are found, TFs that bind to the region of 1kb upstream to the TSS (i.e. the miRNA promoter), EST evidence and literature evidence for the TSS (Figure 3B).
Simple search by cell line returns a table where all the pre-miRNAs of which TSSs are found in this cell line, are listed. Additional information in the table includes miRNA location, TSS location of the miRNA, host gene of the miRNA and the location of the host gene (Figure 3C). More details can be retrieved when a miRNA or TSS in table is clicked (Figure 3C).
Simple search by TF returns a table where all the TF-miRNA regulations in which the TF participates are listed. Associated information includes the miRNA that the TF regulates, the cell line where the TF-miRNA regulation happens, the TFBS where the TF binds, affinity score that describes the binding strength of the TF to the TFBS, conservation score of the TFBS sequence, and literature evidence (Figure 3D). Detail information about the literature that supports the TF-miRNA regulation can be referred when the links in ‘Details’ are followed (Figure 3D). In particular, a filter option is offered, with which customers are free to retrieve either the whole results or only the literature-supported ones (see the bottom-left button in Figure 3D). Download of the return table is associated with the filter option (see the bottom-right button in Figure 3D).
Advanced search enables a flexible query with any combinations of the three keywords used in simple search (Figure 3A). The results of the advanced search will be returned in the way similar to that of simple search.
Data download
mirTrans provides two ways of data download, i.e. customized download and the complete download. The customized download is associated with the query that the customer submits. Every result of the query can be downloaded by clicking the ‘download’ button at the bottom of the return page (Figure 3B–D). The complete download is specifically offered in the download page (Figure 3E). Customers are allowed to separately download the complete data in the form of the returned table when miRNA/cell line/TF is inquired. A collective download packing all the forms of data is also provided.
Database maintenance
A new version of mirTrans is released every 6 months in regular. The update includes the refresh of supportive data (e.g. H3K4me3, DHS, EST and pre-miRNA), core data (e.g. cell-specific miRNA TSSs and TF-miRNA regulation), EST evidence and literature evidence. The whole updating process is developed in a semi-automatic way. The supportive data are automatically collected from the related database, the core data and EST evidence are automatically produced through the workflow of our own, and the literature curation are manually made after related literatures are collected from PUBMED (https://www.ncbi.nlm.nih.gov/pubmed).
CONCLUSION
The wide participations in gene regulation of biological processes and clinical application potentials of miRNAs intrigue a great demand on the knowledge of cell-specific regulations of miRNAs. mirTrans offers abundant and high-quality information about cell-specific transcriptions of human miRNAs. Compared with the current databases reported so far, mirTrans specially advances in that, a significant amount of data is provided with sufficient evidence from high throughput data and literature reports manually curated. Firstly, the information for a total of 54 cell lines covering most of the key biological systems ensures the database applicable to a large variety of biological and medical investigations in different tissues or processes; Secondly, nearly all the human miRNAs known so far residing in either intergenic genomic region or intragenic region are included in mirTrans, while many of the other databases only provide intergenic miRNAs which generally account for <50% of the total miRNAs encoded by human genome; Finally, quality evaluation on either miRNA TSSs or TF-miRNA regulations were made by EST data and literature reports. Concerned with these aspects, mirTrans is very helpful for both the experimental biologists and the system biologists, who are interested in gene regulations, pathways and gene networks where miRNAs are involved.
Recent years have shown an ever rapid increase in the genomic and epigenomic data for diverse samples of tissues and cells. Accordingly, mirTrans will be updated regularly by employing new experimental data and information from literature mining. It will also incorporate cell-specific miRNA transcriptional networks by integrating all the regulations between miRNAs and TFs in the future.
ACKNOWLEDGEMENTS
The authors would acknowledge the Center of High Performance Computation of Nanjing University for the support of computational resources.
FUNDING
China Scholarship Council, DZHK (German Center for Cardiovascular Research); National Science Foundation of China [81250044, 31500674]; NJU-Yangzhou Institute of Optoelectronics. Funding for open access charge: National Science Foundation of China [81250044, 31500674].
Conflict of interest statement. None declared.
REFERENCES
Author notes
These authors contributed equally to this work as first authors.

{kind=link}
{kind=link}
{kind=link}
Comments