





Abstract
An R-loop is a DNA:RNA hybrid formed during transcription when a DNA duplex is invaded by a nascent RNA transcript. R-loops accumulate in nucleoli during RNA polymerase I (RNAP I) transcription. Here, we report that mammalian RNase H1 enriches in nucleoli and co-localizes with R-loops in cultured human cells. Co-migration of RNase H1 and R-loops from nucleoli to perinucleolar ring structures was observed upon inhibition of RNAP I transcription. Treatment with camptothecin which transiently stabilized nucleolar R-loops recruited RNase H1 to the nucleoli. It has been reported that the absence of Topoisomerase and RNase H activity in Escherichia coli or Saccharomyces cerevisiae caused R-loop accumulation along rDNA. We found that the distribution of RNase H1 and Top1 along rDNA coincided at sites where R-loops accumulated in mammalian cells. Loss of either RNase H1 or Top1 caused R-loop accumulation, and the accumulation of R-loops was exacerbated when both proteins were depleted. Importantly, we observed that protein levels of Top1 were negatively correlated with the abundance of RNase H1. We conclude that Top1 and RNase H1 are partially functionally redundant in mammalian cells to suppress RNAP I transcription-associate R-loops.
INTRODUCTION
The movement of an RNA polymerase (RNAP) along duplex DNA during transcription generates positive torsional stress ahead of the enzyme and negative torsional stress in its wake (1). The accumulation of positive torsional stress ahead of the transcription bubble prevents further unwinding of DNA duplex and impedes transcription elongation by the RNAP, whereas the negative torsional stress behind the transcription bubble leads to the separation and unwinding of the DNA duplex. In this unwound region, nascent RNA can hybridize with template DNA, leaving the non-template DNA single stranded. Such a structure is termed an R-loop (2).
R-loops were first described in bacteria and mitochondria where they are required for the initiation of DNA replication (3–6). It now appears that R-loops regulate diverse cellular processes including transcription initiation and termination, immunoglobulin class switching, alterations in chromatin structure, and DNA repair (7–14). However, uncontrolled accumulation of R-loops is linked to DNA damage and genome instability (15–18). Studies suggest that structural elements of R-loops can make regions of the DNA strands particularly vulnerable to nucleases (19,20). These structural elements include the exposed single-stranded non-template DNA and the DNA flaps that form at either end of the R-loops. In addition, collision between R-loops and the replication machinery can lead to double-strand breaks (DSB) (21,22). Recently, accumulation of R-loops has been linked to cancer and neurodegenerative disorders (16,23).
Several mechanisms are known to resolve R-loops or prevent their formation. R-loops can be removed by RNase H endonucleases, which cleave RNA within DNA:RNA hybrids (12,24–26). R-loops can also be resolved by DNA:RNA helicases such as SETX and AQR (16,27). It has also been suggested that R-loop formation is suppressed by topoisomerases Top1 and Top3B, which resolve the negative torsional stress behind the transcription bubble to prevent the annealing of nascent RNA with template DNA (22,25,28,29). Moreover, RNA binding proteins, such as SF2 and THO, also preclude R-loop formation by coating nascent RNA as it is transcribed (30).
There are two main types of RNase H, RNase H1 and RNase H2. The N-terminal domain of RNase H1 is responsible for binding to DNA:RNA hybrids, and the C-terminal domain catalyzes RNA cleavage (31–33). The functions of RNase H1 and H2 are suggested by their subcellular localization. RNase H1 is present mostly in mitochondria and nuclei (31,34). Depletion of Rnaseh1 in mice results in embryonic lethality due to failure to replicate mitochondria DNA (35). We also recently reported hepatic apoptosis and mitochondrial R-loop accumulation in a liver-specific Rnaseh1 knockout mouse (26). In nuclei, RNase H1 is implicated in R-loop resolution, Okazaki fragment processing, DSB repair mediated by homologous-recombination (HR), and telomere elongation in cells in which the alternative lengthening of telomeres pathway is active (12,36). In addition, RNase H1 plays a major role in the activity of DNA-like antisense oligonucleotides in both the nucleus and cytoplasm (26,37,38).
RNase H2 is a heterotrimer composed of the catalytic unit H2A and auxiliary units H2B and H2C (31). RNase H2 is localized predominantly in nuclei in most cultured cells, although cytoplasmic localization of RNase H2 was reported in 15PC3 cells (39). RNase H2 overlaps with RNase H1 in nuclear functions such as R-loop resolution and Okazaki fragment processing (29). Unlike RNase H1, which requires at least four consecutive perfect DNA:RNA base pairs in order to cleave an RNA strand, RNase H2 can recognize a single ribonucleotide inserted in a DNA duplex and is responsible for removing misincorporated ribonucleotides from nascent DNA (40–44). Rnaseh2 knock-out in mice is embryonically lethal due to ribonucleotide accumulation in genomic DNA and DNA damage. Involvement of RNase H2 in ribonucleotide excision repair pathway is supported by its unique 5′-junction ribonuclease activity which supports the cleavage on the 5′-side of the ribonucleotide at a DNA-RNA-DNA junction (45).
Although RNase H enzymes have been linked to R-loop resolution in both nuclei and mitochondria, little has been known for subnuclear distribution and functions of the RNase H enzymes, especially RNase H1, due to its limited abundance. Here we show that in the nuclei, endogenous human RNase H1 localizes throughout the nucleoplasm and is enriched in nucleoli. Nucleolar enrichment of RNase H1 is due to its function in nucleolar R-loop processing. We report that RNase H1 and Top1 suppress the accumulation of transcriptional R-loops in nucleoli and that the levels of Top1 protein negatively correlate with the abundance of RNase H1 protein.
MATERIALS AND METHODS
Cell culture and treatment
HeLa and HEK293 cells were grown at 37°C, 7.5% CO2 in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. For siRNA treatment, cells at 70% confluency were transfected with 3 nM siRNA using Lipofectamine RNAiMax (Thermo Fisher Scientific) at a 6 μg/ml final concentration. siRNAs targeting human Rnaseh1 (s48357 and s48358), human TOP1 (siRNA1: 111294 and siRNA2: 111295), and luciferase were purchased from Thermo Fisher Scientific.
For plasmid transfection, 7.5 × 105 HeLa cells were transfected with 1 μg of plasmid using TurboFect Transfection Reagents (Thermo Fisher Scientific). pRNase H1-Flag was purchased from OriGene. Adenoviruses used in this study were produced as described previously (37). Treatments with actinomycin D (ActD) (Sigma-Aldrich), CX5461 (Selleck Chemicals), camptothecin (CPT) (Sigma-Aldrich), or pladienolide B (Santa Cruz Biotechnology) were performed as specified.
RNA isolation, Quantitative reverse transcription PCR (qRT-PCR), and northern blot
RNAs were isolated using TRIzol (Thermo Fisher Scientific) or RNeasy 96 Kits (Qiagen) according to protocols supplied by the manufacturers. TaqMan One-step qRT-PCR was performed using AgPath-ID™ One-Step RT-PCR Reagents (Thermo Fisher Scientific). Briefly, total RNAs (|$\sim$|50–100 ng) in 5 μl nuclease-free water were mixed with 0.3 μl primer probe sets containing forward and reverse primers (10 μM of each) and fluorescently labeled probe (3 μM), 10 μl 2 × RT-PCR Buffer, 0.8 μl 25 × RT-PCR Enzyme Mix, 1.7 μl Detection Enhancer and 2.2 μl nuclease-free water in a 20 μl reaction. Reverse transcription was performed at 45°C for 10 min, the reactions were then denatured at 95°C for 10 min, and forty cycles of PCR reactions were conducted at 95°C for 15 s and 60°C for 60 s within each cycle, using Applied Biosystems StepOnePlus Real-Time PCR system. Expression levels of target RNA were normalized to total RNA quantified using Quant-iT RiboGreen RNA Reagent (Thermo Fisher Scientific). Primer-probe sets for human Rnaseh1 (Hs00161407_m1), human TOP1 (Hs00243257_m1), mouse Cdkn1a (Mm01303209_m1), and mouse Tnfrsf10b (Mm00457866_m1) mRNAs were purchased from Thermo Fisher Scientific. Primer sequences for quantification of NCL1 mRNA precursor (NCL1 pre-mRNA) are: forward, 5′-CTC TGT CAC TGG TAT CTT TTC CC-3′; reverse, 5′-CAA AAC CAA ACC TAG AAC ACC AAA TG-3′; and probe, 5′-CAA GGC TAC TTT CTG TGG GAT GGC T-3′. Expression levels of 47S pre-rRNA precursor and mature 18S rRNA were examined using EXPRESS One-Step SYBR GreenER Kit (Thermo Fisher Scientific). Levels of 47S pre-rRNA precursor were normalized to the levels of 18S rRNA. Primer sequences for 47S pre-rRNA are: forward, 5′-TGT CAG GCG TTC TCG TCT C-3′ and reverse, 5′-AGC ACG ACG TCA CCA CAT C-3′. Primer sequences for 18S rRNA are: forward, 5′-GCA ATT ATT CCC CAT GAA CG-3′ and reverse, 5′-GGG ACT TAA TCA ACG CAA GC-3′. Northern analysis was performed as described previously (38). Briefly, total RNAs (5 μg) were resolved on a 1% agarose gel buffered with 1 × MOPS and transferred onto a nylon membrane. Hybridization was performed at 42°C using 5′-end [γ-32P] labeled oligonucleotide probes. The probe for detecting pre-rRNA spans the boundary between 18S and ITS1. Probe sequence: 5′-CCT CGC CCT CCG GGC TCC GGG CTC CGT TAA TGA TC-3′.
Western analysis and dot blot
Cell extracts were isolated in RIPA buffer supplemented with 1 × Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and were separated on a 4–12% gradient SDS-PAGE gel. Proteins were transferred to a nitrocellulose membrane using iBlot Gel Transfer Device (Thermo Fisher Scientific). The membranes were blocked at room temperature for 30 min with blocking buffer containing 5% (w/v) nonfat dry milk in 1 × PBS and incubated with primary antibodies in blocking buffer at 4°C overnight. After washing three times with washing buffer (0.1% Tween-20 in 1 × PBS) for 5 min each wash, membranes were incubated with secondary antibodies in blocking buffer at room temperature for 1 h. After washing three times with washing buffer for 5 min each wash, proteins were detected based on Enhanced chemiluminescence (Abcam) using ChemiDoc XRS+ (Bio-Rad). The signal intensities of the protein bands were measured and quantitated using Image Lab software (Bio-Rad). Antibodies for western blotting used in this study are listed in Supplemental Table S1. For dot blots, 5 μg of purified genomic DNA was treated with E. coli RNase H (NEB) for 20 min at 37°C. Treated or untreated DNA samples were spotted onto a nitrocellulose membrane. DNA:RNA hybrids were detected using the S9.6 antibody. Total DNA was detected by SYBR™ Gold (Thermo Fisher Scientific).
Neutral comet assay
HeLa cells were infected with AdV[Null], AdV[H1], and AdV[H1-E186Q] at a multiplicity of infection (moi) of 100 for 48 h. The neutral comet assay was performed using the TREVIGEN Comet Assay kit according to the manufacturer's instructions with 103 HeLa cells plated per comet slide. DNA was stained using SYBR Green (Thermo Fisher Scientific) and visualized using a confocal laser scanning FV1000 Fluoview microscope (Olympus).
Chromatin immunoprecipitation-Quantitative PCR (ChIP-qPCR)
Protein-DNA complexes were crosslinked by treatment with 1% formaldehyde for 10 min; reactions were quenched by addition of glycine to 125 mM and incubation for 5 min at room temperature. Cells were lysed, and chromatin fragmented by MNase and sonication to an average length of 250–500 bp. ChIP assays were performed using SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling), according to the manufacturer's instructions. Antibodies used for ChIP in this study are listed in Supplemental Table S1. The purified immunoprecipitated and input DNAs were used as template for TaqMan Real-Time PCR. Primer and probe sequences used in this study are listed in Supplemental Table S2. Briefly, purified immunoprecipitated or input DNAs in 2 μl nuclease-free water were mixed with 0.3 μl primer probe sets containing forward and reverse primers (10 μM of each) and fluorescently labeled probe (3 μM), 10 μl 2 × TaqMan® Gene Expression Master Mix (Thermo Fisher Scientific), and 7.7 μl nuclease-free water in a 20 μl reaction. The reactions were denatured at 95°C for 10 min and forty cycles of PCR reactions were then conducted at 95°C for 15 s and 60°C for 60 s within each cycle, using Applied Biosystems StepOnePlus Real-Time PCR system. The percent input was calculated as: 100 × (quantity [immunoprecipitated by specific antibody] – quantity [immunoprecipitated by IgG])/quantity[total input]. With this method, signals obtained from each immunoprecipitation are expressed as a percent of the total input chromatin.
Immunofluorescence (IF)
Cells seeded on glass-bottom culture dishes (MatTek) were treated as specified. To stain for DNA:RNA hybrids and RNase H2A, cells were fixed and permeabilized in ice-cold 100% methanol at –20°C for 15 min. For staining other proteins, cells were fixed at room temperature for 30 min with 4% formaldehyde and permeabilized for 5 min with 0.1% Triton X-100 in 1× PBS. Fixed cells were then blocked in blocking buffer (1 mg/ml BSA in 1× PBS) at room temperature for 30 min, and incubated in blocking buffer with primary antibodies at room temperature for 2 h. After washing three times with washing buffer (0.1% Tween-20 in 1× PBS; 5 min per wash), cells were incubated in blocking buffer with secondary antibodies at room temperature for 1 h. Finally, cells were washed three times in washing buffer for 5 min each wash and mounted using Prolong Gold anti-fade reagent with DAPI (Molecular Probes). Tyramide Signal Amplification (TSA) staining was performed using Tyramide SuperBoost™ Kits with Alexa Fluor™ Tyramides (Thermo Fisher Scientific), according to the manufacturer's instructions. Briefly, fixed and permeabilized cells were incubated with 3% hydrogen peroxide for 60 min at room temperature to quench the endogenous peroxidase activity of the samples. Cells were rinsed three times with 1× PBS and blocked in blocking buffer (1 mg/ml BSA in 1× PBS) at room temperature for 30 min. Cells were then incubated in blocking buffer with primary antibodies at room temperature for 2 h. After washing three times with 1× PBS (5 min per wash), cells were incubated in blocking buffer with HRP-conjugated secondary antibodies at room temperature for 1 h. After rinsing the cells three times in 1× PBS for 10 min each wash, cells were then incubated in tyramide working solution for 5 min. The reaction was stopped by adding stop reagent. Finally, cells were washed three times in 1× PBS for 5 min each wash and mounted using Prolong Gold anti-fade reagent with DAPI (Molecular Probes). Both tyramide working solution and stop reagent were made following the manufacturer's instructions. Antibodies used for immunofluorescent staining in this study are listed in Supplemental Table S1. Images were generated by confocal laser scanning microscopy using an FV1000 Fluoview (Olympus) and analyzed using Fluoview Ver. 2.0b Viewer (Olympus). Some of the images were recorded with different settings or adjusted to allow the optimal representation of detailed subcellular localization pattern.
Liver perfusion and hepatocyte isolation
Animal experiments were conducted according to American Association for the Accreditation of Laboratory Animal Care guidelines and were approved by the institutions Animal Welfare Committee (Cold Spring Harbor Laboratory's Institutional Animal Care and Use Committee guidelines). Liver perfusion and hepatocyte isolation were performed as described previously (26).
RESULTS
Mammalian RNase H1 localizes to nucleoli in an RNAP I transcription-dependent manner
To evaluate the subcellular distribution of RNase H1, we transiently infected HeLa cells with an adenoviral vector encoding full-length human RNase H1 (AdV[H1]) and stained for RNase H1 by indirect immunofluorescence with an anti-RNase H1 antibody raised in mouse against a region of the C-terminal catalytic domain of human RNase H1 (amino acids 189–287). As expected, RNase H1 localized to both mitochondria and nuclei. Significant amounts of RNase H1 were also observed in nucleoli as shown by the co-localization of nucleolar markers including nucleophosmin (NPM1) and fibrillarin (Figure 1A and Supplementary Figure S1A). A similar distribution pattern of RNase H1 was observed using a different antibody raised in rabbit against truncated human RNase H1 (amino acids 47–287). Using this antibody, overexpressed RNase H1 was also observed in mitochondria, nucleoplasm, and nucleoli. In nucleoli, RNase H1 co-localized with nascent ribosomal RNAs (rRNAs) labeled by 5′-bromodeoxyuridine (BrU) pulsing (Figure 1B). Co-staining of RPA194, an RNAP I subunit and RNase H1 (rabbit anti-RNase H1 Ab) also confirmed nucleolar enrichment of RNase H1 (Supplementary Figure S1B). Moreover, nucleolar localization was observed for Flag-tagged RNase H1 using the anti-Flag antibody in HeLa cells transiently transfected with the RNase H1-Flag plasmid (pRNaseH1-Flag) (Figure 1C).
![RNase H1 localizes to nucleoli in a RNAP I transcription-dependent manner. (A and B) Representative confocal immunofluorescence images of HeLa cells infected with AdV[H1] for 24 h. Cells were stained with (A) an antibody raised in mice against amino acids 189–287 of human RNase H1 and an anti-NPM1 antibody or (B) with an antibody raised in rabbit against amino acids 47–287 of human RNase H1 and an anti-BrdU antibody. Cells in panel B were pulsed with 1 mM BrU for 30 min immediately prior to fixation. (C) Representative confocal immunofluorescence images of HeLa cells transiently transfected with the pRNase H1-Flag for 48 h and stained using an anti-Flag antibody. (D) Representative confocal microscopy images of HeLa cells stained for endogenous RNase H1 with signal amplified using TSA. (E) Levels of 47S pre-rRNA and NCL1 pre-mRNA were quantified using qRT-PCR in extracts of HeLa cells treated with 0.02 μg/ml ActD for 2 h, 250 nM CX5461 for 6 h, or vehicle (DMSO). The error bars represent standard deviation from three parallel experiments. (F) HeLa cells infected with AdV[H1] for 24 h were treated with DMSO, 0.02 μg/ml ActD for 2 h, or 250 nM CX5461 for 6 h. Cells were stained with the anti-RNase H1 antibody.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/45/18/10.1093_nar_gkx710/3/m_gkx710fig1.jpeg?Expires=1716333752&Signature=qnS6BXbh2a4OJ~lJ-~DSu0MhW1Sb0pc2c~OT1eup1~6PoCo91J2QR4Rqzj-fqaW7jBmxXFYrHjwyiI1tXu1vIk7nL4sXE~ScozjCpgUSrwfA~wkVeLdOAP1ehVi5ClBy-Nfm3Vu7XzzjyCgzdeqJMPLolUIX0Em20o2Nf6r~ZVkgqzQhEDxF7fn~aYSVMRWBf3HgKeDr3G5F9haKr1qqvtkkQ3yJnY68w2eOhts5Zn84Hc1J3icF3ZeIkjbvDUatNGMESYC6aJ-weVEvtvfbDBowWDaqZin7Rw5ucL1g0gWOfcXzOhSzRT8HZ8z9VZjy~0fOhFiRKEP-eDA-Sm5rBw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
RNase H1 localizes to nucleoli in a RNAP I transcription-dependent manner. (A and B) Representative confocal immunofluorescence images of HeLa cells infected with AdV[H1] for 24 h. Cells were stained with (A) an antibody raised in mice against amino acids 189–287 of human RNase H1 and an anti-NPM1 antibody or (B) with an antibody raised in rabbit against amino acids 47–287 of human RNase H1 and an anti-BrdU antibody. Cells in panel B were pulsed with 1 mM BrU for 30 min immediately prior to fixation. (C) Representative confocal immunofluorescence images of HeLa cells transiently transfected with the pRNase H1-Flag for 48 h and stained using an anti-Flag antibody. (D) Representative confocal microscopy images of HeLa cells stained for endogenous RNase H1 with signal amplified using TSA. (E) Levels of 47S pre-rRNA and NCL1 pre-mRNA were quantified using qRT-PCR in extracts of HeLa cells treated with 0.02 μg/ml ActD for 2 h, 250 nM CX5461 for 6 h, or vehicle (DMSO). The error bars represent standard deviation from three parallel experiments. (F) HeLa cells infected with AdV[H1] for 24 h were treated with DMSO, 0.02 μg/ml ActD for 2 h, or 250 nM CX5461 for 6 h. Cells were stained with the anti-RNase H1 antibody.
To examine if endogenous RNase H1 localizes to nucleoli, we stained for RNase H1 in HeLa (Figure 1D) and Hek293 cells (Supplementary Figure S1C). Since RNase H1 is present in cells at very low abundance, we used tyramide signal amplification (TSA) to enhance the staining of the endogenous protein. Similar to the overexpressed RNase H1, endogenous RNase H1 localized to mitochondria, nucleoplasm, and nucleoli. Depletion of RNase H1 by specific siRNA resulted in the reduction in signals of endogenous RNase H1 by immunofluorescence staining (Supplementary Figure S1D). Additional evidence that RNase H1 functions in nucleoli comes from our previous observation that DNA-like antisense oligonucleotides, which harness RNase H1 to cleave the complementary RNA, sequence-specifically induce degradation of small nucleolar RNAs (46). In contrast to RNase H1, RNase H2A, the catalytic subunit of RNase H2, did not significantly present in nucleoli (Supplementary Figure S1E).
The nucleolus is the site of rRNA synthesis and processing (47). To examine whether the nucleolar localization of RNase H1 requires on-going rRNA synthesis, we inhibited RNAP I transcription by treating HeLa cells overexpressing human RNase H1 with ActD at 0.02 μg/ml for 2 h. A significant reduction in levels of 47S pre-rRNA was observed in cells treated with ActD whereas levels of a precursor mRNA transcribed by RNAP II (NCL1 pre-mRNA) were not affected in treated and untreated cells (Figure 1E). Interestingly, after a 2-h exposure to ActD, RNase H1 was not detected in nucleoli, but the diffuse distribution of RNase H1 in the nucleoplasm was not altered by ActD treatment (Figure 1F). ActD also induced nucleolar clearance of endogenous RNase H1 in HEK293 cells (Supplementary Figure S2). Nucleolar clearance of RNase H1 was also observed in cells treated with another RNAP I inhibitor, CX5461. CX5461 disrupts the SL1-rDNA interaction and induces nucleolar segregation (48,49). A 6-h exposure of HeLa cells to 250 nM CX5461 reduced levels of 47S pre-rRNA by 75% relative to untreated cells (Figure 1E) and induced nucleolar clearance of RNase H1 (Figure 1F). Together, these results suggest that the nucleolar localization of RNase H1 is dependent on the on-going RNAP I transcription.
Time course of nucleolar clearance of RNase H1, Top1 and R-loops in the presence of ActD
R-loops can accumulate in nucleoli during rDNA transcription (25,50). The hybrid-binding domain (HBD) of human RNase H1 supports its binding to DNA:RNA hybrids (33). In addition, RNase H and topoisomerase activities are required for rRNA synthesis in yeast (25). Interestingly, nucleolar localization of Top1 requires ongoing rRNA synthesis (51). We thus hypothesized that nucleolar localization of RNase H1 might also depend on the presence of RNAP I transcriptional R-loops.
To test this hypothesis, we monitored the subcellular distribution of RNase H1, Top1 and R-loops in HeLa cells overexpressing RNase H1 throughout a 2-h time course of ActD treatment (Figure 2A). R-loops were stained using an antibody specific for DNA:RNA hybrids (S9.6). Consistent with the previous reports, in untreated cells, S9.6 signals were observed in both cytoplasm and nucleus (Figure 2A) (19,52,53). The cytoplasmic signals have been found to be composed of mitochondrial and RNAP III-dependent cytosolic DNA:RNA hybrids (53), and are sensitive to the RNase H treatment (19,53). In nuclei, small dots of S9.6 signals were observed to distribute throughout the nucleoplasm and high S9.6 signals were present in nucleoli (Figure 2A) (19). Thirty-minute exposure to ActD caused only approximately 20% reduction in the level of 47S pre-rRNA precursor relative to the level in untreated cells (Figure 2B). At this time point, cells showed no distinguishable changes in nucleolar morphology, as indicated by NPM1 staining (Supplementary Figure S3). Both RNase H1 and R-loops were still present in the nucleoli (Figure 2A); however, Top1 had begun to relocate to crescent-like structures at the nucleolar perimeter (Figure 2A), suggesting that the nucleolar localization of Top1 is highly sensitive to rDNA transcription status. These Top1-containing perinucleolar crescent-like structures became even more pronounced after 60 min of exposure to ActD, and at this time point, little or no Top1 was detected in the nucleoli. After exposure to ActD for 60 min, 47S pre-rRNA was reduced by ∼80% (Figure 2B), the nucleoli became rounded and smaller, and NPM1 was located diffusely in the nucleoplasm (Supplementary Figure S3). RNase H1 was enriched in unique perinucleolar ring structures and in many cells, was not detected in nucleoli at this time point. Residual S9.6 signals were observed in the nucleoli and also enriched in the perinucleolar ring structures observed for RNase H1. After a 120-min exposure to ActD, little 47S pre-rRNA was detected (Figure 2B), and NPM1 showed an even distribution between nucleoli and nucleoplasm. Additionally, in many cells, S9.6 signals were not observed, confirming that the nucleolar accumulated R-loops are dependent on ongoing RNAP I transcription (Figure 2A). At this time point, Top1 formed distinct perinucleolar caps, and no nucleolar RNase H1 were observed in most of the cells (Figure 2A).
![Nucleolar localization of RNase H1, DNA:RNA hybrids and Top1 depends on the ongoing transcription of rRNA. (A) Representative images of immunofluorescence staining for RNase H1 (overexpressed from AdV[H1]), Top1, and R-loops (using antibody S9.6) in HeLa cells treated with 0.02 μg/ml ActD for indicated times. (B) qRT-PCR quantification of 47S pre-rRNA and NCL1 pre-mRNA during the time course of ActD treatment in HeLa cells. (C) qRT-PCR quantification of RNase H1 and Top1 mRNA during the time course of ActD treatment in HeLa cells. The error bars represent standard deviation from three parallel experiments. (D) Western analysis of RNase H1 and Top1 during the time course of ActD treatment in HeLa cells.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/45/18/10.1093_nar_gkx710/3/m_gkx710fig2.jpeg?Expires=1716333752&Signature=ED-i0s7ND3oyNbhy0xzdvHNjijJ800Mkv6OYRxrK0fnjeMcxs-649FYKzkdtB5hpNSmxynW21jmlKXzMqch5Px21CCVz~WYztmHl62Rsoc49iEPsUdYYL~Yv3Dt5mc-KndIpjdZa-npu-6JuFkU84oluBQZvFJXQSW~Acs7HEUlPHiPyJtkUV3Qhc~BbTDuYq7bWUn2R7RpBWaF~AI6OBd1dbZ9bWEgToUugjHORdC0BAP48hMDOoc~RsOX7rL1P68mkhR8K~hfMc3QZsENiJ-TB85a-YvOSZfM2D~eM7kJbhcN-oRxYUIVPGqpOVK7irwjEMlNxQZkv4QMvqkbKcg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Nucleolar localization of RNase H1, DNA:RNA hybrids and Top1 depends on the ongoing transcription of rRNA. (A) Representative images of immunofluorescence staining for RNase H1 (overexpressed from AdV[H1]), Top1, and R-loops (using antibody S9.6) in HeLa cells treated with 0.02 μg/ml ActD for indicated times. (B) qRT-PCR quantification of 47S pre-rRNA and NCL1 pre-mRNA during the time course of ActD treatment in HeLa cells. (C) qRT-PCR quantification of RNase H1 and Top1 mRNA during the time course of ActD treatment in HeLa cells. The error bars represent standard deviation from three parallel experiments. (D) Western analysis of RNase H1 and Top1 during the time course of ActD treatment in HeLa cells.
It has been reported previously that the S9.6 antibody does not bind significantly to single-stranded DNA, double-stranded DNA and RNA, and ribosomal RNA (54). However, minor cross-reaction has been observed for a 23-mer AU-rich (%AU: 82.6) double-stranded RNA with the single chain Fv fragment of S9.6 antibody (55). To confirm the specificity of S9.6 staining, we stained HeLa cells using S9.6 antibody pre-incubated with synthetic DNA:RNA hybrids (Supplementary Figure S4A). The RNA strand of the hybrid is 6-carboxyfluorescein (6-FAM) labeled to allow the direct visualization of the hybrids in cells. The overall staining pattern was not significantly affected, but the staining signals of S9.6 in HeLa cells decreased when S9.6 antibody was pre-incubated with increasing concentrations of synthetic DNA:RNA hybrids. Increasing signals of the excess labeled synthetic DNA:RNA hybrids that were not bound by the S9.6 antibody were observed in cells. The unbound hybrids were found to localize to cytoplasm, nucleoplasm, and significantly enriched in nucleoli. In contrast to DNA:RNA hybrids, pre-incubation of S9.6 antibody with synthetic double-stranded RNA did not significantly decreased the S9.6 staining signals in HeLa cells (Supplementary Figure S4B). Duplex RNA distributed evenly throughout cells in cytoplasm, nucleoplasm and nucleoli. Together, our results suggest that the S9.6 antibody prefers the DNA:RNA hybrids, and the S9.6 staining pattern in cells is sensitive to the presence of excess amount of DNA:RNA hybrids but not double-stranded RNA of the same sequence. In addition, dot blots on isolated genomic DNA suggested that the recognition of DNA:RNA hybrids by S9.6 antibody was highly sensitive to the treatment with RNase H, further validating the specificity of the antibody (Supplementary Figure S4C).
To further confirm the staining pattern obtained with S9.6 was primarily due to R-loops binding, we stained the cells with a different antibody against DNA:RNA hybrids, D5H6. Similar to S9.6 staining, D5H6 signals were observed in both cytoplasm and nucleus including nucleoli. Nucleolar signals detected by D5H6 began to decline after 60-min exposure to ActD, and no nucleolar D5H6 signals were observed after 120-min (Supplementary Figure S3). Thus, the distribution of cellular DNA:RNA hybrids, as well as the RNAP I-dependent nucleolar localization of DNA:RNA hybrids were confirmed by two different antibodies.
RNA and protein levels of RNase H1 and Top1 were examined in cells treated with ActD, and no significant changes were observed (Figure 2C and D). This result indicated that the loss of Top1 and RNase H1 from nucleoli was not due to protein degradation. This suggests that nucleolar/nucleoplasmic partitioning of these proteins occurs in response to changes in cellular transcription status.
RNase H1 co-migrates with DNA:RNA hybrids to perinucleolar ring structures upon RNAP I transcriptional arrest
After inhibition of transcription, both overexpressed RNase H1 and S9.6 temporally accumulated in distinct nucleolar ring structures (Figure 2A). To evaluate whether endogenous RNase H1 co-migrated with DNA:RNA hybrids to the perinucleolar regions before diffusing into the nucleoplasm, we co-stained for RNase H1 and S9.6 in DMSO-treated HeLa cells or cells exposed to agents that can cause RNAP I transcriptional arrest and nucleolar segregation. The RNase H1 and S9.6 signals co-localized in nucleoli in control DMSO treated cells (Figure 3A). Upon the treatment of cells with RNAP I transcription inhibitors CX5461 or ActD, endogenous RNase H1 was cleared from nucleoli and localized to ring-shaped structures in the perinucleolar regions (Figure 3A) (see the structures in the highlighted box). Co-localization of RNase H1 and S9.6 staining was observed in these perinucleolar rings, suggesting the co-migration of RNase H1 and DNA:RNA hybrids. Nucleolar localization of RNase H1 and DNA:RNA hybrids was not affected by DRB, a RNAP II inhibitor (Supplementary Figure S5).
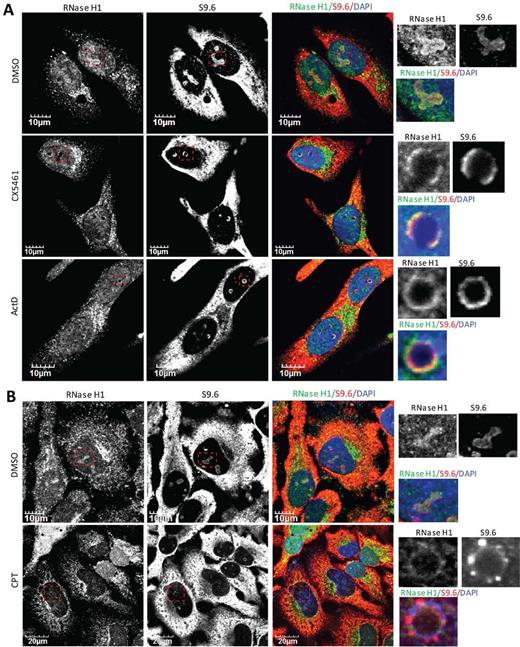
RNase H1 and DNA:RNA hybrids co-migrate to perinucleolar regions upon RNAP I transcriptional arrest or nucleolar segregation. (A and B) Representative images of co-immunofluorescent staining of endogenous RNase H1 and R-loops (using S9.6) in HeLa cells treated with (A) RNAP I transcriptional inhibitors CX5461 (250 nM, 6 h), ActD (0.02 μg/ml, 2 h), or (B) Top1 inhibitor CPT (10 μM, 4 h). Images were adjusted to allow the optimal representation of nucleolar ring structures.
We also observed co-migration of RNase H1 and R-loops from nucleoli to the perinucleolar regions in cells treated with the Top1 poison CPT (Figure 3B) and with splicing inhibitor pladienolide B (Supplementary Figure S6A). CPT inhibits the religation step of the DNA cleavage-religation process and thus stabilizes the covalently linked Top1-DNA intermediate, leading to the inhibition of RNAP I transcription (51). Pladienolide B targets the SF3b subunit of the U2 snRNP and has been reported to induce R-loop accumulation (56). Treatment with pladienolide B increased cellular S9.6 signals especially in the nucleoli of HeLa cells (Supplementary Figure S6B). Nucleolar segregation, as indicated by the smaller and more rounded nucleoli compared to vehicle-treated cells, was observed with prolonged pladienolide B exposure (24 h). Treatment with either CPT or pladienolide B resulted in clearance of RNase H1 from nucleoli and co-enrichment of RNase H1 and R-loops in perinucleolar rings (Figure 3B and Supplementary Figure S6A). These results exemplified the qualitative changes in the nucleolar localization patterns of RNase H1 and DNA:RNA hybrids upon RNAP I transcriptional inhibition, suggesting that the nucleolar localization of RNase H1 is dependent on RNAP I transcription and is sensitive to the morphology of nucleoli.
Hybrid binding domain is required for the nucleolar localization of RNase H1
To determine the domain requirement for the nucleolar localization of RNase H1, we transiently infected HeLa cells with adenoviral vectors encoding the full-length human RNase H1 (AdV[H1]), the catalytically inactive form of RNase H1 (AdV[H1-E186Q]), the nuclear form of RNase H1 (AdV[H1-Nuc]) with mitochondrial targeting signal (MTS) deleted, or the truncated form of RNase H1 (AdV[H1-HBD deleted]) with both MTS and HBD deleted (Figure 4A and B). We identified previously by mutagenesis studies that residues D145, E186 and D210 were essential for the enzymatic activity of human RNase H1 (57). A single point mutation, E186Q, that abolished RNase H1-mediated cleavage without affecting the binding of the RNase H1 to DNA:RNA hybrids (57), did not alter subcellular-localization pattern compared to wild-type RNase H1 (Figure 4C). The truncated RNase H1 with MTS deleted, as expected, did not have the mitochondrial localization but localized throughout the nucleoplasm and nucleoli (Figure 4C).
![HBD of RNase H1 is required for the nucleolar localization of human RNase H1 in HeLa cells. (A) Schematic representation of WT and mutant RNase H1 constructs. (B) Western blot of HeLa cells infected with control virus (AdV[Null]) or adenoviruses encoding WT RNase H1 (AdV[H1]), catalytically inactive RNase H1 (AdV[H1-E186Q]), MTS-deleted nuclear RNase H1 (AdV[H1-Nuc]), or MTS/HBD-deleted RNase H1 (AdV[H1-HBD deleted]). GAPHD serves as a loading control. (C) Representative images of immunofluorescent staining of overexpressed WT or mutant RNase H1. (D) Representative images of co-immunofluorescent staining of R-loops (using S9.6) and overexpressed MTS/HBD-deleted RNase H1 (AdV[H1-HBD deleted]). Upper panel: overexpressed RNase H1 (white box). Lower panel: signal intensified to visualize endogenous RNase H1 signals (green box). Highlighted box (red box): co-localization of endogenous RNase H1 with R-loops in the nucleoli.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/45/18/10.1093_nar_gkx710/3/m_gkx710fig4.jpeg?Expires=1716333752&Signature=GHBc-ETBkyuQ0rPR8bS7AOy-aydfv4MUA0RPnUNrOL-nDtL7r7g6GGEh6URcNhFOcOwykOHTGolggUFiTSNevdqtIgCEDfCNKm9fBusUbZ4tOJtlDNhblykR8y0u~3r3KMNkH0G8u5YY1TY1WeYlwJ5mJPy0-SDA3REiAldlX9xbaJBctLaZ~SuCY1NEkrs-msiwGAlra5hBiOFpwxZODbpbXHr3XlsMfMuQtwSBq-nF7qEzrwP6Z8S2MWV9TBqQ832siLVn5uPFpkeXfL1pD-mBhmd5osAPb0hL0s2NOvBb6UOWNV03WyVWfKTHeK75FhU1GqCPahAnwORo8mgeiQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
HBD of RNase H1 is required for the nucleolar localization of human RNase H1 in HeLa cells. (A) Schematic representation of WT and mutant RNase H1 constructs. (B) Western blot of HeLa cells infected with control virus (AdV[Null]) or adenoviruses encoding WT RNase H1 (AdV[H1]), catalytically inactive RNase H1 (AdV[H1-E186Q]), MTS-deleted nuclear RNase H1 (AdV[H1-Nuc]), or MTS/HBD-deleted RNase H1 (AdV[H1-HBD deleted]). GAPHD serves as a loading control. (C) Representative images of immunofluorescent staining of overexpressed WT or mutant RNase H1. (D) Representative images of co-immunofluorescent staining of R-loops (using S9.6) and overexpressed MTS/HBD-deleted RNase H1 (AdV[H1-HBD deleted]). Upper panel: overexpressed RNase H1 (white box). Lower panel: signal intensified to visualize endogenous RNase H1 signals (green box). Highlighted box (red box): co-localization of endogenous RNase H1 with R-loops in the nucleoli.
The binding affinities of HBD to a 12 bp DNA:RNA hybrid, dsRNA and dsDNA of the same sequence have been evaluated previously, and the Kd values are 0.2, 4.9 and 23.0 μM, respectively (33). Both the catalytic domain and HBD of RNase H1 bind DNA:RNA hybrids with comparable Kd values (|$\sim$|0.1–0.2 μM). However, the Kd of the full-length mouse RNase H1 is |$\sim$|0.04 μM, suggesting that the presence of both domains can further facilitate the hybrid binding of RNase H1 (33,58). This result is consistent with our observation that binding affinity of the DNA:RNA heteroduplex to full-length human RNase H1 was ∼9-fold greater than to the HBD-deleted RNase H1 (59). Indeed, HBD contributes dramatically to the specificity and processivity of RNase H1 (33). Interestingly, the truncated RNase H1 lacking both MTS and HBD (AdV[H1-HBD deleted]), distributed throughout the nucleoplasm but no longer localized to the nucleoli (Figure 4C). In a representative image containing HeLa cells with or without overexpression of mutant RNase H1 (AdV[H1-HBD deleted]) in the same view, the endogenous RNase H1 (green box) localized to mitochondria, nucleoplasm, and nucleoli, with the nucleolar population of RNase H1 co-localizing with nucleolar DNA:RNA hybrids (lower panel, signal intensified to visualize endogenous RNase H1), while the overexpressed MTS/HBD-deleted RNase H1 localized only to the nucleoplasm (upper panel), suggesting that the HBD of RNase H1 is required for its nucleolar localization (Figure 4D). In addition, these results further confirm that the nucleolar localization of RNase H1 is related with its binding to nucleolar DNA/RNA hybrids.
Top1 moves from nucleoli to RPA194-containing perinucleolar caps upon transcriptional inhibition
Top1 localizes predominantly to the nucleoli, and loss of Top1 leads to R-loop-mediated transcriptional blocks during rRNA synthesis in yeast (25). rRNA processing defects are also observed in HeLa cells depleted of Top1 (60). We found that reduction of Top1 by two different siRNAs targeting TOP1 resulted in a morphological change to smaller, more rounded nucleoli (Figure 5A, and Supplementary Figure S7). qRT-PCR and western analysis confirmed the reduction in levels of Top1 RNA and protein by both siRNAs (Supplementary Figure S7A and B). Upon Top1 depletion, nucleolar proteins, NPM1 and NCL1, appeared as sparse dots throughout the nucleoplasm, a pattern strongly reminiscent of the stepwise nucleolar segregation observed upon inhibition of transcription of rRNA by CX5461 (Figure 5A, B and Supplementary Figure S7C and D). These results support the important role of Top1 in rRNA synthesis in mammalian cells.
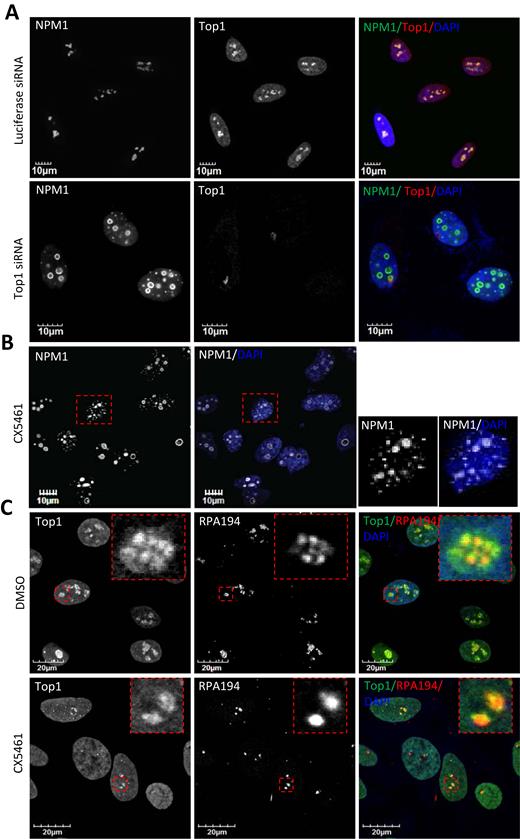
Top1 and RPA194 co-migrate to perinucleolar LNCs upon RNAP I transcriptional arrest and nucleolar segregation. (A) Confocal immunofluorescence imaging of HeLa cells 48 h after transfection with luciferase siRNA or Top1 siRNA. Cells were stained for Top1 and nucleolar marker NPM1. (B) HeLa cells treated with 250 nM CX5461 for 6 h were stained for NPM1. (C) Co-immunofluorescent staining of Top1 and RPA194 in HeLa cells treated with DMSO or 250 nM CX5461 (6 h).
On-going transcription of rRNA is crucial for the positioning of Top1 in the nucleolus. Top1 is observed in bead-like structures corresponding to fibrillar centers in control DMSO-treated HeLa cells, co-localizing with RNAP I subunit RPA194 (Figure 5C). Transcriptional arrest resulted from treatment of cells with ActD or CX5461 induced nucleolar segregation and the movement of Top1 and RPA194 to distinct light nucleolar caps (LNCs) (Figure 5C and Supplementary Figure S8A). In contrast to Top1, Top2 was not significantly enriched in nucleoli in DMSO-treated HeLa cells (Supplementary Figure S8B). In the presence of ActD, Top2 did not co-localize with Top1 in the perinucleolar caps. Thus, Top1, RNase H1 and R-loops all delocalize from nucleoli to perinucleolar regions upon RNAP I transcriptional inhibition, but not RNase H2 or Top2.
Upon transcriptional arrest, Top1 and RNase H1 move to different perinucleolar regions
Although co-localized in the nucleoli in the absence of RNAP I inhibitor, no significant co-localization was observed between R-loops and Top1 after exposure of HeLa cells to RNAP I inhibitor (Figure 6A). The co-localization of Top1 with RPA194 in LNCs in cells in which transcription is inhibited is consistent with a previous study showing that nucleolar/nucleoplasmic partitioning of Top1 is regulated, at least partially, by its interaction with RNAP I (51). In contrast to Top1, nucleolar localization of RNase H1 seems to be governed by direct binding to DNA:RNA hybrids since it co-localizes and co-migrates with R-loops before and during RNAP I transcription inhibition (Figures 2 and 3). To further confirm that RNase H1 and Top1 localize to different perinucleolar sites after RNAP I inhibition, we co-stained RNase H1 and RPA194 in HeLa cells treated with DMSO or ActD. RNase H1 localized, as expected, to perinucleolar ring structures in the presence of ActD, while RPA194 accumulated in LNC (Figure 6B). Staining signals of RNase H1 and RPA194 were mutually exclusive, suggesting that RNase H1 did not co-migrate with RPA194 to the LNC. These results suggest that RNase H1 co-migrates with DNA:RNA hybrids and Top1 co-migrates with RNAP I during RNAP I transcriptional arrest.
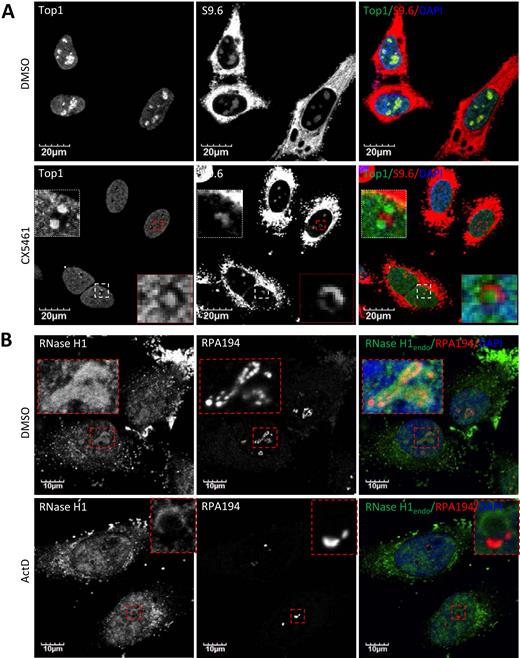
Top1 and RNase H1 localize to different perinucleolar sites upon RNAP I transcriptional arrest. (A) Co-immunofluorescent staining of Top1 and R-loops in HeLa cells treated with DMSO or 250 nM CX5461 (6 h). (B) Co-immunofluorescent staining of endogenous RNase H1 and RPA194 in HeLa cells treated with DMSO or 0.02 μg/ml ActD (2 h).
CPT induces nucleolar clearance of Top1 leading to transient accumulation of R-loops then nucleolar segregation and clearance of RNase H1
The rates of nucleolar clearance of Top1 in response to ActD and to CPT are different. ActD causes a slow relocation (30 min) of Top1 due to inhibition of rRNA synthesis and nucleolar segregation, whereas CPT induces a rapid clearance (2 min) of Top1 from nucleoli independent of the state of transcription (51). Due to the loss of Top1, CPT can induce rRNA transcriptional arrest and nucleolar segregation upon prolonged exposure; in line with this, nucleolar clearance of RNase H1 was observed upon prolonged treatment of CPT (Figure 2D). To further examine mechanisms for nucleolar clearance of RNase H1, we performed a time course experiment in HeLa cells after treatment with CPT. Images were recorded with the same settings to allow the comparison of signal intensity. After 15 min in the presence of CPT, Top1 was not observed in nucleoli (Figure 7) as was reported (51). However, significant nucleolar clearance of RNase H1 was only observed after 180-minute exposure to CPT, correlating with the loss of nucleolar S9.6 signals (Figure 7). Staining of NCL1 suggested that nucleolar segregation occurred between 60 and 180 min of exposure to CPT (Figure 7). Consistent with the occurrence of nucleolar segregation, northern analysis suggested that the levels of pre-rRNA reduced moderately at 60 minutes of exposure to CPT, and reduced almost completely after 180 minutes (Supplementary Figure S9A). Since the nucleolar-localization of RNase H1 and R-loops depends on the on-going RNAP I transcription, RNase H1 and R-loops delocalized out of nucleoli around the same time as the occurrence of nucleolar segregation and transcriptional inhibition. These results suggest that nucleolar clearance of RNase H1 induced by CPT is a result of the RNAP I transcriptional arrest and the loss of transcriptional R-loops. In addition, as was reported previously (61), CPT induced a time-dependent degradation of Top1, but levels of RNase H1 protein were not affected (Supplementary Figure S9B). This suggests that the observed nucleolar loss of RNase H1 in the presence of CPT is due to the protein delocalization not degradation.
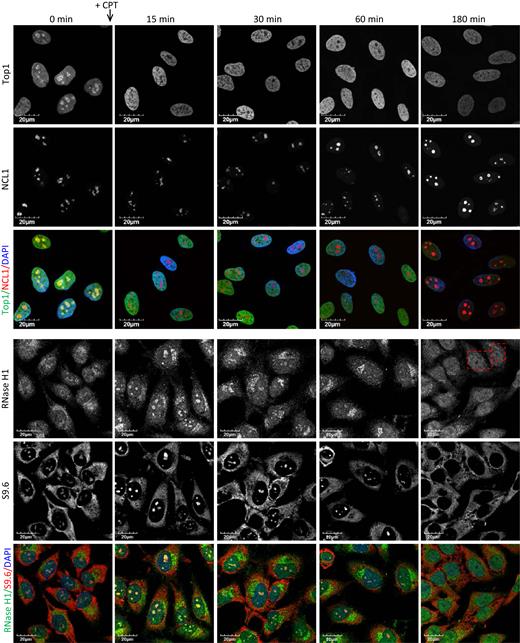
CPT treatment results in transiently increased accumulation of nucleolar DNA:RNA hybrids and RNase H1, followed by nucleolar segregation and clearance of RNase H1. Immunofluorescent staining of Top1, NCL1, endogenous RNase H1 and R-loops (S9.6) in HeLa cells treated with DMSO or 20 μM CPT for indicated times.
After 15-min exposures to CPT, the nucleolar-to-nucleoplasm ratio of RNase H1 staining was significantly increased relative to levels in cells treated with DMSO. Nucleolar signals of RNase H1 began to decrease after 30 min and diminished completely in some cells after the 180-min exposure to CPT (Figure 7). It has been reported that CPT can cause a transient stabilization of mitochondrial and nuclear R-loops in HCT116 cells (28), and we observed moderately elevated nucleolar S9.6 signals after 15- to 30-min exposures to CPT, which coincided with the time course for the nucleolar clearance of Top1 and accumulation of RNase H1 (Figure 7). Upon treatment of cells with CPT, two events happen. An early nucleolar clearance of Top1 leads to a transient accumulation of R-loops that appears to recruit RNase H1 into the nucleoli. The loss of Top1 then results in nucleolar segregation and transcriptional arrest of RNAP I, which subsequently results in fewer R-loops and nucleolar clearance of RNase H1. The dynamics of RNase H1 re-localization in the presence of CPT strongly suggest that the nucleolar/nucleoplasmic distribution of RNase H1 depends on the presence of R-loops that form during RNAP I-catalyzed transcription.
RNase H1 and Top1 are both involved in resolving transcriptional R-loops along rDNA
RNAP I transcription-dependent localization of RNase H1 and Top1 to the nucleoli suggests that these proteins could be involved in suppressing the formation of DNA:RNA hybrids generated as byproducts of rRNA synthesis. To profile the distribution of DNA:RNA hybrids, RNase H1 and Top1 along the rDNA, ChIP-qPCR experiments were performed. Efficient immunoprecipitation of endogenous RNase H1 and Top1 was confirmed by western analysis (Supplementary Figure S10A). S9.6 antibody has been validated in many previous studies to be suitable for ChIP experiments (50,62,63). Nine primer-probe sets were designed to cover different regions along the human rDNA (Figure 8A). Primer probes sets #1 and #2 are located upstream of the rRNA transcription start site to serve as negative controls for S9.6 ChIP. In HeLa cells, DNA:RNA hybrids were detected by the primer-probe sets located within the 5′ external transcribed spacer (ETS) region (set #3), the boundary between 18S and internal transcribed spacer 1 (ITS1) (set #5), and the boundary between 5.8S and ITS2 (set #7) (Figure 8B). Signals of DNA:RNA hybrids declined over 28S (set #8) and 3′-ETS (set #9) regions. Endogenous RNase H1 occupancy and Top1 occupancy were highest over the 5′-ETS regions (set #3), where accumulation of R-loops was detected with the S9.6 antibody (Figure 8B–D). Significantly higher occupancies of both RNase H1 and Top1 were detected over the two other regions with detectable DNA:RNA hybrids, the boundary between 18S and ITS1 (set #5) and the boundary between 5.8S and ITS2 (set #7) (Figure 8B–D). These results suggest that the distributions of both RNase H1 and Top1 along the rDNA units correlates with the occurrence of DNA:RNA hybrids.
![RNase H1 and Top1 suppress the formation of RNAP I transcriptional R-loops in mammalian cells. (A) Schematic representation of rDNA with primer-probe sets indicated. (B–D) ChIP-qPCR profiles of the distribution of (B) R-loops (detected with S9.6), (C) endogenous RNase H1 and (D) Top1 along the rDNA in HeLa cells. (E) ChIP-qPCR profiles of the distribution of R-loops along rDNA when RNase H1, Top1 or both proteins were depleted from HeLa cells using siRNAs. (F) ChIP-qPCR profiles of the distribution of R-loops along rDNA in mock-treated cells, RNase H1-overexpressing cells (AdV[H1]), Top1-depleted cells, and RNase H1-overexpressing cells that are depleted of Top1. (G) ChIP-qPCR profiles of the distribution of R-loops (left panel) and RNase H1 (right panel) along rDNA in HeLa cells treated with DMSO or CPT (15 min). The error bars represent standard deviation from three parallel experiments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/45/18/10.1093_nar_gkx710/3/m_gkx710fig8.jpeg?Expires=1716333752&Signature=W-26n5tWPgh~KBg1WNtB45KaHRfPfdc0x8rGHGPJbrWkg4P4O2XNRKSWDzvClR1MoFrv4NQ0MSbdseFsI1M5iarH1RyisfwJZgo4bP6c1NF26pRKG6Wg56pB3WGT8FZZLhj~aruwNP2eiJN4zOy76TcLXVSlHswAQKlCR8OvK-LXqOfjAbnnPNfpjbYysmHLwnY025WqDo4zJiMVqr8QRfhpI14wKQ1g0AZbqptqixcK~TYhajCgiQEN9scxUx0wY2RVVnR7AGncb02m~50mWspzE6i4nSYNMIBUn5OtaLhq0StNAsUYSJ8cu1xsddREHznm8R4fjyAZdi5D5zEVTA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
RNase H1 and Top1 suppress the formation of RNAP I transcriptional R-loops in mammalian cells. (A) Schematic representation of rDNA with primer-probe sets indicated. (B–D) ChIP-qPCR profiles of the distribution of (B) R-loops (detected with S9.6), (C) endogenous RNase H1 and (D) Top1 along the rDNA in HeLa cells. (E) ChIP-qPCR profiles of the distribution of R-loops along rDNA when RNase H1, Top1 or both proteins were depleted from HeLa cells using siRNAs. (F) ChIP-qPCR profiles of the distribution of R-loops along rDNA in mock-treated cells, RNase H1-overexpressing cells (AdV[H1]), Top1-depleted cells, and RNase H1-overexpressing cells that are depleted of Top1. (G) ChIP-qPCR profiles of the distribution of R-loops (left panel) and RNase H1 (right panel) along rDNA in HeLa cells treated with DMSO or CPT (15 min). The error bars represent standard deviation from three parallel experiments.
To assess the effects of RNase H1 and Top1 on R-loop formation, S9.6-ChIP was performed in cells that were depleted of these enzymes using siRNAs (Supplementary Figure S10B). siRNA-mediated depletion of either RNase H1 or Top1 only moderately elevated DNA:RNA hybrids at regions with positive S9.6 signals (set #5 and set #7), but the elevation of S9.6 signals was significantly more prominent in cells double-depleted of both RNase H1 and Top1 (Figure 8E). This suggests that RNase H1 and Top1 may partially compensate for each other in suppressing R-loop formation. Only when both proteins were depleted did R-loops accumulate significantly along the rDNA. Other proteins found in the nucleolus, such as XRN2 and WRN, which were previously shown to suppress formation of R-loops (11,64), are probably responsible for the R-loop resolution in the absence of RNase H1 and Top1.
To test if RNase H1 can compensate for the loss of Top1, we overexpressed RNase H1 in Top1-depleted cells and performed S9.6-ChIP. Overexpression of RNase H1 in mock-treated cells reduced the presence of R-loops at regions with positive S9.6 signals, especially for set #7 where levels of R-loops are high (Figure 8F), validating the role of RNase H1 in suppressing R-loops. R-loops accumulated in Top1-siRNA treated cells but overexpression of RNase H1 in Top1-depleted cells reduced R-loop signals. These data suggest that both RNase H1 and Top1 act over the same regions along the rDNA to suppress R-loop formation and that RNase H1 can at least partially compensate for the loss of Top1.
To confirm our observation that CPT induces transient stabilization of nucleolar DNA:RNA hybrids that recruit RNase H1 (Figure 7), we performed S9.6-ChIP and RNase H1-ChIP in HeLa cells treated with DMSO or CPT for 15 min. Treatment with CPT significantly increased S9.6 occupancy over rDNA, especially around the boundary between 18S and ITS1 (Figure 8G). Importantly, increased RNase H1 occupancy over the same region was observed during CPT exposure, suggesting that short exposure of cells to CPT can stabilize DNA:RNA hybrids in some regions along rDNA which can recruit RNase H1 to these locations.
Overexpression of RNase H1 causes significant DNA damage and reduces levels of Top1 protein
Uncontrolled accumulation of R-loops often results in DNA breaks and genome instability. Depletion of proteins that are involved in R-loop prevention or resolution, such as SETX and THO, can lead to the formation of both single-strand and double-strand breaks (DSBs) in cells (65). Depleting endogenous RNase H activity impairs R-loop removal in budding yeast, causing DNA damage that occurs preferentially in the repetitive rDNA locus (12). Liver apoptosis and regeneration were observed in hepatocyte-specific Rnaseh1-knockout mice, presumably due to both mitochondrial defects and nuclear DNA damage (26). However, little is known about cellular responses to RNase H1 overexpression. We found that although RNase H1 overexpression suppressed the formation of DNA:RNA hybrids, structures that are potential threats to genome integrity, a significant increase in DSBs, quantified using a neutral comet assay, was observed in cells overexpressing RNase H1 compared to control cells (Figure 9A–C). DSBs were found only in cells infected with adenovirus encoding catalytically active RNase H1 but not the inactive E186Q mutant or control viruses. The intensity of the comet tail relative to the head was quantified for ∼200 cells per group. A 5-fold increase in median tail-to-head ratio was observed for cell overexpressing catalytic active RNase H1 compared with the control groups (Figure 9B and C).
![Overexpression of RNase H1 causes DNA damage and reductions in levels of Top1 and other components of the NHEJ pathway. (A–C) Neutral comet assays were performed in control untreated HeLa cells, cells that are infected with control virus (AdV[Null]), cells that express full-length RNase H1 (AdV[H1]), and cells that express catalytically inactive RNase H1 (AdV[H1-E186Q]). HeLa cells were infected with adenoviruses for 48 h at 100 moi. (A) Representative microscopy images. (B) Tail-to-head ratios calculated from ∼200 cells per each treatment. (C) Calculations for minimal, 25th percentile, median, 75th percentile and maximum tail-to-head ratio in each group were listed. (D) Western analysis of Top1 and NHEJ factor levels from HeLa cells transiently infected with specific concentrations of AdV[H1] to express full-length RNase H1 for 48 h. (E) Western analysis of indicated proteins from HeLa cells transiently infected with control adenoviral vector (AdV[Null]), or with vectors for expression of full-length (AdV[H1]) or truncated RNase H1 with MTS deleted (AdV[H1-Nuc]) at 100 moi for 48 h. (F) Western analysis of indicated protein levels in HeLa cells transiently infected with specified concentrations of adenoviral vector encoding catalytically inactive RNase H1 (AdV[H1-E186Q]) for 48 h. (G) Western analysis of indicated protein levels from control HEK293 cells and HEK293 cells that stably overexpressing full-length human RNase H1.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/45/18/10.1093_nar_gkx710/3/m_gkx710fig9.jpeg?Expires=1716333752&Signature=zjdwP-Pz2X-8aGEdQmCI6-y6U~WI-NmGi6oGC8j~IUW48j4PH22JE4E11ZlONlM4MZOhW80W9zyXFq-yD4HwQoRZRYTaU6vHQp19uc0rXfEVYmsCqddmWWu7pLloaPPEujnOzk05-q1o5HuNLLQU27BOYdU65uoTG4ugZgWJzn~Tqu8r7um6OPAGoY79qw6Xz1KWObyTNwgEbMA0wAU1ciE9mlH~ItEA~Z716TKwe9qZ050spz-K4vJy9MdY6SZ-Epy3pfnFuwXs3Gqxp-oXOzfNZvFZAl3YsAyySb2rlnclyjfqFXKeFWtKsCkwY1G3UB6eksah~qCNy2oolK9dvw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Overexpression of RNase H1 causes DNA damage and reductions in levels of Top1 and other components of the NHEJ pathway. (A–C) Neutral comet assays were performed in control untreated HeLa cells, cells that are infected with control virus (AdV[Null]), cells that express full-length RNase H1 (AdV[H1]), and cells that express catalytically inactive RNase H1 (AdV[H1-E186Q]). HeLa cells were infected with adenoviruses for 48 h at 100 moi. (A) Representative microscopy images. (B) Tail-to-head ratios calculated from ∼200 cells per each treatment. (C) Calculations for minimal, 25th percentile, median, 75th percentile and maximum tail-to-head ratio in each group were listed. (D) Western analysis of Top1 and NHEJ factor levels from HeLa cells transiently infected with specific concentrations of AdV[H1] to express full-length RNase H1 for 48 h. (E) Western analysis of indicated proteins from HeLa cells transiently infected with control adenoviral vector (AdV[Null]), or with vectors for expression of full-length (AdV[H1]) or truncated RNase H1 with MTS deleted (AdV[H1-Nuc]) at 100 moi for 48 h. (F) Western analysis of indicated protein levels in HeLa cells transiently infected with specified concentrations of adenoviral vector encoding catalytically inactive RNase H1 (AdV[H1-E186Q]) for 48 h. (G) Western analysis of indicated protein levels from control HEK293 cells and HEK293 cells that stably overexpressing full-length human RNase H1.
We found that upon RNase H1 overexpression, a dose-dependent reduction was observed for Top1 protein (Figure 9D). In addition, protein levels of the RNase H2A subunit were slightly elevated, presumably as a consequence of Top1 reduction, due to the overlapping function of RNase H2 and Top1 in ribonucleotide excision repair. Moreover, reductions that depended on RNase H1 dose were also observed for P54nrb protein. P54nrb and its heterodimer partner PSF interact directly with Top1 and substantially stimulate Top1-mediated relaxation of DNA supercoils (66). P54nrb and PSF proteins share similar domain arrangements and structures, and, with PSPC1, form a family known as the Drosophila behavior human splicing (DBHS) factors (67). The three DBHS proteins can form homo- or heterodimers and are enriched in nuclear paraspeckle bodies (68). It seemed that only the fraction of P54nrb proteins that form P54nrb–PSF–Top1 trimers were reduced in cells that overexpress RNase H1, since protein levels of PSPC1 were not affected by RNase H1 overexpression. Importantly, p54nrb and PSF are involved directly in DSB repair through the non-homologous end joining (NHEJ) pathway (69). Simultaneous depletion of p54nrb and PSF induce p53-dependent apoptotic cell death (48).
Other proteins in NHEJ pathway were also affected by RNase H1 overexpression. Levels of two major components in NHEJ pathway, Ku70 and PARP1, were significantly increased in an RNase H1 dose-dependent manner. Disruption of the NHEJ pathway due to RNase H1 overexpression explains, at least partially, the increase in DSBs observed upon overexpression of catalytically active form of RNase H1 (Figure 9A–C). Interestingly, it was recently reported that depletion or overexpression of RNase H inhibits HR-mediated DSB repair in S. pombe (70). Thus, levels of Ku70 and PAPR1 may increase in response to defects in the HR pathway since PAPR1 was observed to be upregulated when the HR pathway is impaired as it is in BRCA1-mutated tumors (71).
Considering the potential involvement of RNase H1 in DNA replication in processing Okazaki fragments, protein levels of FEN1, PCNA and RPA70 were also examined. Overexpression of RNase H1 had no effect on these proteins (Figure 9D). Finally, levels of P32, a previously reported RNase H1-associated protein (72), were not significantly affected by transient overexpression of RNase H1 (Figure 9D). Due to the compensatory changes in levels of proteins belonging to NER and NHEJ pathways, the increase in DSBs observed in RNase H1-overexpressed cells is likely due to the impaired DSB repair pathways.
To ensure that the changes in the levels of these proteins are indeed as a consequence of the increased RNase H1 levels, we overexpressed either full-length RNase H1 (AdV[H1]) or a truncated version (Adv[H1-Nuc]) which lacks the MTS and localizes only to nuclei in HeLa cells. A control adenoviral vector (AdV[Null]) was included in these experiments to exclude secondary effects of virus infection. Indirect immunofluorescent staining indicated dual localization of the full-length RNase H1 to mitochondria and nuclei (Figure 4C), as reported previously due to the differential translation initiation at two different in frame AUGs of the same mRNA (31,34). The truncated RNase H1 lacking the MTS was almost exclusively observed in the nuclei (Figure 4C). Although overexpression of both forms of RNase H1 reduced nuclear-localized Top1 levels, only overexpression of the full-length RNase H1 reduced mtSSBP1, a protein that mainly localizes to mitochondria (Figure 9E), suggesting that these proteins are reduced as a consequence of the changes in abundance of RNase H1 protein in corresponding subcellular compartments.
Next, we examined whether enzymatic activity of RNase H1 is required for the reduction of Top1 protein. Infection of HeLa cells with adenovirus encoding this catalytic inactive mutant (AdV[H1-E186Q]) caused no reduction in levels of Top1 or Ku70 proteins, suggesting that changes in levels of Top1 protein are linked to the increase cellular RNase H1 activity (Figure 9F). This is also consistent with our observation that only the expression of the catalytically active form of RNase H1 induced DSBs (Figure 9A–C).
To confirm the above phenotypes obtained from transient overexpression of RNase H1, we also examined the protein levels of Top1 in a HEK293 cell line stably overexpressing RNase H1. As was observed in the transient overexpression system, both Top1 and P54nrb were reduced in the HEK293 cell line stably overexpressing RNase H1 as compared with the control HEK293 cells (Figure 9G). These results suggest that overexpression of the catalytically active RNase H1 causes reductions in levels of Top1 protein and induces DSBs. Considering the overlapping functions of RNase H1 and Top1 in R-loop resolution, it is possible that levels of Top1 are regulated by the abundance of RNase H1 through sensing the amounts of cellular DNA:RNA hybrids.
Levels of Top1 increase in liver-specific Rnaseh1-knockout mice
We tested the hypothesis that levels of Top1 are affected by the abundance of RNase H1 due to their partial functional redundancy in R-loop processing by analysis of hepatocyte-specific Rnaseh1-knockout mice. Loss of RNase H1 specifically in mouse liver was previously shown to induce liver toxicity (26). In hepatocytes isolated from Rnaseh1-knockout mice, the p53 pathway was activated relative to control wild-type hepatocytes (Figure 10A). Transcripts downstream in the p53 pathway, including Cdkn1a/p21 and Tnfrsf10b, were upregulated by 10-fold and 7-fold, respectively, in hepatocytes isolated from 6–7-week-old Rnaseh1-knockout animals (Figure 10A). Elevated levels of p21 were also observed in Rnaseh1-knockout mice compared to controls (Figure 10B). Significantly elevated signals of S9.6 staining in nucleoli were observed in isolated primary hepatocytes from 6–7-week-old knockout animals compared with controls (Figure 10C). In livers of the 9–10-week-old Rnaseh1-knockout animals we observed an increase in Top1 by western analysis (Figure 10B). Likely due to the increase in Top1, no significant elevation in levels of nucleolar R-loops were observed in the 9–10-week-old knockout hepatocytes compared with those from control animals (Figure 10C). It has been reported that overexpression of Top1 reduced the formation of R-loops (73). These results suggest that protein levels of Top1 are elevated as a consequence of the loss of RNase H1 and that at least one of the mechanisms that results in this elevation is sensing of DNA:RNA hybrids.
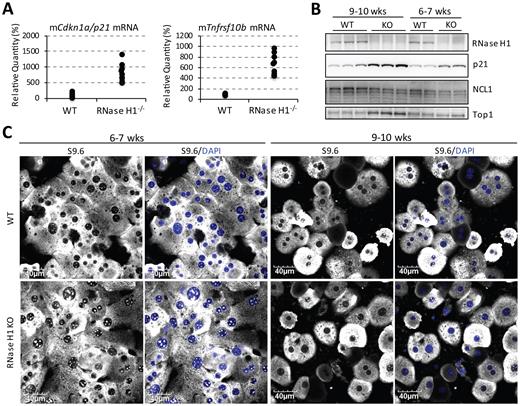
Top1 levels are increased in Rnaseh1-knockout mice to reduce nucleolar accumulation of R-loops. (A) qRT-PCR of Cdkn1a/p21 and Tnfrsf10b from isolated primary mouse hepatocytes. 10 animals per group were assayed. (B) Western analysis of RNase H1, p21 and Top1 in liver lysates from control mice and Rnaseh1-knockout mice that were either 6–7 weeks old (two animals/group) or 9–10 weeks old (three animals/group). (C) Representative images of immunofluorescent staining of S9.6 in isolated mouse hepatocytes.
DISCUSSION
We demonstrated here that both RNase H1 and Top1 participated in suppression of R-loops that form during RNAP I-mediated transcription in mammalian cells. We found that the nucleolar/nucleoplasmic partitioning of RNase H1 was regulated by RNAP I transcription and the presence of R-loops. RNase H1 co-migrated with DNA:RNA hybrids during rDNA transcriptional arrest, suggesting that the nucleolar localization of RNase H1 occurred at least partially due to its direct interaction with DNA:RNA hybrids. R-loop staining using DNA:RNA hybrid-specific antibodies S9.6 and D5H6 revealed that nucleoli are enriched for DNA:RNA hybrids, likely the result of R-loops formation due to active transcription of the GC-rich rDNA loci, compared with nucleoplasm. In the nucleoplasm, scattered dots of S9.6 signals were observed presumably representing sites of RNAP II transcription. The subcellular localization of RNase H1 correlated with the distribution of DNA:RNA hybrids in mitochondria, nucleoplasm, and nucleoli; RNase H1 is probably responsible for suppressing the formation of R-loops in all these cellular compartments. As a result, when RNAP I was inhibited and no RNAP I transcriptional R-loops were present in the nucleoli, RNase H1 delocalized to the nucleoplasm to engage in suppressing R-loops from RNAP II transcription.
In our experiments with cultured human cells and in mice, levels of Top1 protein depended on the abundance of RNase H1. Further, both the decreased abundance of Top1 protein and the occurrence of DSBs upon RNase H1 overexpression required the catalytic activity of RNase H1. As both Top1 and RNase H1 are enriched in nucleoli and process transcriptional R-loops, it is possible that sensing of DNA:RNA hybrids results in regulation of Top1 levels. In HeLa cells, RNase H1 and Top1 co-occupied the same regions along rDNA that were also bound by antibody that recognizes DNA:RNA hybrids. It is possible that overexpression or depletion of RNase H1 results in corresponding decreases or increases in amounts of DNA:RNA hybrids, which in turn influence the abundance of Top1.
The increased levels of Top1 in Rnaseh1-knockout mice support our hypothesis of compensatory regulation. Elevated amounts of DNA:RNA hybrids were observed in hepatocytes isolated from Rnaseh1-knockout mice at 6–7 weeks, but not at 9–10 weeks compared with the control animals. Levels of Top1 protein were higher in knockout hepatocytes from 9–10-week-old mice relative to those from 6–7-week-old mice and controls. These results demonstrate the correlation between the levels of RNase H1, Top1, and the accumulation of DNA:RNA hybrids. Although RNase H1 was completely absent from 6–7-week-old knockout animals, increased levels of Top1 protein were only observed in the 9–10-week-old mice. This lag between RNase H1 depletion and Top1 upregulation implies that cells need to sense the loss of RNase H1 or its consequences, such as R-loop accumulation and genome instability, and response by increasing Top1 protein levels. As both RNase H1 and Top1 were observed in regions of DNA:RNA hybrid formation, this may be the link between the compensatory regulation of these two proteins. However, we cannot exclude the possibility that DNA damage responses related to the overexpression or depletion of RNase H1 feedback to influence levels of Top1, since Top1 has been implicated in DNA repair pathway function (74).
The cellular abundance of endogenous RNase H1 protein is very limited. However, levels of RNase H1 do not seem to be regulated at transcriptional level, as indicated by the moderately abundant levels of mRNA. In addition, mRNA levels of RNase H1 do not seem to change during the cell cycle. This is different from RNase H2; its mRNA levels increase during S phase, consistent with its involvement in nuclear DNA replication (31,75). The limited abundance of RNase H1 protein is probably due to dual controls at the translational level: A weak Kozak signal context for both AUGs and a potent upstream open reading frame (uORF) result in inefficient protein translation (34,76). The two in-frame ORFs and the uORF of Rnaseh1 also fine-tune the ratio between mitochondrial versus nuclear RNase H1. In addition, multiple PI shifts were observed for RNase H1 by two-dimensional gel electrophoresis, suggesting that the protein is post-translationally modified (72). Whether these post-translational modifications impact localization, abundance, and functions of RNase H1 await further investigation. All these observations support the conclusion that abundance of RNase H1 is tightly regulated in mammalian cells.
The existence of mechanisms to fine-tune the levels of RNase H1 protein is consistent with our observation in mammalian cells and those of others in yeast that depletion or overexpression of RNase H1 induces DNA breaks (70). The induction of DNA breaks upon loss of RNase H1 is easily rationalized since the relationship between accumulation of R-loops and genome instability has been well-established in E. coli and yeast (24,25). Activation of the p53 pathway and spontaneous liver apoptosis occur in animals that are deficient in RNase H1 (Figure 10) (26,35). Mitochondrial dysfunction contributes significantly to the liver toxicity observed in Rnaseh1-knockout mice since the impaired mitochondrial DNA replication is observed in 6-week-old RNase H1 knockout animals (26). However, since we observed that the levels of nucleolar DNA:RNA hybrids were elevated significantly in 6–7-week-old knockout hepatocytes, it is possible that the loss of RNase H1 in both mitochondria and nucleus contribute to p53 activation and hepatocyte death.
It was reported recently that overexpression of RNase H1 caused DNA breaks in S. pombe. This study also demonstrated a beneficial role of DNA:RNA hybrids in facilitating HR-mediated DSB repair (70). In the model proposed, overexpression of RNase H1 impairs HR-mediated DSB repair. By destabilizing DNA:RNA hybrids, overexpression of RNase H1 causes excessive strand resection and loss of repeat regions around DSBs. In HeLa cells, we also observed significant accumulation of DSBs upon overexpression of RNase H1. In addition, levels of many NHEJ factors, including p54nrb, Ku70 and PARP1, were altered. DSBs can be repaired through two pathways: the rapid but error-prone NHEJ or the more accurate HR pathway. There is evidence for competition between the HR and NHEJ pathways, and it is possible that overexpression of RNase H1 impacts HR-mediated DSB repair, which results in changes in the abundance of NHEJ factors. Although direct involvement of RNase H1 in the NHEJ pathway has not been reported, this requires further evaluation.
Understanding the biological functions of RNase H1 has long been limited by a lack of tools to monitor subcellular distribution and dynamics due to the extreme low abundance of the protein. Using an antibody raised against human RNase H1 (amino acids 47–287), we were able to demonstrate the subcellular distribution of endogenous human RNase H1 to mitochondria, nucleoplasm, and nucleoli. Dynamic localization of RNase H1 between nucleoli and nucleoplasm in response to RNA transcription and transcriptional R-loops was observed, consistent with a role in suppressing nucleolar R-loop formation. In addition, overexpression or depletion of RNase H1 in mammalian cells or mice resulted in DNA breaks, presumably due to the changes in the abundance of Top1 as well as proteins in NHEJ pathway. Our results suggest that subcellular distribution and levels of RNase H1 are fine-tuned in cells to maintain genome integrity.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Timothy A. Vickers and Shiyu Wang for insightful discussions.
FUNDING
Ionis Pharmaceuticals, Inc. Funding for open access charge: Ionis Pharmaceuticals, Inc.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments