





Abstract
The Topology Data Bank of Transmembrane Proteins (TOPDB, http://topdb.enzim.ttk.mta.hu) contains experimentally determined topology data of transmembrane proteins. Recently, we have updated TOPDB from several sources and utilized a newly developed topology prediction algorithm to determine the most reliable topology using the results of experiments as constraints. In addition to collecting the experimentally determined topology data published in the last couple of years, we gathered topographies defined by the TMDET algorithm using 3D structures from the PDBTM. Results of global topology analysis of various organisms as well as topology data generated by high throughput techniques, like the sequential positions of N- or O-glycosylations were incorporated into the TOPDB database. Moreover, a new algorithm was developed to integrate scattered topology data from various publicly available databases and a new method was introduced to measure the reliability of predicted topologies. We show that reliability values highly correlate with the per protein topology accuracy of the utilized prediction method. Altogether, more than 52 000 new topology data and more than 2600 new transmembrane proteins have been collected since the last public release of the TOPDB database.
INTRODUCTION
Every cell and organelle is surrounded by a double lipid layer, which separates the internal side from the environment. Integral membrane proteins form the most prevalent protein class. However, the structure determination of transmembrane proteins (TMPs) is one of the most challenging tasks for structural biologists. While 25–30% of the genomes encode TMP, only 2% of the solved structures in Protein Data Bank (PDB) belong to this type of proteins (1–3).
To overcome the difficulties of structure determination, molecular biologists often try to determine the localization of TMP segments relative to the membrane. These experiments are time-consuming and provide only limited information about the topologies. Pioneering studies used random insertion of alkaline-phosphatase by transposons (4), followed by the application of other reporter enzymes or proteins like β-galactosidase (LacZ) (5) and β-lactamase (BlaM) (6) in bacteria, invertase and histidinol dehydrogenase in yeast (7) or various kinds of fluorescence proteins (GFP, roGFP) (8,9). Novel molecular biology techniques were also developed to insert the reporter enzyme or protein to a specific position of the investigated protein. Besides fusions with reporter proteins, immunolocalization of the outer or inner part of a TMP was also used to determine the topology of TMPs, either by generating monoclonal antibodies against a certain part of the TMP (10) or by inserting known epitopes into various positions of TMPs (11–15). While the former immunolocalization techniques did not alter the native sequence of the investigated TMP, epitope insertions might result in structural and hence functional alteration of the protein. Still, these variations are less detrimental than in the case of fusion proteins, where a part of the protein is completely removed. A method to introduce smaller variations to TMPs can be the insertion/deletion of asparagine-linked glycosylation sites. N-linked glycosylation occurs on the luminal/extracytosolic site of the TMPs, if there is a common sequence motif (NxS/T, x can be any amino acids except proline) more than 10 amino acids in sequence from the border of transmembrane segments (16). The glycosylation events can be monitored by molecular weight shift on the gel, or recently by tandem mass spectrometry techniques (17–20). An even smaller topological signal can be generated by chemical modification of cysteine residues either by membrane permeable or impermeable chemical reagents (21–23).
Certainly, the most accurate way to determine the topology of TMPs is solving their 3D structure by nuclear magnetic resonance or x-ray crystallography. Besides the technical difficulties of applying these methods on TMPs, vital components, the co-ordinates of the membrane bilayer itself are missing from the final structure files deposited to PDB. With the exception of a few tightly bound lipid or detergent molecules, the deposited experimental data have no direct indication on how the protein is immersed into the membrane under native conditions, and do not provide information about the exact location of the lipid bilayer (24). Therefore, this information has to be supplied later using the 3D co-ordinates of the proteins. Currently, there are only three publicly available algorithms addressing this problem: TMDET (1,25), Positioning of Proteins in Membrane (PPM) (26,27) and Ez-potential (28). TMDET uses a geometrical algorithm to determine the possible orientation of the membrane proteins in the lipid bilayer, PPM utilizes a more sophisticated biophysical model (minimizing TMPs’ transfer energies from water to the lipid bilayer) to calculate the orientation of TMPs in the double lipid bilayer. Ez-potential is a knowledge-based potential for positioning not just TMPs but all membrane associated structures. All these algorithms were applied to the whole PDB database to extract TMPs and to determine the topography of these TMPs, which resulted in the establishment of PDBTM (2,3), OPM (27,29) and nrDB (28) databases, respectively. However, the 3D co-ordinates can only determine the topography, and not the topology. Therefore, topology has to be generated using complementary information about the localization of the inside and outside protein parts.
Experimental topology data can be used to predict the full topology of TMPs by constrained predictions. Hidden Markov model based prediction methods can be easily modified to incorporate experimentally determined topology data into the prediction as constrains by the modification of the Baum–Welch and Viterbi algorithms (see Bagos et al. (30) for details). The first such application, which was able to make constrained prediction on TMPs, was HMMTOP2 (31) in 2001, followed by two other hidden Markov model based methods, TMHMM and Phobius (32–35). It was shown that constrained prediction increases both the accuracy and the reliability, first in the case of the human multidrug resistance-associated protein 1 (MRP1) (31). The optimal placement of constraints was also investigated and it was shown that the accuracy could be increased by 10% if the N- or C-terminal of the polypeptide chain is locked in the prediction. More significant increase (maximum 20%) in the prediction accuracy can be obtained by the fixation of loop or tail residues in turns to their experimentally annotated location (36). Recently we have developed a constrained consensus prediction method called CCTOP, and mapped the complete human genome in order to determine topology of TMPs in the human transmembrane proteome (HTP) (37).
Here we present a major update on our TOPDB database originally published seven years ago. Topology data were gathered from several sources including literature using PubMed; annotated sequence databases like UniProt; the PDBTM database by extending topography information with topology data described in the original article of the solved structure; results of high-throughput determination of N- or O-glycolysation sites; and by the comparison of the UniProt, PDB and PDBTM databases. The collected experimental topology data were utilized as constraints by the CCTOP algorithm to give the most probable topologies of TMPs. Altogether, more than 75 000 topology data were collected by extracting information from about 4200 publications that resulted in topology for 4190 proteins. The database is available at http://topdb.enzim.ttk.mta.hu.
MATERIALS AND METHODS
Data resources
Four sources of data were utilized during the building of the database: Pubmed (up to August, 2014), UniProt (2014_07 release), PDB and PDBTM (both up to 22 August 2014).
CCTOP algorithm
CCTOP incorporates the prediction results of 10 topology prediction methods, the experimental constraints of the given and homologous entries in the TOPDB and other bioinformatical evidences from the TOPDOM database (38). The 10 selected prediction methods are: HMMTOP (31,39), Membrain (40), MEMSAT-SVM (41), Octopus (42), Philius (43), Phobius (32), Pro-TMHMM (44), Prodiv-TMHMM (44), Scampi-MSA (45) and TMHMM (46). To determine the homologous sequences, the BLAST algorithm was used against the TOPDB database itself with the parameter E-value 10−10. Hits were accepted if the following clauses were all true: (i) the hit's length was above 80% of the query sequence's length; (ii) all TM helices were covered in the homologous TOPDB entry by the alignment; (iii) sequence identity was above 40% within HSPs (high-scoring segment pair). Topology data of the homologous proteins in the TOPDB database were used in the constrained prediction by mirroring their sequential positions according to the position of the HSPs. The search engine of the TOPDOM homepage (38) was used to locate those domains/motifs in the sequences that were found conservatively on the same side of TMPs, and we used the position and topology localization of the result(s) as constraint(s). For more details see (37) and the home page of the TOPDB database.
Calculating reliability
The source code of the HMMTOP program was modified in order to calculate the sum of the posterior probabilities along the Viterbi path. According to the unique hidden structure of the HMMTOP, the posterior probabilities were summed up for each main hidden state type (inside, membrane, loop and outside) in each position of the amino acid sequence, then these probabilities were summed up along the most probable state sequence provided by the Viterbi algorithm. We use this sum divided by the length of the protein to measure the reliability (37).
RESULTS AND DISCUSSION
Data processing
Literature search
We have analyzed all publicly available articles containing the keywords ‘topology’ and ‘transmembrane’. To achieve the most reliable result, all search results were manually processed. Characterization and classification of experiments are described in the TOPDB homepage under the Documents menu. The main classes are: (i) fusions with reporter enzymes or proteins, (ii) determination or alteration of post translational modifications, (iii) experiments based on protease digestions, (iv) immunolocalization of TMPs segments, (v) chemical modification. In addition, we checked publications that can be found in UniProt entries as literature sources containing the ‘TOPOLOGY’ keyword in lines starting with the ‘RP’ label. Altogether 1438 entries, containing one or more experimental topology data have been collected.
Converting data from PDBTM
The PDBTM database is automatically updated every week following the updates of PDB database. Since the last update of the TOPDB database, more than 1300 TMP structures have been solved, submitted to PDB and processed by the TMDET algorithm resulting in topography information for these TMPs. The PDBTM entries do not contain topology data, just the Side1 and Side2 notations to distinguish the two parts of a TMP which are on the two sides of the double lipid layer. Therefore, the topology information (if it was obtainable) had to be added manually by checking the original publications for the new structures. For the recent update of the TOPDB database we processed all publicly available original papers describing the structures to produce the side definitions. In the cases when we had no access to a publication, or the PDB entry contains the ‘To be published’ phrase, we used the side definitions of homologous entries. In case the sidedness of the TMP could not be determined, only the sequential localizations of the transmembrane helices were used in the final constrained prediction.
Combining data from the UniProt, PDB and PDBTM databases
In the first release of the TOPDB database all structures, which correspond to the soluble fragments of TMPs, were also gathered from the PDB database, as these cases also contain information about the topology. All entries in the UniProt database containing ‘FT TRANSMEM’ lines or membrane subcellular localization and cross-references to one or more PDB entries, which have not been filtered out as transmembrane by the TMDET algorithm, and therefore in the PDBTM database is classified as not TMP, were processed in this step. For each UniProt entry selected this way, available evidences of the exact localization of the structures were manually extracted from the publications. These data were incorporated as additional experimental data, specifying the inside/outside localization of a given part of a TMP. Altogether, more than 898 UniProt entries, containing 2797 PDB cross-references were processed in this step resulting in additional topology information for 653 new TOPDB entries.
High-throughput topology data
The rapid development of biotechnology tools and techniques in the last decade has facilitated the spreading of high-throughput proteomics research. The results extracted from these experiments can be used to enhance topology predictions. Two types of post-translational modifications, the N- or O-glycosylation and the ubiquitination are side-specific modifications, and can be detected by high-throughput techniques like trypsin digestion followed by the sequencing of the peptide fragments by various tandem mass-spectrometry methods (47–49). While glycosylation can occur only on the extra-cytosolic side, ubiquitination can occur only on the cytoplasmic side. We collected these types of topology data from the literature and the UniProt database. Altogether almost 1400 TOPDB entries contain topology data generated by a high-throughput technique.
Putting all the pieces together: predicting the topology by CCTOP
Recently, we have developed a novel prediction algorithm called CCTOP (37), which is a consensus and constrained prediction method based on the HMMTOP algorithm. The CCTOP method combines the results of (i) 10 different topology or topography prediction methods; (ii) the deposited experimental topology data both of the investigated TOPDB entry and its homologous entries (if any) in the TOPDB database; (iii) the sequential information of any homologous TMP in the UniProt database; and (iv) conservatively localized protein domains and sequence motifs form TOPDOM database. All of these data were integrated into the probabilistic framework of the hidden Markov model. The per protein topology prediction accuracy of the CCTOP algorithm was shown to be the highest among the other currently available state-of-the-art methods (37,45,50–51). Since the current version of the TOPDB database integrates topology data generated by high-throughput methods, as well as the results of the various global topology determination techniques (52–54), there are lot of entries which contain only one experimental topology data. We expect that by using the CCTOP algorithm, the accuracy of topology prediction in these entries can be increased, resulting in a more reliable database.
Current statistics of the TOPDB database
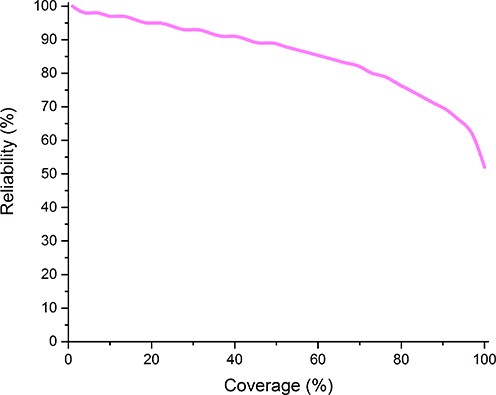
The distribution of the occurrences of the various experiment types among TOPDB entries in the first and the current release is shown in Table 1. The largest increase regarding the number of entries was in the case of the post-translational modification experimental type, due to the incorporation of high-throughput proteomics experiments, while the largest increase regarding the number of topology data was in the case of structure determination, since in this type of experiment one structure produces abundant topology data.
Distribution of experiment types over the TOPDB entries and over the total topology data in the first (purple bars) and the current (red bars) release of the TOPDB database
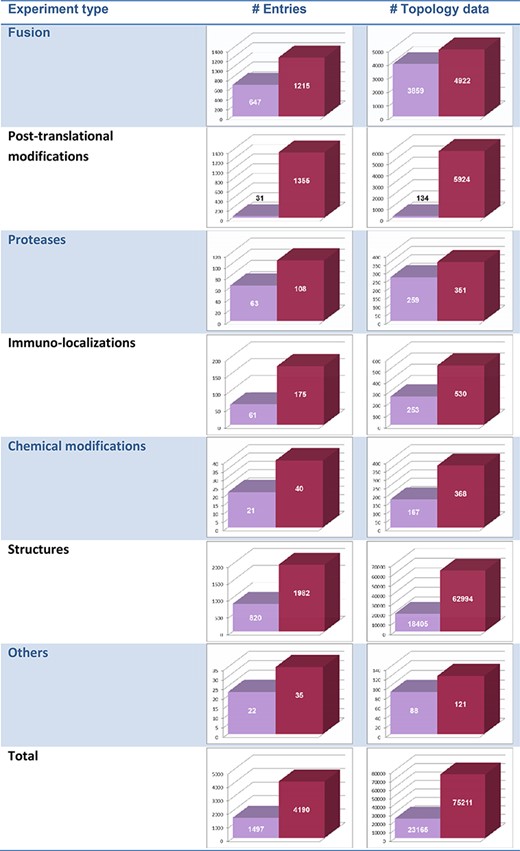
The distribution of the number of transmembrane segments among the TOPDB entries is shown in Figure 1. The most prevalent class is the bitopic TMP class; about 40% of α-helical TMPs belong to this class. We have found similar distribution in the HTP (http://htp.enzim.hu) (37), which indicates that the α-helical bitopic TMPs are not over-represented in the TOPDB database. However, we have to note that while in the HTP database (37) the second more abundant class of proteins contain 7 TM segments, in the TOPDB the second and third most prevalent classes of TMPs are those that contain 2 or 12 TM segments. This difference is probably due to the different organism sources in the two databases. The TOPDB database contains several bacterial TMPs, and it was shown that bacterial transmembrane proteomes contains less 7 TM proteins than eukaryote proteomes (41).
Distribution of proteins with different number of transmembrane segments in the TOPDB database.
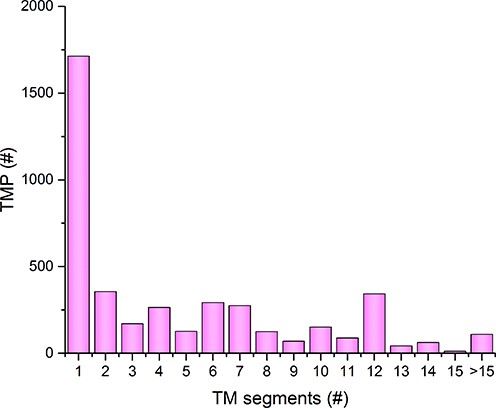
The calculated reliability distribution over the coverage is shown in Figure 2. The lower values (as compared to the HTP database) are probably the result of the conflicting experimental data.
Distribution of the calculated reliability in the TOPDB database. Entries were sorted according to the reliability, and plotted the order number divided by the size of the TOPDB database (coverage) vs reliability of the protein in that position of the sorted list.
Comparing the TOPDB to the UniProt database, we have found that in 966 cases the UniProt does not provide topology data, just the localization of TMHs, and for 415 proteins we could not found the localization of TMHs in the Uniprot. In 465 cases, when the UniProt entries contain topology data, one or more TMHs are missing compared to the TOPDB data.
Conclusions and future directions
The uses of the TOPDB database are quite widespread. It is a good starting point to develop benchmark sets for prediction methods (55); to develop TMP related databases (56–58); and for experimental characterization of individual TMPs (22,59). In the current release of the TOPDB database we focused on the quality and volume of the data. We used several ways to gather as much topology data as available from the various databases and the literature. These were (i) topography defined by the TMDET algorithm using the 3D structure from PDBTM database, extended by topology information from articles containing the description of the original 3D structures; (ii) solved structures of soluble domains of TMPs; (iii) experimental data published in the last couple of years; (iv) global topology analysis of transmembrane proteome of Saccharomyces cerevisiae and Escherichia coli (53,54) and; (v) topology data generated by high throughput techniques, like the sequential positions of N- or O-glycosylations. A new topology prediction algorithm, the CCTOP algorithm was applied to put the collected experimental topology data and other information into a unified topology model.
In this release, we did not enhance the functionality of the TOPDB homepage, the web engine remained the old PHP-based engine. In the future, in addition to regularly updating the data, we will develop a new engine, based on the Wt Web Toolkit C++ library, which was utilized with success in our previous works (3,60). Besides these technical improvements, we will continue to develop data mining algorithms to automatically collect experimental topology information from the literature.
We thank Zsuzsanna Gergely, Dániel Kozma, Lajos Kalmár and Gergely Szakács for the critical reading of the manuscript.
FUNDING
Hungarian Scientific Research Fund [K104586, http://www.otka.hu]. ‘Momentum’ Program of the Hungarian Academy of Sciences [to G.E.T.; LP2012-35]. Funding for open access charge: ‘Momentum’ Program of the Hungarian Academy of Sciences.
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
Comments