





Abstract
The ability of proteins to establish highly selective interactions with a variety of (macro)molecular partners is a crucial prerequisite to the realization of their biological functions. The availability of computational tools to evaluate the impact of mutations on protein–protein binding can therefore be valuable in a wide range of industrial and biomedical applications, and help rationalize the consequences of non-synonymous single-nucleotide polymorphisms. BeAtMuSiC (http://babylone.ulb.ac.be/beatmusic) is a coarse-grained predictor of the changes in binding free energy induced by point mutations. It relies on a set of statistical potentials derived from known protein structures, and combines the effect of the mutation on the strength of the interactions at the interface, and on the overall stability of the complex. The BeAtMuSiC server requires as input the structure of the protein–protein complex, and gives the possibility to assess rapidly all possible mutations in a protein chain or at the interface, with predictive performances that are in line with the best current methodologies.
INTRODUCTION
The formation of protein complexes plays an essential role in the regulation of numerous biological processes. The rational design or modification of the affinity and specificity of protein–protein interactions is a challenging issue that stimulated considerable efforts, as it presents many promising applications, notably for therapeutical purposes (1,2).
The characteristics of protein interfaces have been thoroughly investigated (3–10). Even if the diversity of binding modes precludes the identification of a simple set of general rules, a number of common features have been underlined, such as the importance of hydrophobic contacts and electrostatic interactions at the interface. Importantly, it has also been shown that a small fraction of the residues participating to the protein–protein interface are generally responsible for most of the binding affinity (11–13). These critical residues, commonly referred to as ‘hotspots’, are usually defined as positions where a mutation would cause an increase of the binding free energy of at least 2.0 kcal/mol. Alanine scanning mutagenesis has been widely used to experimentally characterize protein–protein interfaces and identify these hotspots, which constitute prime targets for the modulation of protein–protein interactions (14,15).
Considerable attention has been devoted to the development of computational methods for the identification of hotspot residues in protein–protein interfaces (16–29). Most rely on a machine learning technique to integrate a variety of features characterizing each residue and its environment. These features typically include information about sequence conservation, as well as physicochemical (e.g. residue hydrophobicity, electrostatic charge), structural (e.g. solvent accessibility, number of contacts, secondary structure), or energetic parameters. Although knowledge of the structure of the complex is generally required, methods have also been implemented to predict the localization of hotspots directly from the sequence (18), or from docking simulations (22).
Besides the binary classification of hotspot residues, a more general challenge consists in the estimation of the impact of mutations on the free energy of binding. Molecular mechanics combined to continuum solvent models, MM-PBSA or MM-GBSA (MM: molecular mechanics, PB: Poisson-Boltzmann, GB: generalized Born, SA: surface area), have been exploited for that purpose (30–33). Less computationally intensive approaches, based on empirical energy functions coupled with a somewhat simplified representation, have also been described (34–37). With a few exceptions (34,37), these methods have so far been mainly focused on evaluating the effects of mutations into alanine, but not into other types of amino acids.
We present here a webserver for the prediction of changes in protein–protein binding affinity on mutations. BeAtMuSiC is based on a set of statistical potentials adapted to a coarse-grained representation of protein structures, which allows the fast assessment of all possible mutations in the protein complex. Originally parametrized on the basis of a data set of mutations into alanine (38), our approach is here validated on a much larger data set including mutations into any kind of amino acid (39). In addition, our method stood among the top performers during the 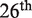 round of the blind prediction experiment Critical Assessment of PRedicted Interactions (CAPRI) (40), which consisted in the evaluation of ∼2000 mutations in two designed inhibitors of influenza hemagglutinin.
round of the blind prediction experiment Critical Assessment of PRedicted Interactions (CAPRI) (40), which consisted in the evaluation of ∼2000 mutations in two designed inhibitors of influenza hemagglutinin.
METHODOLOGY
Binding models
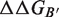 ) resulting from a mutation is then expressed as follows:
) resulting from a mutation is then expressed as follows: 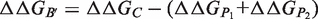
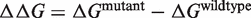 ,
, 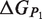 and
and 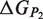 are the respective folding free energies of the two partners, and
are the respective folding free energies of the two partners, and 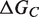 is the folding free energy of the complex as a whole (Figure 1). In the second model, the partners are unable to fold independently, and the change in binding free energy on mutation is thus given by
is the folding free energy of the complex as a whole (Figure 1). In the second model, the partners are unable to fold independently, and the change in binding free energy on mutation is thus given by 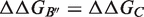
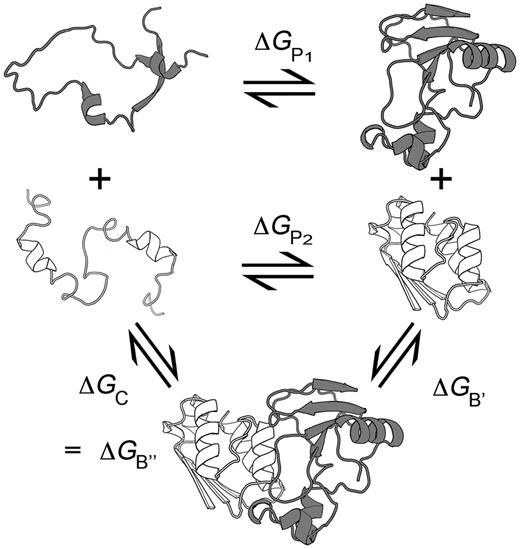
Schematic representation of the binding and folding free energies. 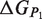 and
and 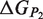 are the folding free energies of the two partners of the interaction.
are the folding free energies of the two partners of the interaction. 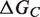 is the folding free energy of the complex as a whole. In the first binding model, the complex is formed from the association of two individually folded partners, and the binding free energy is
is the folding free energy of the complex as a whole. In the first binding model, the complex is formed from the association of two individually folded partners, and the binding free energy is 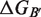 . In the second binding model, the proteins are unable to fold independently, and the binding free energy (
. In the second binding model, the proteins are unable to fold independently, and the binding free energy (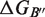 ) is thus equal to the folding free energy of the complex (
) is thus equal to the folding free energy of the complex (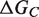 ). The figure was made using PyMOL.
). The figure was made using PyMOL.
Change in folding free energy on mutation
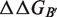 and
and 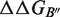 can be obtained from the estimation of the impact of the mutation on the folding free energy of the partners (
can be obtained from the estimation of the impact of the mutation on the folding free energy of the partners (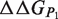 and
and 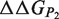 ) and of the complex (
) and of the complex (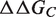 ). These contributions are computed as follows:
). These contributions are computed as follows: 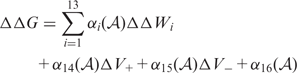
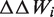 corresponds to the energetic change induced by the mutation, according to 1 of 13 different statistical potentials extracted from a data set of known protein structures (41). These potentials describe the correlations between amino acid types, pairwise inter-residue distances, backbone torsion angles and solvent accessibilities. The inter-residue distances are evaluated from the coordinates of the average geometric centers of the side chains. In addition, the terms
corresponds to the energetic change induced by the mutation, according to 1 of 13 different statistical potentials extracted from a data set of known protein structures (41). These potentials describe the correlations between amino acid types, pairwise inter-residue distances, backbone torsion angles and solvent accessibilities. The inter-residue distances are evaluated from the coordinates of the average geometric centers of the side chains. In addition, the terms 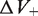 and
and 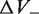 were introduced to account for the possible creation of packing defects:
were introduced to account for the possible creation of packing defects: 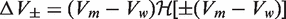 , where
, where 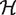 is the Heaviside function, and Vw and Vm the volume of the wild-type and mutant amino acid, respectively. The weights
is the Heaviside function, and Vw and Vm the volume of the wild-type and mutant amino acid, respectively. The weights 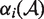 (
(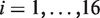 ) are sigmoid functions of the solvent accessibility
) are sigmoid functions of the solvent accessibility 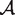 of the mutated residue, and were identified on the basis of a data set of experimentally measured changes in folding free energy resulting from 2648 mutations (42).
of the mutated residue, and were identified on the basis of a data set of experimentally measured changes in folding free energy resulting from 2648 mutations (42).Change in binding free energy on mutation
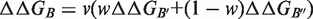
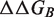 values reached 0.55 on the full data set and 0.76 on 90% of the data set (Table 1).
values reached 0.55 on the full data set and 0.76 on 90% of the data set (Table 1). BeAtMuSiC performances
| Data set | 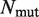 | All mutations | Exclusion of 10% outliers | ||
|---|---|---|---|---|---|
| R | σ (kcal/mol) | R | σ (kcal/mol) | ||
| Ala scans | 362 | 0.55 | 1.01 | 0.76 | 0.72 |
| SKEMPI | 2007 | 0.40 | 1.80 | 0.68 | 1.19 |
| SKEMPIa | 1991 | 0.47 | 1.59 | 0.70 | 1.18 |
| Data set | | All mutations | Exclusion of 10% outliers | ||
|---|---|---|---|---|---|
| R | σ (kcal/mol) | R | σ (kcal/mol) | ||
| Ala scans | 362 | 0.55 | 1.01 | 0.76 | 0.72 |
| SKEMPI | 2007 | 0.40 | 1.80 | 0.68 | 1.19 |
| SKEMPIa | 1991 | 0.47 | 1.59 | 0.70 | 1.18 |
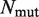 is the number of mutations in the considered data set. R is the Pearson correlation coefficient and σ the root mean square error, between predicted and measured
is the number of mutations in the considered data set. R is the Pearson correlation coefficient and σ the root mean square error, between predicted and measured 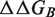 values. aThis data set was obtained after removal of 16 mutations of the Lysine residue at position I15 in the BPTI–BT complex (PDB: 2FTL).
values. aThis data set was obtained after removal of 16 mutations of the Lysine residue at position I15 in the BPTI–BT complex (PDB: 2FTL).
BeAtMuSiC performances
| Data set | | All mutations | Exclusion of 10% outliers | ||
|---|---|---|---|---|---|
| R | σ (kcal/mol) | R | σ (kcal/mol) | ||
| Ala scans | 362 | 0.55 | 1.01 | 0.76 | 0.72 |
| SKEMPI | 2007 | 0.40 | 1.80 | 0.68 | 1.19 |
| SKEMPIa | 1991 | 0.47 | 1.59 | 0.70 | 1.18 |
| Data set | | All mutations | Exclusion of 10% outliers | ||
|---|---|---|---|---|---|
| R | σ (kcal/mol) | R | σ (kcal/mol) | ||
| Ala scans | 362 | 0.55 | 1.01 | 0.76 | 0.72 |
| SKEMPI | 2007 | 0.40 | 1.80 | 0.68 | 1.19 |
| SKEMPIa | 1991 | 0.47 | 1.59 | 0.70 | 1.18 |
is the number of mutations in the considered data set. R is the Pearson correlation coefficient and σ the root mean square error, between predicted and measured values. aThis data set was obtained after removal of 16 mutations of the Lysine residue at position I15 in the BPTI–BT complex (PDB: 2FTL).
Because conformational rearrangements on binding are not explicitly modeled, 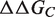 and
and 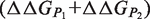 are rigorously equal for mutations outside of the interface region.
are rigorously equal for mutations outside of the interface region. 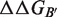 is thus focused on the impact of the mutation on the interactions established at the interface [Equation (1)], whereas
is thus focused on the impact of the mutation on the interactions established at the interface [Equation (1)], whereas 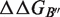 describes the effect of the mutation on the overall stability of the complex [Equation (2)]. Interestingly, if we consider the subset of 216 mutations into alanine that occur at the protein–protein interface, the optimal value of w remains close to 0.7. Accounting for the variation in stability is thus important for the evaluation of the change in binding affinity, even in the case of mutations occurring at the interface.
describes the effect of the mutation on the overall stability of the complex [Equation (2)]. Interestingly, if we consider the subset of 216 mutations into alanine that occur at the protein–protein interface, the optimal value of w remains close to 0.7. Accounting for the variation in stability is thus important for the evaluation of the change in binding affinity, even in the case of mutations occurring at the interface.
VALIDATION
SKEMPI data set
We applied our prediction method to a recently published data set, named SKEMPI, in which the experimentally measured effects of mutations on protein–protein binding affinity were compiled (39). This data set is not limited to the results of alanine scanning experiments, and thus includes mutations in any type of amino acid. Out of the 3047 entries in the original data set, we removed 717 multiple mutations, 87 reverse mutations (i.e. from the mutant protein back to the wild type) and 236 redundant entries (i.e. when several experimental values of 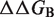 are available for the same mutation in the same protein). In case of redundant entries, the average value of the change in binding free energy was used. The final data set contains 2007 mutations, with
are available for the same mutation in the same protein). In case of redundant entries, the average value of the change in binding free energy was used. The final data set contains 2007 mutations, with 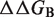 values ranging from −3.8 to 12.3 kcal/mol. These values are compared with the predictions of BeAtMuSiC on Figure 2.
values ranging from −3.8 to 12.3 kcal/mol. These values are compared with the predictions of BeAtMuSiC on Figure 2.
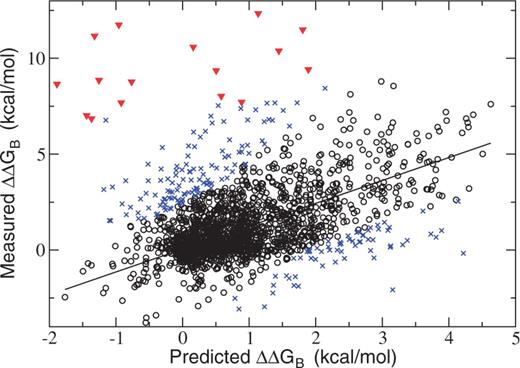
Correlation between predicted and measured changes in binding free energies in the SKEMPI data set. (Black circle) Main data set. (Blue cross) 10% outliers. (Red triangle) Mutations of the lysine at position I15 in the BPTI–BT complex (PDB: 2FTL).
Strikingly, significant errors in the predictions are observed for a set of 16 mutations of the lysine residue in position 15 of the bovine pancreatic trypsin inhibitor (BPTI), in complex with bovine β-trypsin (BT). Although these mutations severely disrupt the protein–protein interaction, they are predicted to increase or mildly decrease binding affinity (Figure 2). The interaction between BPTI and BT relies mainly on a single residue, Lys 15, which gets inserted into a small cavity on the surface of the protease (Figure 3). The binding affinity is largely determined by the shape complementary of the lysine side-chain and the surface of the cavity, and the highly specific interactions that are created (44). It is not surprising that our coarse-grained approach may sometimes fail to provide an accurate description of the consequences of mutations in such an extreme situation, especially because no modeling and optimization of the conformations of side-chains are performed.
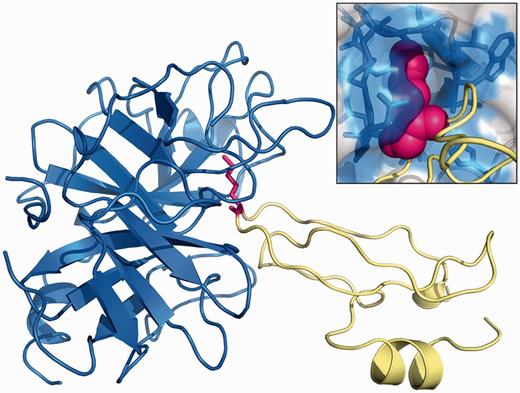
Structure of the complex formed by BPTI (yellow) and bovine BT (blue) (PDB: 2FTL). The lysine residue in position 15 of BPTI is depicted in magenta. The figure was made using PyMOL.
If we discount these 16 mutations, the performances are relatively similar—although somewhat lower—than those observed in the data set of mutations into alanine used to devise our method. The Pearson correlation coefficient between prediction and experiments reaches 0.47 on the full data set and 0.70 on 90% of the data set (Table 1). Note that the 362 mutations into alanine used to identify two parameters of the model are also included in the SKEMPI data set. However, the performances are not affected by the removal of these mutations (correlation coefficient of 0.48 instead of 0.47).
CAPRI experiment
The  round of the blind prediction experiment CAPRI (40) was the opportunity to assess the performances of our method, in comparison with various approaches developed by other groups. The challenge consisted in the evaluation of the effect of a large number of single-site mutations in two de novo designed influenza inhibitors, HB36.4 and HB80.3 (45), on the binding affinity with their target, hemagglutinin. More precisely, predictions were requested for 1007 mutations at 53 positions in the sequence of HB36.4 (target 55), and 855 mutations at 45 positions in the sequence of HB80.3 (target 56).
round of the blind prediction experiment CAPRI (40) was the opportunity to assess the performances of our method, in comparison with various approaches developed by other groups. The challenge consisted in the evaluation of the effect of a large number of single-site mutations in two de novo designed influenza inhibitors, HB36.4 and HB80.3 (45), on the binding affinity with their target, hemagglutinin. More precisely, predictions were requested for 1007 mutations at 53 positions in the sequence of HB36.4 (target 55), and 855 mutations at 45 positions in the sequence of HB80.3 (target 56).
Out of the 22 participating groups, 18 and 15 submitted predictions for the complete set of mutations in HB36.4 and HB80.3, respectively. The predictions were compared with previously unreleased experimental data concerning the effect of these mutations on the binding properties of the two inhibitors. This data was obtained using deep sequencing of mutant libraries before and after selection for binding (46). Kendall’s τ rank correlation coefficient between the predictions of the different groups and the experimental measurements is reported in Figure 4.
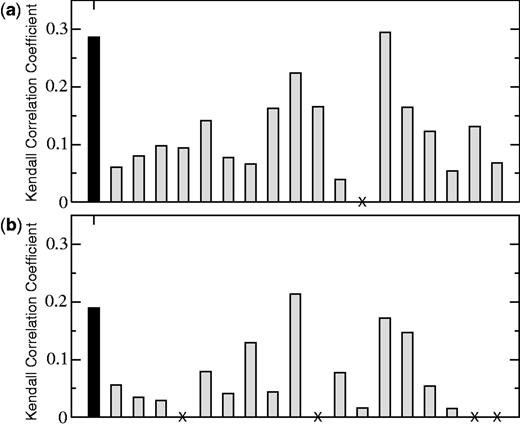
Kendall’s τ rank correlation coefficient between predictions and experiments, during the  round of the CAPRI experiment (http://www.ebi.ac.uk/msd-srv/capri/round26). The results of our method are depicted in black, and those of other participating methods in gray. Groups that did not submit predictions for the complete set of mutations are not considered here. The symbol ‘X’ is used when a group submitted a full set of predictions for one target but not for the other. (a) Target 55: hemagglutinin-HB36.4. (b) Target 56: hemagglutinin-HB80.3. A detailed analysis of the results of this experiment, along with a description of the different prediction methods, will be reported elsewhere (Moretti et al., manuscript submitted).
round of the CAPRI experiment (http://www.ebi.ac.uk/msd-srv/capri/round26). The results of our method are depicted in black, and those of other participating methods in gray. Groups that did not submit predictions for the complete set of mutations are not considered here. The symbol ‘X’ is used when a group submitted a full set of predictions for one target but not for the other. (a) Target 55: hemagglutinin-HB36.4. (b) Target 56: hemagglutinin-HB80.3. A detailed analysis of the results of this experiment, along with a description of the different prediction methods, will be reported elsewhere (Moretti et al., manuscript submitted).
The comparison of the results for the two target proteins underlines the difficulty in establishing consistently a detailed ranking of the different approaches. Yet, in both cases, our method stands among the top performers, indicating that the predictive power of BeAtMuSiC is in line with the most efficient state-of-the-art methodologies. The average performance of our method on the two CAPRI targets is somewhat lower than on the other data sets: Kendall’s τ reaches 0.36 for the 362 mutations into alanine and 0.29 for the 1991 mutations of the SKEMPI data set. This may be due to the fact that the experimental data used during the CAPRI experiment does not consist in direct measurements of the changes in binding affinity, and that other sources of error may therefore be present.
WEBSERVER
Server input
The main input of the webserver is the structure of the protein–protein complex, in PDB format. The user may either upload his own structure file, or provide the 4-letter PDB code of the structure, which will then be automatically downloaded from the Protein Data Bank (47).
Once the structure has been correctly retrieved, the server will display a summary of the protein chains present in the structure file. The second step consists in the definition of the two partners of the protein–protein interaction. Each chain must be assigned to either the first or second partner, or discarded. Obviously, for the server to provide meaningful predictions, each partner must contain at least one protein chain, and the two partners must be in contact.
The user may then choose to evaluate the effect a few specific mutations, or to perform a systematic scan of all possible mutations in a protein chain or at the protein–protein interface. If several protein chains of identical sequence are present, the mutations will be introduced simultaneously in all of them.
Server output
The main output of the webserver is the change in binding free energy resulting from each mutation. Users should be aware that when several chains of identical sequence are present, the mutation is introduced simultaneously in each one of those. In such cases, the predictions are not normalized with respect to the number of chains concerned by the mutation, and the reported  value corresponds thus to the total change in binding free energy between the two selected partners.
value corresponds thus to the total change in binding free energy between the two selected partners.
The webserver also reports the solvent accessibility of the mutated residue, in the complex and in the individual partners. The solvent accessibility is defined as the ratio of the solvent-accessible surface in the considered structure, as computed by DSSP (48), and in an extended tripeptide Gly-X-Gly (49). BeAtMuSiC identifies a residue as part of the protein–protein interface if its solvent accessibility in the complex is at least 5% lower that in the individual partner. This latter information is provided to the user for convenience, but not used during the computations.
The results may be downloaded as a plain text file, or browsed interactively on the Web site (Figure 5). In particular, if systematic mutations have been performed, the user may choose to display the predictions for mutations at a given position in the sequence, or for mutations with the strongest predicted increase or decrease in binding affinity.
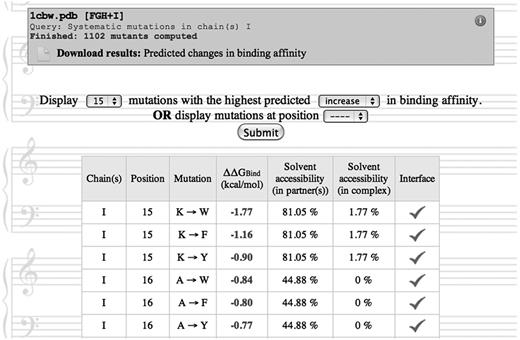
Example output of the BeAtMuSiC server.
DISCUSSION
The prediction of the impact of a mutation on protein–protein binding affinity is more challenging than the prediction of the change in folding free energy. For instance, the method that we previously developed for the latter purpose, on the basis of a model with the same level of coarse-graining, yielded a Pearson correlation coefficient between prediction and experiments of 0.63 on a data set of 2648 mutations, and 0.79 on 90% of the data set (42,50).
In both cases, the accuracy of the predictions ultimately hinges on the quality of the energy function. However, because coarse-grained models do not describe explicitly all of the structural and energetic consequences of a mutation, a number of new obstacles arise when the question of protein–protein binding is considered. Firstly, evaluating mutations occurring outside of the interface region can be challenging, as their impact on binding may be related to an effect on the overall stability of the complex, to an alteration of the dynamical properties and flexibility of one of the interacting partners, or to conformational changes affecting the shape complementarity of the two partners. On the other hand, protein–protein interfaces have been shown to possess distinctive properties from core or surface regions (3–10), and the highly specific nature of many interactions established at those interfaces may be difficult to render accurately without modeling side-chain conformations at atomic resolution. Finally, further investigations would be needed to evaluate whether a single coarse-grained model is sufficient to encompass the large variety of binding modes that characterize protein–protein interactions (individually folded or unfolded partners, transient or permanent interfaces, occurrence of structural rearrangements on binding, etc).
Although these considerations suggest that there is still room for improvement, the results of the  round of the CAPRI experiment demonstrated that the predictive power of our method compares well with that of other approaches developed for the same purpose, including predictive models based on a much more detailed structural representation. In addition, the coarse-grained nature of our method provides unique advantages in terms of computational speed, with the possibility to assess rapidly the impact of all possible mutations in a protein chain or at the protein–protein interface. The coarse-graining also ensures that the predictions are robust to imperfections in the structural data. Therefore, a similar level of performance should be expected when using structural models rather than experimentally determined structures (51), provided the relative positioning of the two partners of the interaction is correctly defined.
round of the CAPRI experiment demonstrated that the predictive power of our method compares well with that of other approaches developed for the same purpose, including predictive models based on a much more detailed structural representation. In addition, the coarse-grained nature of our method provides unique advantages in terms of computational speed, with the possibility to assess rapidly the impact of all possible mutations in a protein chain or at the protein–protein interface. The coarse-graining also ensures that the predictions are robust to imperfections in the structural data. Therefore, a similar level of performance should be expected when using structural models rather than experimentally determined structures (51), provided the relative positioning of the two partners of the interaction is correctly defined.
The BeAtMuSiC server should thus prove useful in a wide range of applications. Typically, protein engineering projects aiming at the design or modification of protein–protein interactions would benefit from the possibility to identify a restricted number of mutations that would constitute ideal candidates for further investigations using more detailed computational approaches and/or experimental tests. Given the importance of binding for the proper biological functioning of many proteins, BeAtMuSiC may also participate to a better understanding of the pathological consequences of some non-synonymous single-nucleotide polymorphisms.
FUNDING
Belgian Fonds de la Recherche Scientifique (F.R.S.-FNRS) through an FRFC grant. Y.D. and M.R. are Postdoctoral Researcher and Research Director, respectively, at the F.R.S.-FNRS. Funding for open access charge: Belgian F.R.S.-FNRS (FRFC grant).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments