





Abstract
RNase E initiates the decay of Escherichia coli RNAs by cutting them internally near their 5′-end and is a component of the RNA degradosome complex, which also contains the 3′-exonuclease PNPase. Recently, RNase E has been shown to be able to remove poly(A) tails by what has been described as an exonucleolytic process that can be blocked by the presence of a phosphate group on the 3′-end of the RNA. We show here, however, that poly(A) tail removal by RNase E is in fact an endonucleolytic process that is regulated by the phosphorylation status at the 5′- but not the 3′-end of RNA. The rate of poly(A) tail removal by RNase E was found to be 30‐fold greater when the 5′-terminus of RNA substrates was converted from a triphosphate to monophosphate group. This finding prompted us to re-analyse the contributions of the ribonucleolytic activities within the degradosome to 3′ attack since previous studies had only used substrates that had a triphosphate group on their 5′-end. Our results indicate that RNase E associated with the degradosome may contribute to the removal of poly(A) tails from 5′‐monophosphorylated RNAs, but this is only likely to be significant should their attack by PNPase be blocked.
Received January 25, 2001; Revised and Accepted March 6, 2001.
INTRODUCTION
The decay of many if not most RNA transcripts in Escherichia coli appears to be initiated by a series of endonucleolytic cleavages that are generated primarily by ribonuclease E (RNase E), an enzyme that has also been shown to have a role in the processing of rRNA and tRNA (for recent reviews see 1–6). Subsequently, the 3′→5′ exonucleases polynucleotide phosphorylase (PNPase) (7) and/or RNase II (8) convert the decay intermediates produced by endonucleolytic cleavage into mononucleotides and short oligonucleotides. The latter are further digested by oligoribonuclease (9,10). RNase E cleavage of intact primary transcripts, which have a triphosphate group at their 5′-end, generates downstream intermediates that have a 5′-monophosphate group (11). Recently it has been shown in vitro that RNase E preferentially cleaves RNAs that have a monophosphate group at their 5′-end. This suggests that when RNase E makes a decay-initiating cleavage in a 5′-triphosphorylated transcript it should prefer to cut any remaining site(s) in that RNA over attack on another intact transcript (12). This ‘5′-end dependency’, for which there is now evidence in vivo (13), may provide a mechanism to ensure that RNA decay in E.coli occurs in general without the accumulation of significant levels of decay intermediates (2). Another contributing factor to this so-called ‘all or nothing’ phenomenon may be the physical coupling of RNase E and PNPase within the multi-component RNA degradosome (14–17), which is likely to be the major centre for mRNA processing and decay in E.coli (18).
Rapid 3′-exonucleolytic decay in E.coli is facilitated by poly(A) tails on the 3′-end of the RNA (for reviews see 19–22). It is known that poly(A) tails are added primarily by poly(A) polymerase I (PAPI) (23–26), the product of the pcnB gene (27); however, the precise mechanism(s) by which poly(A) tails regulate 3′-exonucleolytic decay remains to be determined. Recently it has been reported that RNase E can cleave poly(A) tails on the 3′-end of RNAI, the antisense RNA regulator of ColE1-type plasmid replication (for a review see 28), to generate a ‘remnant’ of 5–7 A residues (29). However, cleavage of the poly(A) tail was not observed when the RNAI substrate was labelled at its 3′-end (29) by a process that incorporated a 3′-monophosphate group (30). This observation, together with the finding that mononucleotides and short oligonucleotides were the detectable products of RNase E cleavage of RNAI that had an internally labelled poly(A) tail, has been interpreted as evidence for the poly(A) nuclease activity of RNase E being 3′-exonucleolytic (2,4,31). We show here that the processing of poly(A) tails by E.coli RNase E is in fact an endoribonucleolytic process that is stimulated by the generation of a 5′-monophosphate group and is not blocked by 3′‐phosphorylation. We have also investigated the contribution of RNase E and PNPase activity within the RNA degradosome to the removal of 3′-poly(A) tails in vitro. Our results indicate that the exposed 3′-poly(A) tails of 5′-monophosphorylated, as well as 5′-triphosphorylated, substrates (31) are attacked primarily by PNPase.
MATERIALS AND METHODS
Synthesis of RNA substrates
Oligoribonucleotides A40 and A29:G21 (Fig. 1) were synthesised using phosphoramidite chemistry, deprotected and purified by reverse phase chromatography as described previously (32). RNAI substrates were synthesised using the T7-MEGAshortscript kit from Ambion and gel purified (32). When internally labelled RNA was required (Figs 3, 4, 6 and 7) 60 µCi [α‐32P]UTP (ICN) was included in the reaction. The template for the synthesis of adenylated pppRNAI–5 was a PCR product generated using forward and reverse primers with the sequences 5′-GGATCCTAATACGACTCACTATAGGGATTTGGTATCTGCGCTCTG and 5′-(T40)AACAAAAAAACCAC, respectively. The PCR product used to synthesise 5′-extended RNAI–5 that was subsequently cleaved by RNase H to generate pRNAI–5 (Fig. 2) was made using 5′-ATCCTAATACGACTCACTATAGGGATCCCGGGATTTGGTATCTGCGCTC as the forward primer. The T7 promoter sequence in each of the forward primers is underlined. Site-directed RNase H cleavage was done as described previously (33,34). Briefly, a 3-fold excess of the DNA/2′-O-methyl RNA chimera (5′-CmCmCmdGdGdGdAUmCmCmCm) was annealed to 5′-extended RNAI–5 and then the hybrid was incubated with RNase H (Life Technologies). As shown previously (35), RNase H cleaved the RNA at a single position immediately 5′ to the first complementary deoxynucleotide.
5′-Radiolabelling of RNA
Four to five pmoles of RNA that had been either chemically synthesised or transcribed in vitro and then dephosphorylated was radiolabelled at the 5′-end by incubating with 23 pmol (160 µCi) [γ-32P]ATP (ICN) and 10 U T4 polynucleotide kinase (MBI Fermentas) as described previously (32). The reactions were quenched with urea loading buffer [7 M urea, 0.1% (w/v) bromophenol blue and xylene cyanol] and the 5′‐labelled RNAs were gel purified (32).
3′-Radiolabelling of RNA
Six to seven pmoles of RNA was radiolabelled at the 3′‐terminus in a volume of 20 µl containing 50 mM HEPES (pH 7.5), 3.3 mM DTT, 10 mM MgCl2, 10% (v/v) DMSO, 10 U T4 RNA ligase (Pharmacia) and 33 pmol (100 µCi) [5′-32P]pCp (ICN). After incubation at 4°C for 2 h the reaction was stopped and the RNAs were gel purified as done for the 5′‐labelled substrates.
Purification of RNase E and the RNA degradosome
Full-length RNase E and its N-terminal ribonucleolytic domain (NTD-RNase E, residues 1–498) that were His-tagged at their N-terminal end were purified under denaturing conditions (7 M urea) from cultures of E.coli BL21 (DE3) cells harbouring pFUS1500 or pNSTOP, respectively (35), using the pET-based system of immobilised metal affinity chromatography (Novagen) as described previously (32). Imidazole was used as the elutant. The preparation of RNA degradosomes was purified as described previously (14,16) from BRL2288 cells (Life Technologies). The identities of RNase E, PNPase, RhlB and enolase in the preparation were confirmed by MALDI MS analysis of trypsin-generated fingerprints of each of the major polypeptide species. The activities of RNase E and PNPase were also confirmed using assays described previously (14).
RNA cleavage reactions
RNase E was incubated with substrate RNA in 20 mM Tris–HCl (pH 7.6), 100 mM NaCl, 10 mM MgCl2, 0.1% (v/v) Triton X-100 and 1 mM DTT containing 20 U RNase inhibitor (Pharmacia) at 37°C as described previously (32). The latter was added as a precaution to prevent cleavage by RNase A‐type enzymes that can contaminate components of the buffer or plastic ware during their use. Samples were taken at regular intervals and quenched using urea loading buffer before aliquots were run in polyacrylamide sequencing-type gels (36). The reaction products were identified by phosphorimaging and analysed using the TINA package (Raytech). The concentration of RNase E and substrate and the length of incubation are given in the figure legends. Assays using degradosomes were done as described for RNase E except that sodium phosphate and ATP were present at 10 and 1 mM, respectively, unless indicated otherwise. RNA and protein were quantitated by measuring the absorbance of stocks at 260 and 280 nm, respectively. When appropriate, the amount of radioactivity incorporated into an RNA substrate was also used to determine its concentration.
RESULTS
When using an A40 oligonucleotide (32) to assay the recently reported poly(A) nuclease activity of RNase E we found that, in contrast to what had been reported previously for RNAI (29), when labelled at its 3′-end using 32pCp this substrate could be cleaved provided it was monophosphorylated at its 5′‐end (Fig. 1). Additionally, the distributions of the sizes of 3′- and 5′-labelled products (Fig. 1A and B, respectively) were broadly similar, suggesting that cleavage of this substrate occurred endonucleolytically at random positions rather than exonucleolytically from the 3′-end. To confirm this notion, we synthesised another oligonucleotide substrate, A29:G21, which contained a 3′-poly(G) segment. Such sequences are known to form stable quartet structures (37–39) that can block exonucleolytic decay (for a review see 40). As shown in Figure 1D, this substrate, which was labelled at the 5′-end, was cleaved by RNase E at positions throughout the poly(A) segment. Purified PNPase was unable to degrade this substrate (data not shown). The above results combined show that the cleavage of short poly(A) substrates by RNase E occurs endonucleolytically.
Our finding that RNase E cleavage of an A40 oligonucleotide is dependent on the presence of a monophosphate group on its 5′-end raised the possibility that the reason 3′-labelled polyadenylated RNAI was found previously to be resistant to RNase E attack at the 3′-end (29) was not the presence of a 3′‐phosphate, but a consequence of not replacing the triphosphate at the 5′-end with a monophosphate group. To test this hypothesis we synthesised two polyadenylated RNAI species that were identical in sequence but had either a monophosphate or triphosphate group at their 5′-end. The 5′-triphosphorylated species was purified directly from a standard in vitro transcription reaction, whereas its 5′-monophosphorylated counterpart was generated by RNase H cleavage (33,34) of a species that had an extension on its 5′-end (Fig. 2). Both these RNAI species were internally labelled using [α-32P]UTP and lacked the first 5 nt of the major RNase E site (41–42; Fig. 2); the latter eliminated cleavage at the 5′-end, thereby simplifying the analysis of processing of the poly(A) tail on the 3′-end. Additionally, we chose to further analyse poly(A) tail removal (Fig. 3) using NTD-RNase E, the catalytically active N‐terminal domain (amino acids 1–498) (35), as this is the polypeptide that was used previously by Huang and co-workers (29). NTD-RNase E, like the full-length enzyme, has been shown by us to prefer cleaving 5′-monophosphorylated RNA (32).
We found that the level of poly(A) tail removal (Fig. 3B) from RNAI–5A40 correlated precisely with the amount of NTD-RNase E in multiple fractions across a peak of NTD-RNase E (Fig. 3A) that had been purified under denaturing conditions (see Materials and Methods). This indicated that the poly(A) nuclease activity in our preparation was indeed that of NTD-RNase E and not an overlapping peak of a minor contaminant (Fig. 3). Consistent with this conclusion, we were unable to detect poly(A) nuclease activity when the purification procedure was repeated using an extract from cells that did not produce NTD-RNase E (data not shown). Under conditions where the poly(A) tail of 5′-monophosphorylated RNA was cleaved by RNase E (Fig. 3B) we were unable to identify cleavage of RNA that was triphosphorylated at the 5′-end (Fig. 3C). Cleavage of the latter substrate could be detected, however, when the amount of enzyme added to the reaction was increased 10-fold (Fig. 3D).
To quantitate the difference in the rate at which poly(A) tails were removed from these substrates by RNase E we carried out a time course experiment using the peak fraction of the NTD-RNase E polypeptide (Fig. 3, lane 4). As shown in Figure 4, poly(A) tails were removed from 50% of the 5′-monophosphorylated RNA within 2 min, whereas 90% of the 5′-triphosphorylated RNA retained its poly(A) tail after 80 min (compare Fig. 4A and B). We calculated that the difference in the initial rates is at least 30‐fold (Fig. 4C). A similar difference in the initial rate of poly(A) tail removal from 5′-monophosphorylated and 5′‐triphosphorylated RNAI was observed using a preparation of full-length RNase E (data not shown). These results support the notion that the presence of a 5′-triphosphate group on 3′‐labelled RNAI could have prevented Huang and co‐workers (29) detecting poly(A) tail removal. A small amount (<10%) of RNA in the 5′-monophosphorylated preparation was resistant to RNase E (Fig. 4). This is most likely 5′-extended RNA that was carried over from the RNase H cleavage reaction and gel purification (see Materials and Methods).
To confirm that the presence of a 3′-phosphate group per se does not block poly(A) tail removal by RNase E, 5′-monophosphorylated RNA that was generated by RNase H cleavage was labelled at the 3′-end using [32P]pCp and T4 RNA ligase and incubated with NTD-RNase E. As anticipated, RNase E efficiently removed the poly(A) tail from this substrate (Fig. 5). Consistent with the observations of Huang and co-workers (29), RNase E cleavage of the poly(A) tail of a 5′‐triphosphorylated substrate was not detected (Fig. 5, right). Figure 5 also showed that the initial downstream products of RNase E cleavage are oligomers, the most abundant initial products being between 31 and 37 A residues. With further incubation these products were converted to smaller species (data not shown), as was observed using the A40 oligonucleotide (Fig. 1). In contrast, Huang and co-workers (29) reported that the initial products of cleavage of an internally labelled poly(A) tail were mononucleotides and short oligonucleotides. However, as their studies used a substrate that was triphosphorylated at its 5′-end, RNase E cleavage of the poly(A) tails would have been blocked and it is possible that the detected products were generated by a minor 3′-nuclease activity in their preparation.
The results of the experiments using 3′-labelled, 5′-monophosphorylated RNAs (Fig. 5) also revealed that RNase E is unable to cleave (or at least cleave efficiently) phosphodiester bonds within 8 nt of the 5′-end of the A40 substrate: 3′-labelled products of 33–39 A residues were not detected (Fig. 5, lane M). In agreement with this notion, the smallest detectable 5′-labelled product of RNase E cleavage of A40 was 8 nt (Fig. 1). Additionally, close examination of the 5′-labelled products of RNase E cleavage of A40 reveals that species of 37–39 A residues may also be under-represented (Fig. 1), indicating that RNase E also prefers to cleave phosphodiester bonds that are >3 nt from the 3′-end of an RNA.
We next investigated the contribution of RNase E activity associated with the degradosome to the 3′ decay of polyadenylated RNAs that are monophosphorylated at their 5′‐ends. Previous studies of the 3′-nuclease activities of the degradosome had only used substrates that had a 5′-triphosphate group (31), which we have shown here can block 3′‐processing by RNase E. We synthesised polyadenylated RNAI–5 that was internally labelled and had either a triphosphate or monophosphate group at the 5′-end (see Fig. 2). Both of these substrates were then incubated with a degradosome preparation using a buffer in which both RNase E and PNPase were active (see Materials and Methods). As controls we also included 5′-monophosphorylated RNAI–5 that lacked a 3′‐poly(A) tail or had a 3′-remnant generated by RNase E cleavage of RNAI–5A40. We found that the presence of a 5′‐triphosphate on RNAI–5A40 did not significantly reduce the rate of poly(A) tail removal by the degradosome (compare Fig. 6A and B), in contrast to what was found above for purified RNase E (Fig. 4). Moreover, we found that the 3′-stem–loop of RNAI provides an extremely effective barrier to degradosome-mediated decay (Fig. 6), as reported previously for other substrates (31,43). There was no measurable difference in the relative rate of poly(A) tail removal from the 5′-triphosphorylated and 5′-monophosphorylated RNA when the amount of degradosome was reduced 10-fold to slow 3′ processing (data not shown). We estimate that no more than 5% of the adenylated RNAI was degraded [beyond poly(A) tail removal] after incubating with the degradosome for 60 min (Fig. 6). The degradosome appeared to degrade polyadenylated RNAI to within a few nucleotides of the base of the 3′-stem–loop of RNAI (compare the mobility of the 3′-cleaved products with unadenylated RNAI–5) and then it seems that it can dissociate, since poly(A) tails were removed from the bulk of the substrate, which was in excess. We also found that the degradosome was similarly able to attack the poly(A) tail remnant generated by RNase E cleavage, but that neither the presence of this remnant nor an A40 tail dramatically increased the overall rate of decay of RNAI.
Our finding that the 5′-phosphorylation status has no detectable effect on the rate of poly(A) tail removal by the degradosome (Fig. 6), even though it can dramatically affect the rate of poly(A) tail removal by purified RNase E (Fig. 4), suggested that under the conditions used PNPase is the major activity responsible for removing 3′-poly(A) tails. To confirm this, we investigated the effect of removing inorganic phosphate, which is required for PNPase activity, from the reaction buffer. As shown in Figure 7, the rate of poly(A) tail removal was reduced by at least 20-fold, supporting the notion that when PNPase is exonucleolytically active it is the primary 3′-nuclease within the degradosome that removes exposed 3′-poly(A) tails (31). RNAI species that had poly(A) remnants of the size generated by RNase E were detected, however, in the absence of inorganic phosphate (compare Fig. 7A and B).
DISCUSSION
In the experiments described above we have demonstrated that RNase E processing of 3′-poly(A) tails is endonucleolytic and can be stimulated by the generation of a monophosphate group on the 5′-end of the RNA substrate (Figs 1–4). Moreover, in contrast to a previous report (29), we find that the cleavage of poly(A) tails by RNase E is not blocked by the presence of a phosphate group on the 3′-end of the RNA (Figs 1 and 5). We suggest that the presence of a 5′-triphosphate group on the 3′‐labelled RNAI substrate used previously (29) prevented the detection of poly(A) tail removal. Our analysis of the poly(A) nuclease activity of RNase E using 3′-labelled RNAI–5A40 (Fig. 2) also indicated that the sizes of the initial 3′ products were 31–37mers (Fig. 5). These were then converted to smaller species upon further incubation with RNase E (data not shown). Interestingly, a preference for making initial cleavages toward the 5′-end of a poly(A) segment was not observed using the A40 oligonucleotide substrate (Fig. 1), suggesting that some structural feature(s) of a transcript can affect the cleavage of poly(A) tails by RNase E. Perhaps, for example, there is a maximum distance beyond which a 5′-monophosphate group cannot stimulate cleavage by RNase E (32).
Having established that the removal of 3′-poly(A) tails by RNase E is stimulated by the generation of a monophosphate group on the 5′-end of the RNA, we re-investigated the relative contributions of the ribonucleolytic activities within the degradosome to the attack of decay intermediates from their 3′-end, since a previous study had used substrates that were triphosphorylated at their 5′-end (31). We found, however, that rapid poly(A) tail removal was not significantly affected by the phosphorylation status at the 5′-end (Fig. 6), but did require inorganic phosphate (Fig. 7), which is required by PNPase (7). These findings combined confirm that RNase E cutting of 3′-poly(A) tails is unlikely to have a major influence on the decay of RNA from the 3′-end when degradation by PNPase is unhindered. However, RNAI species that had poly(A) remnants of the size generated by RNase E were detected when inorganic phosphate was omitted from reactions with the RNA degradosome (compare Fig. 7A and B) and it seems increasingly possible that, as in eukaryotes (for a recent review see 44), the poly(A) tails of E.coli RNAs are not naked. The S1 ribosomal protein has been reported to bind poly(A) tails in vitro and it has been suggested that this interaction may promote translation in vivo (45). Additionally, it has recently been shown that Hfq (46,47), which is known to interact with A-rich segments of several RNAs (48,49), can stimulate the processivity of PAPI (50). This parallels the situation in eukaryotes, where it is known that the processivity of polyadenylation is facilitated by a poly(A)-binding protein (44). Therefore, in E.coli the processivity of 3′-exonucleases such as PNPase and RNase II may be impeded by proteins bound to poly(A) tails. RNase E cleavage of the poly(A) tail to produce a short remnant would remove this barrier and generate a 3′-terminus that we have shown here is still a target for at least PNPase (Fig. 6). Moreover, such a 3′-cleavage by RNase E would be stimulated as the result of a decay-initiating cleavage generating a monophosphate at the 5′-end and could thus provide a mechanism for coordinating 5′ and 3′ decay.
The activities of purified PNPase and PAPI have been reported to be stimulated between 3- and 5-fold by the generation of a 5′-monophosphate group (23,51); however, the significance of this level of stimulation with regard to degradosome-mediated decay remains to be established. In light of the work discussed above, any analysis of the contribution of 5′-phosphorylation to 3′ decay by the degradosome needs to take into account effects on RNase E.
Our degradosome preparation, which was purified as described previously (14,16) and contained all of the major components (see Materials and Methods), was insufficient to degrade the bulk of 5′-monophosphorylated or 5′-triphosphorylated substrates beyond removal of the poly(A) tail (Figs 6 and 7). This result was not unanticipated, as it is well established that terminal stem–loop structures can offer an efficient barrier to 3′-exonucleolytic attack (52,53). It should be noted, however, that RNAI could be degraded rapidly when PAPI was added to the reaction (data not shown), as described previously for the mRNA for ribosomal protein S20 (43). Under these conditions it has been suggested that even inefficient 3′ processing events can, through multiple cycles of polyadenylation, produce a significant enhancement of the rate of decay (43). It is also possible that the addition of PAPI, which has recently been shown to interact physically with RNase E (54), stabilises the interaction of the degradosome with its substrates, much in the same way that the association of eukaryotic poly(A) polymerase with the complex that generates the 3′-termini for polyadenylation stimulates cutting of the RNA (44).
Interestingly, it was previously found that a preparation of PNPase could degrade the bulk of a sample of polyadenylated RNAI without the addition of ATP or poly(A) polymerase (23). Clearly our results, obtained using a degradosome preparation, differ from those obtained using PNPase and suggest that the activity of PNPase is moderated when this enzyme is associated with RNase E. Indeed, this possibility was raised initially by the finding that PNPase associated with other degradosomal components could impede the processivity of purified PNPase in vitro (55). It is possible, for example, that RNase E binding of structured regions within RNA via the arginine-rich domain of this enzyme not only promotes cleavage by RNase E (56), but also moderates 3′-exonucleolytic attack.
ACKNOWLEDGEMENTS
We thank Jeff Keen at the Biomolecular Analysis Unit, University of Leeds, for MALDI MS analysis of components of the degradosome, Stan Cohen for open discussion of the poly(A) nuclease activity of RNase E and A.J.Carpousis for clarifying details of the degradosome purification protocol. This work was supported by a grant from the UK BBSRC to K.J.M., who is the recipient of a Royal Society University Research Fellowship.
To whom correspondence should be addressed. Tel: +44 113 233 3109; Fax: +44 113 233 1407; Email: k.j.mcdowall@leeds.ac.uk Present addresses: A. P. Walsh, MRC Laboratory of Molecular Biology, Cambridge CB2 2QH, UKM. R. Tock, Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors
![Figure 1. Cleavage of an A40 oligonucleotide substrate by RNase E is dependent on the presence of a 5′- but not a 3′-monophosphate group. The A40 substrate in (A) was labelled at the 5′-end using [γ-32P]ATP and T4 polynucleotide kinase (PNK), whereas the same oligonucleotide in (B) and (C) was labelled at the 3′-end using [5′-32P]pCp and T4 RNA ligase. The oligonucleotide in (B) was in addition monophosphorylated at the 5′-end using PNK and an excess of non-radioactive ATP. The oligonucleotide in (D) (A29:G21) had a 5′-segment of 29 A residues linked to a 3′-segment of 21 G residues and was labelled at the 5′-end. In each case the concentrations of full-length RNase E and substrate were ∼50 and 750 nM, respectively. Samples were removed immediately prior to the addition of enzyme (lane 1) and after incubation at 37°C for 2, 5, 10 and 20 min (lanes 2–5, respectively) and were analysed on standard (45 cm length) 10% (w/v) polyacrylamide sequencing-type gels. The numbers on the left of (A) to (C) indicate the sizes of 5′-labelled species produced by RNase E cleavage of A40. Similarly, those on the right of (D) indicate the sizes of 5′-labelled species corresponding to A29:G21. The latter were determined from a series of gels in which the 5′-labelled products were run against RNAs of known size (data not shown). The results shown in (D) and those in (A) to (C) were derived from different gels; therefore, the mobility of the bands cannot be compared directly. Each of the panels is derived from a phosphorimage.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/29/9/10.1093_nar_29.9.1864/3/m_gke29701.jpeg?Expires=1716445774&Signature=twz9jcSP14M365NA2Ri6ilozCdNGIw~rjQHqV1RN~yjlPQn0xFQty9B5IMK--Ht4SzDIYBItROTi1tg5ICBnFwSFj6o4m27EE6KtbHAd9Xw1JmLxxIxUlGyxeGwhgKanehT8ix-Yzp6aBS~9058l9yVeRrIZQbWYn-70JiOyZRUkCLPSrqM6ZPAizg2Gf8n7ib3eHlea-lqws86dbUSJYpkIMZ1tFZHlQEfK6ECrwmb0Pt3gPDhzXLnN45TnzDw9H~VQ840KVzAUA7f58QrZwAzYj94LGUab3YqPs86UE8OAjnJxbIfd1Xa7AnE0DoYsILDkjeEZ4LSlSImOf8cdrg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 1. Cleavage of an A40 oligonucleotide substrate by RNase E is dependent on the presence of a 5′- but not a 3′-monophosphate group. The A40 substrate in (A) was labelled at the 5′-end using [γ-32P]ATP and T4 polynucleotide kinase (PNK), whereas the same oligonucleotide in (B) and (C) was labelled at the 3′-end using [5′-32P]pCp and T4 RNA ligase. The oligonucleotide in (B) was in addition monophosphorylated at the 5′-end using PNK and an excess of non-radioactive ATP. The oligonucleotide in (D) (A29:G21) had a 5′-segment of 29 A residues linked to a 3′-segment of 21 G residues and was labelled at the 5′-end. In each case the concentrations of full-length RNase E and substrate were ∼50 and 750 nM, respectively. Samples were removed immediately prior to the addition of enzyme (lane 1) and after incubation at 37°C for 2, 5, 10 and 20 min (lanes 2–5, respectively) and were analysed on standard (45 cm length) 10% (w/v) polyacrylamide sequencing-type gels. The numbers on the left of (A) to (C) indicate the sizes of 5′-labelled species produced by RNase E cleavage of A40. Similarly, those on the right of (D) indicate the sizes of 5′-labelled species corresponding to A29:G21. The latter were determined from a series of gels in which the 5′-labelled products were run against RNAs of known size (data not shown). The results shown in (D) and those in (A) to (C) were derived from different gels; therefore, the mobility of the bands cannot be compared directly. Each of the panels is derived from a phosphorimage.
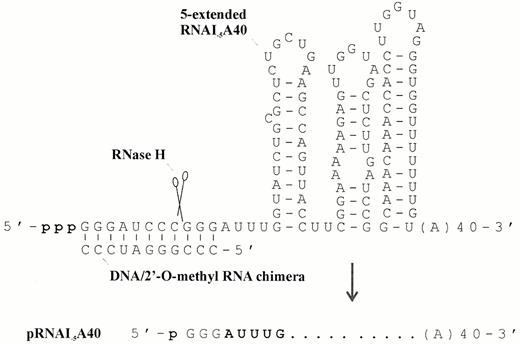
Figure 2. Synthesis, sequence and secondary structure of polyadenylated 5′-monophosphorylated RNAI–5. This RNA substrate lacks the first 5 nt of RNAI that are removed by RNase E and thus models the decay intermediate that is subsequently degraded from the 3′-end (23,42). The details of the primers used to synthesise the template for the in vitro transcription of 5′-extended RNAI–5, which has an A40 tail, can be found in Materials and Methods. After transcription a chimeric oligonucleotide was annealed to the 5′-extended RNA and the annealed complex incubated with RNase H to produce a 5′-monophosphorylated species that was 8 nt shorter and could, therefore, be purified by preparative gel electrophoresis. The secondary structure is as reported previously for pBR322 (57,58). It should be noted that the three G residues at the 5′-terminus of RNAI–5 synthesised in vitro are a consequence of the promoter requirements of T7 RNA polymerase and are not present in the natural decay intermediate. RNAI–5 having an A40 tail is 146 nt in length.
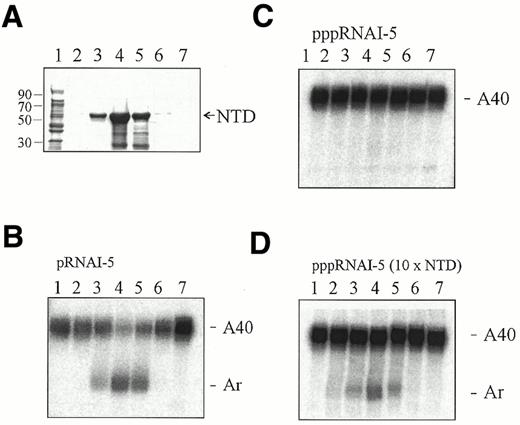
Figure 3. 5′-End dependency of poly(A) tail removal from the 3′-end of RNAI–5 by RNase E. (A) SDS–PAGE analysis of fractions across a peak of N-terminal domain RNase E that was purified by immobilised metal affinity chromatography under denaturing conditions. Lane 1 contains the flow-through, whereas lanes 2–7 contain fractions eluted using 100, 200, 300, 400, 500 and 600 mM imidazole, respectively. The 8% (w/v) polyacrylamide gel shown was stained with Coomassie blue. Numbers to the left of this panel indicate the positions of a selection of components of 10 kDa size markers (Life Technologies), which are not shown. The polypeptides that migrate faster than the major band are truncated forms of the N-terminal domain (NTD) that cross-react with antibodies raised against the first 323 residues of RNase E. (B–D) Assay of poly(A) nuclease activity in fractions across a peak of N-terminal domain RNase E. The substrate used was internally labelled RNAI–5A40 (∼25 nM). In (B) it was monophosphorylated at its 5′-end, whereas in (C) and (D) it had a 5′-triphosphate group. Samples as in (A) except for lane 1, which contained a no enzyme control. Each reaction was incubated at 37°C for 40 min and contained the same volume of each fraction. The concentration of the peak fraction of NTD-RNase E (lane 4) in (B) and (C) was 15 nM, whereas in (D) it was 150 nM. The positions of RNAI–5 species having a 3′-A40 tail (A40) or an RNase E-generated remnant (Ar) of 5–7 adenosines (29; Fig. 6) are indicated. Short (7 cm length) 10% (w/v) polyacrylamide–7 M urea gels were used to allow the remnant species to be quantitated as a single band.
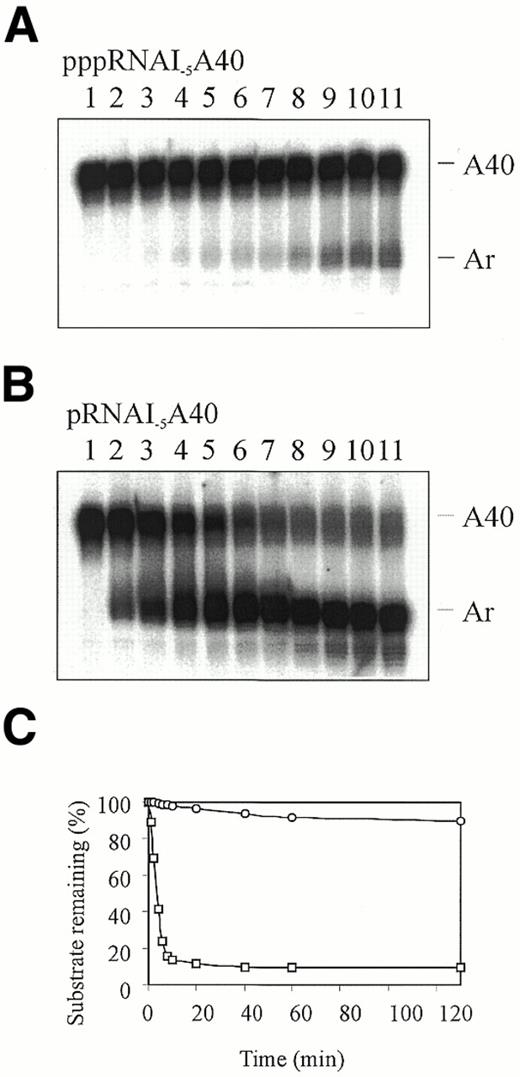
Figure 4. Comparison of the rate of removal by RNase E of poly(A) tails from RNAs that differ only in phosphorylation at their 5′-ends. The concentrations of NTD-RNase E and substrate in each reaction were 15 and 25 nM, respectively. The source of NTD-RNase E was the 300 mM imidazole fraction (see Fig. 3), which we estimated was >95% pure as judged by densitometric scanning of the Coomassie stained gel (Fig. 3). The substrates in (A) and (B) were 5′-triphosphorylated and 5′-monophosphorylated RNAI–5A40, respectively. Samples in lanes 1–11 were taken after 0, 1, 2, 4, 6, 8, 10, 20, 40, 60 and 130 min, respectively. (C) Plot of the amount of substrate remaining (%) with time in each of the above reactions. Data points for 5′-triphosphorylated and 5′-monophosphorylated RNAI–5A40 are represented by circles and squares, respectively.
![Figure 5. Analysis of RNase E cleavage of an A40 tail on 3′-labelled RNAI–5. 5′-Monophosphorylated and 5′-triphosphorylated RNA synthesised as described in Figure 2 and labelled at the 3′-end using [5′-32P]pCp and T4 RNA ligase. The concentration of NTD-RNase E and substrate in each reaction was as described in Figure 4. The source of NTD-RNase E was the 300 mM imidazole fraction (lane 4, Fig. 3). Samples in lanes 1–4 were taken after 0, 15, 30 and 60 min, respectively. The marker (lane M) is 3′-labelled, 5-monophosphorylated A40 that was partially cleaved with RNase E (see Fig. 1). The numbers on the right of the panel indicate the size of the 3′-labelled products.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/29/9/10.1093_nar_29.9.1864/3/m_gke29705.jpeg?Expires=1716445774&Signature=tXOgiuKb0I5ycWifSYyVKziipImX9xnrMtbezFx5f6yc50okvaGNSWFKdij2aMwm0Qz~82KorrV0AtVAYy49UU0D0kV~9Bvi2Eif~foDE9LcGnCRYpcN~e43uj6i7xKhSObqO83FZMm7sLGQDt~Z~cu8ZcCG6VrxR7S2IUjqp3wTzhJbZP15SyiMmV7an3xEq21Li~FYN5u7Z9q7ebO220l8~RzAHb-803lFZMPjpXb5QEJI3Ihyz42xX9nCwz21qXN5FkvX51WT3WCGCGswq1k81DkWhHTTyownZjyRvPezabX8GK4y6xV6PGZa0Ri1rDKc-s1i5ZApmdf8lodceA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 5. Analysis of RNase E cleavage of an A40 tail on 3′-labelled RNAI–5. 5′-Monophosphorylated and 5′-triphosphorylated RNA synthesised as described in Figure 2 and labelled at the 3′-end using [5′-32P]pCp and T4 RNA ligase. The concentration of NTD-RNase E and substrate in each reaction was as described in Figure 4. The source of NTD-RNase E was the 300 mM imidazole fraction (lane 4, Fig. 3). Samples in lanes 1–4 were taken after 0, 15, 30 and 60 min, respectively. The marker (lane M) is 3′-labelled, 5-monophosphorylated A40 that was partially cleaved with RNase E (see Fig. 1). The numbers on the right of the panel indicate the size of the 3′-labelled products.
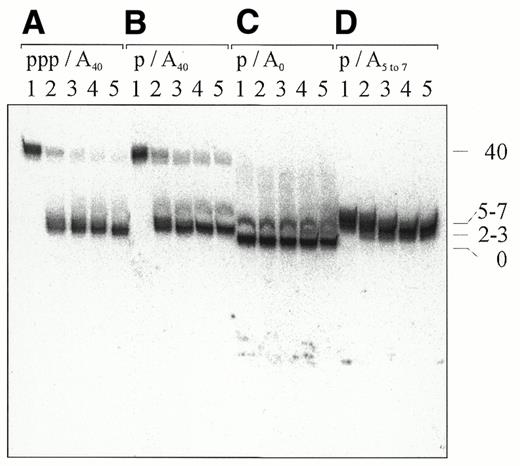
Figure 6. Comparison of poly(A) tail removal from 5′-monophosphorylated and 5′-triphosphorylated RNAI–5 by the degradosome. (A–D) Polyadenylated 5′-monophosphorylated and 5′-triphosphorylated RNAI–5 and 5′-monophosphorylated RNAI–5 that was unadenylated or had a 3′-remnant produced by RNase E cleavage of an A40 tail, respectively. All of the RNA species were internally labelled during in vitro transcription. The concentrations of degradosome and substrate in each reaction were 2 and 20 nM, respectively. Samples in lanes 1–5 were taken after incubation for 0, 2, 5, 15 and 30 min, respectively. The numbers on the right indicate the approximate number of adenosines on the 3′-end of the RNA, as judged by their mobility relative to other RNA species on the same gel. At the top, p and ppp indicate whether the RNA substrate was monophosphorylated or triphosphorylated at the 5′-end, respectively, whereas A0 and A40 indicate whether it was unadenylated or polyadenylated, respectively. The faint band immediately above that of the major products in (B) and (C) is likely a small amount of 5′-extended RNA that we know can be carried over during the preparation of 5′-monophosphorylated RNA substrate (see Fig. 2).
![Figure 7. Comparison of the rate of poly(A) tail removal from 5′-monophosphorylated RNAI–5 by the degradosome in the absence and presence of inorganic phosphate. The substrate in each of the panels is polyadenylated RNAI–5 (20 nM) that was internally labelled. (A, B and D) The substrate was 5′-monophosphorylated (5′ p); (C) the substrate was 5′-triphosphorylated (5′ ppp). The reactions in (A) and (B) did not contain inorganic phosphate, whereas in (C) and (D) it was at a concentration of 10 mM. The concentration of degradosome (Deg.) in reactions (A), (C) and (D) was 2 nM, as in Figure 6. (B) A control incubation with full-length RNase E (Rne) that served to generate a marker for species that had poly(A) tail remnants. Lanes 1–5 contain samples from incubations of 0, 2, 5, 15 and 30 min, respectively. Numbers on the right are as in Figure 6. Samples were analysed as in Figure 1. The digestion products below the major band in (B) and, to a lesser extent, in (A) correspond to RNase E cleavage at internal sites, which have been described previously (59). (E) A plot of the amount of adenylated pRNAI–5 remaining (%) against the time incubated with degradosome in the absence (circles) or presence (squares) of inorganic phosphate [(A) and (D), respectively].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/nar/29/9/10.1093_nar_29.9.1864/3/m_gke29707.jpeg?Expires=1716445774&Signature=BY8nZcycLcI8XOMnjKro6hIpp7iOSJl67MivzDLS1ZMmnZ3JGYVyb7C2qi1UAXz-RIPQ-5tTPE0jElYe1uZdz4L0FVxXGqOuPuXfXv8Sn~uDNVObHukRkZMV-B8HZG2MOUuzcT4C0jwA7Kfe0ZS1gHWS6Vn1A-juo44jLlthyZilkpnD6CuXv8N40BF-ogjZnG4qpdgdWaGWyuoISfgMF4JbCG5ZFpQJATjYmFDGOO6iw8G3COHm~JatneI-r7h9dSxnLhR6OIUNHRnoX8z4dmTDWfnvEDWLCUXt68THBB33-yY7laR6~Oy-VLKU60X68OaylPbaGyE9~1l0MEsKUw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 7. Comparison of the rate of poly(A) tail removal from 5′-monophosphorylated RNAI–5 by the degradosome in the absence and presence of inorganic phosphate. The substrate in each of the panels is polyadenylated RNAI–5 (20 nM) that was internally labelled. (A, B and D) The substrate was 5′-monophosphorylated (5′ p); (C) the substrate was 5′-triphosphorylated (5′ ppp). The reactions in (A) and (B) did not contain inorganic phosphate, whereas in (C) and (D) it was at a concentration of 10 mM. The concentration of degradosome (Deg.) in reactions (A), (C) and (D) was 2 nM, as in Figure 6. (B) A control incubation with full-length RNase E (Rne) that served to generate a marker for species that had poly(A) tail remnants. Lanes 1–5 contain samples from incubations of 0, 2, 5, 15 and 30 min, respectively. Numbers on the right are as in Figure 6. Samples were analysed as in Figure 1. The digestion products below the major band in (B) and, to a lesser extent, in (A) correspond to RNase E cleavage at internal sites, which have been described previously (59). (E) A plot of the amount of adenylated pRNAI–5 remaining (%) against the time incubated with degradosome in the absence (circles) or presence (squares) of inorganic phosphate [(A) and (D), respectively].
References
1 Cohen,S.N. and McDowall,K.J. (
2 Coburn,G.A. and Mackie,G.A. (
4 Nicholson,A.W. (
6 Regnier,P. and Arraiano,C.M. (
7 Littauer,U.Z. and Grunberg-Manago,M. (
8 Shen,V. and Schlessinger,D. (
9 Donovan,W.P. and Kushner,S.R. (
10 Ghosh,S. and Deutscher,M.P. (
11 Ghora,B.K. and Apirion,D. (
13 Mackie,G.A. (
14 Carpousis,A.J., van Houwe,H.G., Ehretsmann,C. and Krisch,H.M. (
15 Py,B., Causton,H., Mudd,E.A. and Higgins,C.F. (
16 Py,B., Higgins,C.F., Krisch,H.M. and Carpousis,A.J. (
17 Miczak,A., Kaberdin,V.R., Wei,C.L. and Lin-Chao,S. (
18 Carpousis,A.J., Vanzo,N.F. and Raynal,L.C. (
20 Manley,J.L. (
23 Xu,F. and Cohen,S.N. (
24 O’Hara,E.B., Checkanova,J.A., Ingle,C.A., Kushner,Z.R. Peters,E. and Kushner,S.R. (
25 Mohanty,B.K. and Kushner,S.R. (
26 Mohanty,B.K. and Kushner,S.R. (
27 Cao,G.J. and Sarkar,N. (
28 Cesareni,G., Helmer,C.M. and Castagnoli,L. (
29 Huang,H.J., Liao,J. and Cohen,S.N. (
30 Uhlenbeck,O.C. and Gumport,R.I. (
31 Blum,E., Carpousis,A.J. and Higgins,C.F. (
32 Tock,M.R., Walsh,A.P., Carroll,G. and McDowall,K.J. (
33 Inoue,H., Hayase,Y., Iwai,S. and Ohtsuka,E. (
34 Lapham,J. and Crothers,D.M. (
35 McDowall,K.J. and Cohen,S.N. (
36 Sambrook,J., Fritsch,E.F. and Maniatis,T. (
38 Sugimoto,N., Ohmichi,T. and Sasaki,M. (
39 Phillips,K., Dauter,Z., Murchie,A.I.H., Lilley,D.M.J. and Luisi,B. (
40 He,W. and Parker,R. (
41 Tomcsanyi,T. and Apirion,D. (
42 Lin-Chao,S. and Cohen,S.N. (
43 Coburn,G.A. and Mackie,G.A. (
44 Wahle,E. and Ruegsegger,U. (
45 Kalapos,M.P., Paulus,H. and Sarkar,N. (
46 Fernandez,F.M., Eoyang,L. and August,J.T. (
47 Franze de Fernandez,M.T., Hayward,W.S. and August,J.T. (
48 Senear,A.W. and Argetsinger-Steitz,J. (
49 Zhang,A.X., Altuvia,S., Tiwari,A., Argaman,L., Hengge-Aronis,R. and Storz,G. (
50 Hajnsdorf,E. and Regnier,P. (
51 Feng,Y.N. and Cohen,S.N. (
52 Belasco,J. and Higgins,C.F. (
53 Higgins,C.F., Peltz,S.W. and Jacobson,A. (
54 Raynal,L.C. and Carpousis,A.J. (
55 Causton,H., Py,B., McLaren,R.S. and Higgins,C.F. (
56 Kaberdin,V.R., Walsh,A.P., Jakobsen,T., McDowall,K.J. and von Gabain,A. (
57 Morita,M. and Oka,A. (
58 Tamm,J. and Polisky,P. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments