





Abstract
Concerns have been raised that posterior probabilities on phylogenetic trees can be unreliable when the true tree is unresolved or has very short internal branches, because existing methods for Bayesian phylogenetic analysis do not explicitly evaluate unresolved trees. Two recent papers have proposed that evaluating only resolved trees results in a “star tree paradox”: when the true tree is unresolved or close to it, posterior probabilities were predicted to become increasingly unpredictable as sequence length grows, resulting in inflated confidence in one resolved tree or another and an increasing risk of false-positive inferences. Here we show that this is not the case; existing Bayesian methods do not lead to an inflation of statistical confidence, provided the evolutionary model is correct and uninformative priors are assumed. Posterior probabilities do not become increasingly unpredictable with increasing sequence length, and they exhibit conservative type I error rates, leading to a low rate of false-positive inferences. With infinite data, posterior probabilities give equal support for all resolved trees, and the rate of false inferences falls to zero. We conclude that there is no star tree paradox caused by not sampling unresolved trees.
Accurately characterizing statistical confidence in phylogenetic hypotheses is an important and long-standing challenge. Bayesian phylogenetics expresses confidence in terms of posterior probability—the probability that a tree or clade is true given the data, an evolutionary model, and prior probability distributions over model parameters (Huelsenbeck et al. 2001). There is growing concern that inferred posterior probabilities may be generally “overcredible” in a frequentist sense, leading to inflated confidence in uncertain relationships and a high rate of incorrect inferences, especially when the true tree has zero or near-zero length internal branches (Suzuki et al. 2002; Cummings et al. 2003; Lewis et al. 2005 [LHH]; Yang and Rannala 2005 [YR]).
Most software for Bayesian phylogenetic inference uses Markov Chain Monte Carlo (MCMC) techniques that sample only resolved trees; unresolved phylogenies are approached by examining very short internal branch lengths (typically 10−6 substitutions/site), but the remaining “hole” in parameter space is not sampled. LHH suggested that when the true tree is unresolved, not sampling unresolved trees causes “disturbingly high” posterior probabilities to be inferred for one or another arbitrarily resolved tree, and this problem gets worse as sequence length increases: “For large data sets, the phylogenetic uncertainty generated by the true polytomy manifests itself as unpredictability in the level of estimated posterior support for arbitrary resolutions of the polytomy, not as increased homogeneity of support for all possible resolutions.” This conclusion was based on a series of simulations using a 4-taxon star tree with equal terminal branch lengths: when replicate sequences of length N = 1 were analyzed, equal posterior probability was always inferred for each possible resolved tree, but when longer sequences (N=100,000) were analyzed, some replicates produced high support for 1 of the 3 resolved trees. YR examined additional sequence lengths and also found that very short sequences (N = 20) always produced roughly equal support for each resolved tree, but longer sequences (N = 200 and N = 1,000) occasionally yielded high support for 1 of the 3 topologies. Both LHH and YR sketched theoretical arguments predicting that “posterior probabilities of particular resolutions of polytomous tree topologies will become more unpredictable with increasing sequence length” (Lewis et al. 2005) and become completely unpredictable as N approaches infinity. If true, this “star tree paradox” is a real concern; it suggests that posterior probabilities on trees with short internal branches may regularly generate inflated confidence in incorrect or uncertain phylogenies, leading to frequent inferences of incorrect evolutionary relationships and increasingly pathological behavior as more data are analyzed.
The prediction that posterior probabilities would become more problematic as sequence length grows has not been directly tested, however. Both LHH and YR examined too few sequence lengths to establish a general trend, and neither explicitly examined how MCMC methods would perform with infinite data. Further, neither study investigated whether at any sequence length high posterior probabilities are observed more often than they should be. Here we use simulation experiments to test the predictions associated with the star tree paradox.
If posterior probabilities become increasingly unpredictable as sequence length increases, then the variance in the posterior probability of a particular resolved tree over replicate data sets should increase as N grows. We tested this prediction by simulating alignments of various lengths on a 4-taxon star tree using conditions similar to those examined by LHH and YR and estimating the posterior probability of a resolved phylogeny using MrBayes v3.1, which does not sample unresolved trees. We found that the mean posterior probability is always close to 1/3 (fig. 1A), and—after an initial increase—the variance remains stable with increasing sequence length (fig. 1B). When N ≤ 10, the variance in posterior probability is close to zero because a resolved tree can only be supported by convergent substitutions on at least two branches; in very small data sets, such low-probability patterns usually do not occur at all. Once sequences are long enough for convergent patterns to appear, however, there is no increase in variance with the amount of data analyzed. The apparent increase in earlier studies was due to a failure to examine enough sequence lengths to distinguish between a long-term trend and the initial increase due to near-zero variance at extremely short sequence lengths.
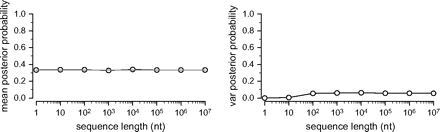
Variance in posterior probability of a resolved tree does not increase with increasing sequence length. The mean (left panel) and variance (right panel) in posterior probability of a particular resolved tree over replicate data sets is shown when data are generated using the JC69 substitution model and a 4-taxon star tree with long terminal branch lengths (0.5 substitutions/site). Gray line indicates expected mean support of 1/3.
Both LHH and YR noticed that, when moderate or long sequences are simulated on a 4-taxon star tree, “disturbingly high” posterior probabilities were occasionally observed, in contrast to very short sequences, for which posterior probabilities were always close to 1/3. The occasional presence of high posterior probabilities is not in itself reason for concern. Although a star tree is expected to generate equal frequencies of state patterns that support each of the 3 trees, stochastic variation with finite data sets causes pattern frequencies to deviate from expectation, leading to spurious support for one tree or another. Usually the stochastic deviation is small, but infrequently it will be larger. Unequal pattern frequencies also occur when the true tree is resolved, producing phylogenetic signal. The purpose of posterior probabilities is to help distinguish these possibilities by expressing the probability that some resolved tree is true given the data. If a method is to have any power to detect a resolved phylogeny when it is true, high posterior probabilities must occur occasionally when finite data are generated on the star tree. The crucial question is whether they occur more often than they should, leading to a high rate of erroneous inferences—an issue not addressed by LHH or YR.
If the posterior probability of a tree accurately estimates the probability that the tree is the true tree (which it has been shown to do when the true tree is resolved, provided the model and priors are correctly specified [Huelsenbeck and Rannala 2004; Yang and Rannala 2005]), a hypothesis with posterior probability 0.95 should have a 0.05 chance of being false, and a group of hypotheses with posterior probability 0.95 should contain 5% incorrect trees. Though unconventional in a Bayesian framework, the use of a decision rule that accepts a phylogenetic hypothesis only if it has posterior probability >0.95 should therefore result in a long-run type I error rate <0.05 if posterior probabilities accurately measure the frequentist probability that a tree is correct. We tested whether use of current MCMC implementations leads to high rates of type I error by simulating replicate sequence alignments on various unresolved trees and observing the proportion of resolved trees with posterior probabilities greater than various cutoff values. Resolved trees with support greater than cutoffs of 0.90, 0.95, and 0.99 were considered type I errors (Swofford et al. 2001), and observed error rates were compared to maximum error rates expected if posterior probabilities are accurate estimators that a tree is true (0.10, 0.05, and 0.01, respectively). We found that type I error rates were lower than the maximum acceptable for all sequence lengths and thresholds used, whether terminal branch lengths were equal or in the more challenging Felsenstein zone and whether data were generated using simple or complex evolutionary models (fig. 2A).
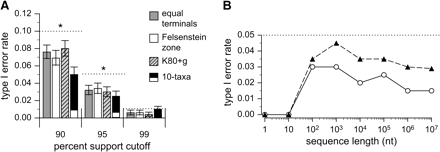
Type I error rates based on posterior probability are conservative. (A) The fraction of star trees incorrectly resolved at support cutoff values of 0.90, 0.95, and 0.99 posterior probability is shown. Sequences were 5,000-nt long. Dotted line indicates maximum permissible error rate. Bars indicate standard error, with a significantly reduced error rate compared with the maximum permissible for each cutoff being indicated by an asterisk (α = 0.01). For 10-taxon trees, we calculated type I error rates when posterior probabilities were summarized on clades in 2 ways: 1) each clade is considered an independent hypothesis (white), and 2) a single resolved clade per replicate is considered a type I error (black). (B) Type I error rates (based on a 0.95 posterior probability cutoff) are shown as sequence length increases when the true 4-taxon star tree has long terminal branches (0.5 substitutions/site, filled triangles) or short terminals (0.05, open circles). Dotted line indicates maximally acceptable type I error rate.
To determine if type I error rates remain low when larger phylogenies are analyzed, we simulated data using a 10-taxon star tree with long terminal branches (0.5 substitutions/site), analyzed these data using MrBayes, and assessed the frequency with which incorrectly resolved phylogenetic hypotheses were strongly supported. First, we found that no fully resolved trees were supported with posterior probability >0.9 when data were generated on a large star tree (not shown). Posterior probabilities are typically not reported on an entire tree, however; more commonly, posterior probabilities are reported on individual clades by summarizing over all sampled trees. Recent concerns have been raised that this practice may introduce a bias in posterior probability estimates because the typical assumption of flat priors over trees places higher prior probability on clades with either few or many taxa (Pickett and Randle 2005). We found that when posterior probabilities are summarized on individual clades—with each clade being considered an independent hypothesis—type I error rates are very low (fig. 2A). Even if a single strongly supported clade per replicate is considered a type I error—an extremely conservative measure of type I error rate—the rate of erroneous inferences is still less than maximally acceptable at all posterior probability cutoffs examined. Taken together, the results of our type I error analyses indicate that current MCMC implementations do not frequently produce excessive confidence in falsely resolved hypotheses when data are generated on a star tree, even though the true phylogeny is never explicitly considered.
Contrary to the prediction that posterior probabilities would become increasingly unreliable as sequence length increases, we found that type I error rates fall with increasing sequence length (fig. 2B). As observed with the variance in posterior probabilities, the trend in type I error with sequence length is not monotonic. With extremely short sequences (N ≤ 10), the error rate is close to zero, presumably due to the lack of any convergent state patterns. The rate of type I error then increases with sequence length as convergent patterns begin to occur, peaking at moderately short lengths (N = 100–1,000) and then falling as sampling error becomes less important with longer sequences. Even when type I error rates are at their maximum, posterior probabilities never produce strong support for incorrectly resolved phylogenies more often than they should.
LHH and YR suggested that, as sequences generated on a star tree approach infinite length, we would like the inferred posterior probability of each possible resolved tree to become equal; however, they predicted that these posterior probabilities would become “completely unpredictable” over replicates. We tested this prediction by analyzing pseudo–data sets that possess the same chararcteristics as infinite data. With infinitely long sequences, the frequency of each possible character state pattern equals the expected frequency, and the variance in pattern frequencies among data sets is zero. To determine the posterior probabilities that would be inferred if infinite data were available, we generated pseudo–infinite data sets that do not deviate from expected character state pattern frequencies and estimated posterior probabilities from these data by MCMC without explicitly sampling the true unresolved tree. Specifically, we calculated the per-site likelihood of an infinite data set by calculating the expected pattern frequencies given the simulation conditions and modifying MrBayes to infer posterior probabilities given a list of patterns and their associated frequencies. When these ideal data were repeatedly analyzed, we observed a mean posterior probability of 0.333 for each possible tree (fig. 3A) and very little scatter about the mean (σ2 = 1.2 × 10−4). From 200 replicates—100 with long terminal branches (0.5) and 100 with short terminals (0.05)—the maximum posterior probability observed for any resolved tree was 0.37. The small amount of variation observed among replicates appears to be due to stochastic error in MCMC sampling: when longer runs are executed, posterior probabilities are even closer to 1/3 (fig. 3B, σ2 = 1.06 × 10−5, maximum posterior probability = 0.34). These results indicate that posterior probabilities do produce equal support for all resolved trees in the infinite case (P = 0.98), which is the desired result. Analysis of ideal data sets does not indicate what will happen when very large data sets with some stochastic error are analyzed, but it does show that when infinite data are generated on a star tree, posterior probabilities are predictable, equally supporting each possible resolved tree.
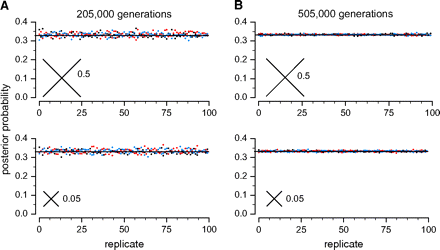
Star tree–generated data with ideal character state pattern frequencies produce equal posterior probability for each possible resolved tree. The inferred posterior probability of each resolved 4-taxon tree (black, blue, and red) is shown for independent Bayesian analyses of an ideal data set with no stochastic variation. In the top panel, the true tree has 4 long terminal branches (0.5 substitutions/site); in the lower panel, the true tree has short terminals (0.05). Solid lines indicate theoretically correct inference of 1/3 support for each tree. MCMC analyses were run for either 205,000 generations (A) or 505,000 generations (B).
LHH and YR both used a coin-flipping analogy to support their contention that posterior probabilities become increasingly unpredictable as sequence length grows. YR demonstrated that when the null hypothesis is true (i.e., the coin is fair), the expected frequency distribution of posterior probabilities for each “resolved” hypothesis (that the coin is biased one way or the other) is uniform. Although the distribution of posterior probabilities on phylogenetic trees is unknown, LHH and YR presented the coin-flipping result as evidence of pathological behavior when the null hypothesis is true but is not explicitly examined. In fact, this behavior is reassuring. The uniform frequency distribution implies that data leading to an inferred posterior probability ≥0.95 on incorrect trees will be observed at most 5% of the time, data leading to a posterior probability ≥0.90 will be observed 10% of the time—precisely the behavior expected if posterior probabilities accurately reflect the probability that the hypothesis is true. The initially appealing intuition that posterior probabilities should converge on equal support for each resolved hypothesis is correct only when data are truly infinite and precisely match the null expectation. If posterior probabilities calculated from finite data were instead concentrated around 1/T (where T is the number of possible resolved hypotheses), we would always infer low posterior probabilities for resolved trees—not only when the null hypothesis is true but also when the true hypothesis is resolved—leading to reduced statistical power to resolve difficult phylogenies.
The implication of our results is that there is no star tree paradox. Even when trees contain zero or near-zero length internal branches, posterior probabilities behave as an appropriate statistical estimator should, providing near-equal support for all possible resolved trees with infinite sequence length and producing strong support for incorrect trees very infrequently when finite data are analyzed. The fact that unresolved trees are not explicitly evaluated has no apparent effect on the accuracy of posterior probability as a measure of statistical confidence. Furthermore, we have shown that evidence previously presented in favor of the star tree paradox has been erroneously interpreted. The occasional support in favor of a falsely resolved phylogeny observed by LHH and YR is the expected result of stochastic error, and the convergence of posterior probabilities to the uniform distribution is a desirable property of a statistical estimator, producing a reasonable balance between power to resolve difficult problems with strong support and a low rate of false inferences. Our results do not imply that posterior probabilities will never be inflated; previous studies have shown that posterior probabilities can be unreliable when either the evolutionary model (Suzuki et al. 2002; Huelsenbeck and Rannala 2004) or prior assumptions about model parameters (Yang and Rannala 2005) are incorrectly specified. That existing methods do not sample unresolved trees, however, does not inflate posterior probabilities inferred by MCMC.
Methods
Posterior probabilities were estimated using MrBayes v3.1 (Ronquist and Huelsenbeck 2003). Four incrementally heated chains (temp = 0.2) were run for 205,000 generations, with samples taken every 100 generations. The first 5,000 generations were discarded as burn-in. Prior probabilities were equal over all tree topologies and uniformly distributed on [0,10] for branch lengths. The shape parameter for gamma-distributed among-site rate variation was given a uniform prior on [0.05,50], and the default prior was used for the transition/transversion ratio. The true evolutionary model was assumed.
Sequence alignments of length 1, 10, 100, 103, 104, 105, 106, and 107 nt were simulated on a 4-taxon star tree. We simulated data using the JC69 substitution model and either equal terminal branches (0.5 or 0.05 substitutions/site) or Felsenstein-zone branch lengths with 2 long terminals (0.75) and 2 short terminals (0.05). Data were also simulated using the K80 + g (κ = 10, α = 0.5) model and equal long terminal branches (0.5). We analyzed 1,000 replicate alignments under each set of experimental conditions as described above. In addition, we analyzed 5,000-nt alignments simulated using a 10-taxon star tree with long terminal branches (0.5), with posterior probabilities on clades summarized over trees using MrBayes. In each case, observed type I error rates were compared with maximum acceptable values using a 1-sided t test.
To examine the accuracy of posterior probabilities with infinite data, ideal pseudo–data sets with no stochastic error were analyzed. We calculated the expected frequency of each character state pattern (f(x)) under a 4-taxon star phylogeny with either long (0.5) or short (0.05) terminal branch lengths and the JC69 substitution model. We modified the source code of MrBayes v3.1 to estimate posterior probabilities given this vector of state pattern frequencies. The per-site likelihood of tree t given any state pattern x is calculated by raising the probability of the pattern, given the tree, to the frequency with which that pattern is expected to occur: L(t|x) = P(x|t)f(x). The total per-site likelihood of the tree is the product of this partial likelihood over all possible state patterns. Each ideal data set was analyzed 100 times to account for variation in Markov chain sampling. We calculated the mean posterior probability over all analyses and assessed deviation from expected support of 1/3 for each possible resolved tree using a t test.
Herve Philippe, Associate Editor
We are grateful to Ziheng Yang and Paul Lewis for helpful comments. Supported by National Science Foundation DEB-0516530, National Institutes of Health GM62351-01, and a Sloan Foundation research fellowship to J.W.T.
References
Cummings MP, Handley SA, Myers DS, Reed DL, Rokas A, Winka K.
Huelsenbeck JP, Rannala B.
Huelsenbeck JP, Ronquist F, Nielsen R, Bollback JP.
Lewis PO, Holder MT, Holsinger KE.
Pickett KM, Randle CP.
Ronquist F, Huelsenbeck JP.
Suzuki Y, Glazko GV, Nei M.
Swofford DL, Waddell PJ, Huelsenbeck JP, Foster PG, Lewis PO, Rogers JS.
Author notes
*Department of Computer and Information Science, University of Oregon and †Center for Ecology and Evolutionary Biology, University of Oregon

{kind=link}
{kind=link}
{kind=link}