




Abstract
The main identified function of BCL2 protein is to prevent cell death by apoptosis. Mouse knock-out for Bcl2 demonstrates growth retardation, severe polycystic kidney disease (PKD), grey hair and lymphopenia, and die prematurely after birth. Here, we report a 40-year-old male referred to for abdominal and thoracic aortic dissection with associated aortic root aneurysm, PKD, lymphocytopenia with a history of T cell lymphoblastic lymphoma, white hair since the age of 20, and learning difficulties. PKD, which was also detected in the father and sister, was related to an inherited PKD1 mutation. The combination of PKD with grey hair and lymphocytopenia was also reminiscent of Bcl2−/− mouse phenotype. BCL2 gene transcript and protein level were observed to be dramatically decreased in patient peripheral blood T-cells and in his aorta vascular wall cells, which was not detected in parents and sister T-cells, suggesting an autosomal recessive inheritance. Accordingly, spontaneous apoptosis of patient T-cells was increased and could be rescued through stimulation with an anti-CD3 antibody. Direct sequencing of BCL2 gene exons, promoter and 3’UTR region as well as BCL2 mRNA sequencing failed in identifying any pathogenic variant. Array-CGH was also normal and whole exome sequencing of the patient, parents and sister DNA did not detect any significant variant in genes encoding BCL2-interacting proteins. miRNA array identified an up-regulation of miR-181a, which is a known regulator of BCL2 expression. Altogether, miR-181a-mediated decrease in BCL2 gene expression could be a modifying factor that aggravates the phenotype of a PKD1 constitutive variant.
Introduction
The survival and death of human cells depend on a complex interplay between anti- and pro-apoptotic proteins of the B-cell lymphoma leukaemia-2 (BCL2) family. BCL2 gene was identified in follicular lymphoma cells in which a chromosomal translocation between chromosome 14 and 18 transfers BCL2 gene to immunoglobulin heavy chain locus, which results in an increased expression of the protein. This event was subsequently shown to prevent apoptosis and BCL2 became the first oncogene involved in tumour development through inhibiting cell death. A series of related genes and proteins were identified through amino acid homologies in BCL2 homology (BH) motifs. Some of these proteins, such as BCL-XL and MCL1, also protect cells from death while others promote apoptosis (1,2). The deregulated expression of proteins of the BCL2 family is observed in most cancers as well as in autoimmune and degenerative diseases (3). Their expression modulates the response of cancer cells to anticancer agents (4) and anti-apoptotic proteins of the family are promising therapeutic targets in oncology (5–7). However, a human genetic disease related to the altered expression and function of BCL2 protein has not been identified so far.
BCL2 gene expression depends on two promoters, with P1 being a TATA-less and GC-rich promoter that is dominant in most cell types, and P2 promoter, located 1.3 kb downstream of P1 and containing a CCAAT box, an ATGCAAAC motif, and a TATA box, being specifically dominant in neuronal cells (8). Gene expression is regulated also by methylation of these promoters (9,10) and by several microRNAs (miRNAs) such as miR-15, miR-16 (11) and miR-181a (12,13).
Mice knock-out for Bcl2 gene complete their embryonic development but display growth retardation, with most of the smallest mice dying prematurely after birth (14,15). Bcl2−/− mouse kidneys are morphologically and histologically abnormal with many cysts, a phenotype rescued by crossing Bcl2−/− mice with Bim+/− mice, indicating a central role for the interplay between Bcl2 and the BH3-only domain protein Bim in kidney development (16) by re-expressing Bcl2 in the ureteric bud/collecting duct during kidney development (17). The hematopoietic differentiation of Bcl2−/− mice is normal, with the exception that their lymphocyte production rate is very low, due to massive apoptosis that occurs in both the thymus and the spleen. Finally, the hair coat of Bcl2−/− mice turns gray after the second hair follicle cycle, leading to a complete grey hair coat at 10 weeks of age.
Here, we describe a patient who shares phenotypic similarities with the Bcl2−/− mouse model, including grey hair, severe polycystic kidney disease and lymphocytopenia. Our investigations suggest a constitutive defect in BCL2 expression and function, possibly through miR-181a deregulation.
Results
Increased apoptosis in patient lymphocytes correlates with decreased BCL2 expression
The combination of PKD with grey hairs and lymphocytopenia recapitulated the main features of Bcl2−/− mouse phenotype. Since Bcl2−/− T-cells had shown accelerated spontaneous apoptosis in vivo and in vitro (18), we first collected peripheral blood CD3-positive T-cells from the patient and his relatives as well as from an unrelated, age-matched healthy donor, and compared their susceptibility with apoptosis in culture. We observed that patient CD3+ lymphocytes died more rapidly than the others (Fig. 1A). Since anti-CD3 stimulation had been shown to protect Bcl2−/− T lymphocytes from apoptosis (18), we repeated the experiment by culturing patient CD3+ T cells for 72 and 96 h in presence or absence of an anti-CD3 antibody. As shown in Figure 1B and C, their apoptosis was partially rescued by stimulation with an anti-CD3 antibody. These results supported the hypothesis of a constitutive defect in BCL2. BCL2 mRNA expression was observed to be decreased in the patient CD3-positive T-cells compared with those collected from 20 healthy donors (Fig. 2A). Interestingly, while BCL2 mRNA expression was normal in his sister T-cells, intermediate levels were detected in his father and mother T-cells, suggesting an autosomal recessive inheritance (Fig. 2B). This decrease in BCL2 mRNA level was confirmed at the protein level in patient compared with healthy donor CD3-positive T-cells (Fig. 2C). Of note, two other BCL2-related anti-apoptotic proteins, namely BCL-XL and MCL1, were expressed at the same level in patient T cells than in healthy donor T-lymphocytes (Fig. 2D). We also collected aorta vascular wall when the patient got aortic surgery and quantified BCL2 mRNA in this tissue compared with control surgical samples. Almost no BCL2 mRNA expression was detected in the patient and control aorta tunica adventitia. In the aorta tunica media, we observed an ∼50% decrease in BCL2 gene expression level as compared with control whereas the expression of MCL1 protein was similar in aorta tunica adventitia and media of the patient and the controls (Fig. 2E). Whereas BCL2 is highly expressed in lymphocytes regardless of the activation state of the cells, BCL-XL is expressed upon cell activation only (8,10,19). In patient and healthy donor CD3+ T-cells cultured in the absence or presence of anti-CD3, we observed an up-regulation of BCL-XL mRNA upon CD3 stimulation (Fig. 1D). Of note, this up-regulation was transient in healthy donor cells and more sustained in patient cells, suggesting that patient lymphocytes might compensate low level of BCL2 by accumulating more BCL-XL upon stimulation.
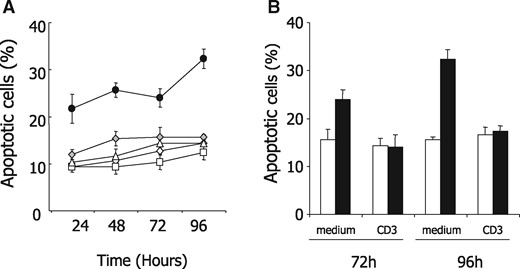
T lymphocytes from the patient are rescued from apoptosis by CD3 stimulation. (A) Peripheral blood CD3+ T cells were sorted from the patient (black dots), an age- and sex-matched control (grey diamond), his sister (white diamond), his mother (white triangle) and his father (white square), and cultured in complete medium for 24, 48, 72 and 96 h before measuring the percentage of apoptotic cells identified cytologically after Hoechst staining. (B) Peripheral blood CD3+ T cells sorted from the patient (black bars) and an age-matched control (white bars) were cultured in complete medium in the absence or presence of an anti-CD3 antibody for 72 or 96 h before measuring the fraction of apoptotic cells by Hoechst staining.
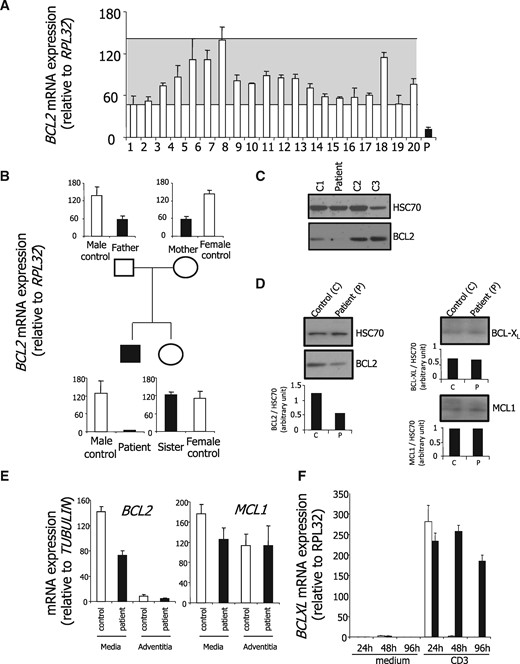
BCL2 mRNA and BCL2 protein decrease in patient cells. Controls, white bars; patient and family, black bars (A) BCL2 mRNA expression was measured by quantitative real-time PCR in CD3-positive T cells sorted from peripheral blood of 20 healthy donors (white bars) and the patient (black bar). The grey zone indicates the variation in healthy donors. Normalizer, RPL32 gene. (B) RT-qPCR analysis of BCL2 mRNA expression in peripheral blood CD3+ T cells from the patient, his parents and his sister, compared with age- and sex-matched healthy donors (controls; one representative of three independent experiment is shown). Normalizer, RPL32 gene. (C) BCL2 protein expression was explored by immunoblotting in CD3-positive T cells sorted from the peripheral blood of the patient and three age-matched healthy donors (C1 to C3). Loading control, HSC70. (D) BCL2, BCL-XL, and MCL1 protein expression was studied by immunoblotting in CD3-positive T cells of the patient and an age-matched healthy donor. Loading control, HSC70. Quantification has been made using ImageJ software and BCL2, BCL-XL and MCL1 normalized to HSC70 histograms are shown under each immunoblot. (E) BCL2 and MCL1 gene expression was quantified by real-Time qPCR in the adventitia and the media of the ascending aorta of the patient compared with a control (patient undergoing vascular surgery). (F) BCL-XL gene expression was quantified by real-Time qPCR in sorted T cells isolated CD3+ lymphocytes of the patient and an age-matched healthy donor cultured in the absence or presence of an anti-CD3 antibody during indicated times. Normalizer, RPL32 gene.
Genetic analysis
The defective expression of BCL2 protein in patient tissues led us to perform direct sequencing of the exons, promoters and 3’UTR region of BCL2 gene, which did not identify any pathogenic variant, including in the two promoters that mediate tissue- and developmental-specific BCL2 gene transcription. Similarly, no pathogenic variation was found in BCL2 mRNA sequence, Computer analysis of the BCL2 TATA-less and GC-rich P1 promoter using MethylPrimer Express (Applied Biosystems) identified a CpG island of 1565 bp with more that 65% of CG in this sequence. The methylation status of this CpG island was explored and no methylation was identified in the patient as well as his sister and parents and two healthy donors. Array-CGH was normal and did not evidence any small deletion or duplication in BCL2 encoding region, nor elsewhere. Whole exome sequencing of the patient, his parents and sister DNA was performed. We detected a total of 494 rare variants affecting the coding sequences in the patient DNA, including 416 missenses, 7 nonsenses, 20 frameshifts, 21 in-frame insertions or deletions, 5 splice-site mutations and 15 variants affecting exon-intron boundaries. We focused our initial analysis on the 103 OMIM variants, which confirmed the PKD1 missense (NM_000296.3: p.Asn1870Asp), inherited from the father and detected also in the sister. We also identified compound heterozygous variants in HERC1 (OMIM 617011): NM_003922.3: p.Arg2419* and p.Ile4329Thr. Abnormalities in this gene were previously reported in five patients with macrocephaly, facial dysmorphism, kypho-scoliosis and severe intellectual disability (20–22). In the studied family, segregation confirmed the heterozygous state in both parents and sister. Based on the phenotype, we could not conclude that the biallelic HERC1 variant was involved in the patient phenotype. To get more insights into the potential role of HERC1 in the observed phenotype of the patients, we used lymphoblastoïd cell lines established from the patient cells as well as his parents and sister cells and down-regulated HERC1 expression using siRNAs but we failed to detect any impact of this down-regulation on BCL2 expression (not shown). Analysis of the other variants failed to detect any alteration in genes encoding proteins that are predicted to interact with BCL2, based on the literature or interaction predictive software (http://genemania.org/ and http://string-db.org/). Rare variants detected were frequently reported in the ExAC database or inherited from parents. The coverage of direct known interactors of BCL2 was directly visualized in IGV software.
miR-181a accumulation could explain BCL2 down-expression
Using total lymphocyte RNA extracted from the patient, sister, father, mother and 10 healthy donors, we performed microarray assays for microRNA and gene expression. The correlation between the biological lymphocyte samples was calculated using principal component analysis (PCA) mapping as shown in Figure 3A and B. Principal component analysis (PCA) of the miRnome and the transcriptome of CD3+ lymphocytes obtained from the father, the mother and 10 healthy donors generated a homogeneous cluster. PCA of the miRnome and transcriptome from patient CD3+ T cells was completely distinct, while those from the sister were intermediate. Among the tested microRNAs, we selected a series of 13 microRNAs known to regulate BCL2 mRNA expression and measured the expression of each of them in the studied samples. Interestingly, miR-181a was strongly up-regulated in the patient lymphocytes and the only identified miRNA whose deregulation could explain the down-regulation of Bcl2 (see crosses in Fig. 3C). The expression of miR-181a and BCL2 was inversely correlated in the patient cells (−0.67; P= 0.008) (Fig. 3D). As miR-181a also targets other BCL2 members like MCL1 and BCL2L11 as demonstrated in astrocytes, (23) we performed qPCR analyses in patient’s T cells and observed that MCL1 and BCL2L11 gene expression were not down-regulated when miR-181a was overexpressed (Fig. 4). Altogether, these results suggest that miR-181a accumulation could be responsible for BCL2 gene down-expression in the patient cells.
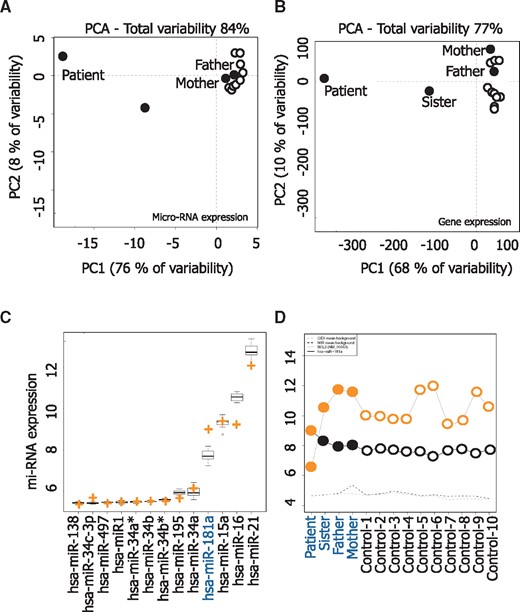
microRNA (mi-RNA) analysis. Micro-array and gene expression analyses were performed on RNA and DNA extracted from peripheral blood CD3+ T cells of the patient, his sister, his mother and his father and 10 healthy donors, respectively. Principal Component Analysis of miRNA expression (A) and gene expression (B). The two principal components and their fraction of the overall variability of the data (%) are shown on the x- and y-axis, respectively. (C) Average expression of 13 mi-RNAs known to regulate BCL2 gene expression; an orange cross indicates the level of expression of each of them in the patient T-cells. (D) Log2 intensity of has-miR-181a (black dots) and BCL2 (NM_000633) gene (orange dots) T-cell sample collected from the family members and control healthy donors. Dotted lines indicate the mean background in miRNA and gene expression arrays.
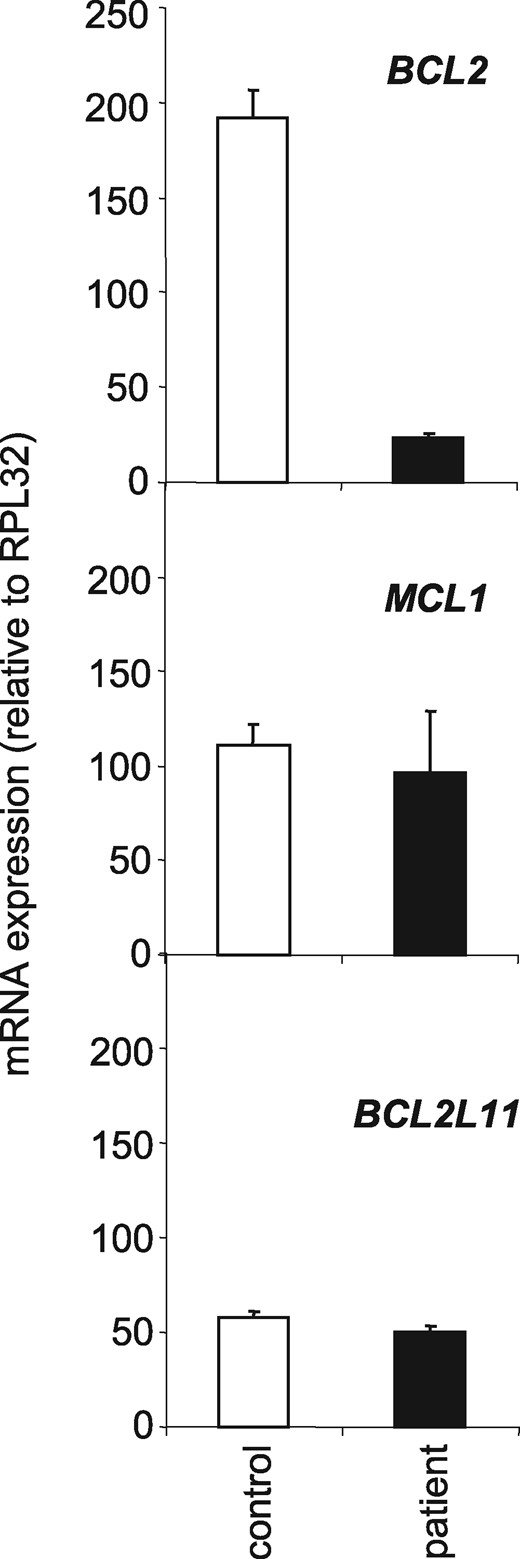
BCL2 mRNA and not MCL1 and BCL2L11 mRNAs decrease in patient T lymphocytes. RT-qPCR analysis of BCL2, MCL1 and BCL2L11 mRNA expression in peripheral blood CD3+ T cells from male control and the patient after one night of culture. Normalizer, RPL32 gene.
Discussion
We have observed a unique clinical phenotype that associates abdominal and thoracic aortic dissection, aortic root aneurysm, PKD, lymphopenia with a history of T cell lymphoblastic lymphoma, white hair since the age of 20 and learning difficulties. This association has never been reported as a human genetic disease. Three of these manifestations, i.e. PKD, lymphopenia and white hair, have been described in Bcl2−/− mice (14,15), which led us to search for a link between the patient phenotype and a constitutive BCL2 haploinsufficiency. Although no constitutive BCL2 gene mutation was detected, we observed a dramatic decrease in the gene and protein expression in patient cells, leading to an increased T-cell sensitivity to apoptosis that recapitulates one of the features detected in Bcl2−/− mice. Our investigations detected an enhanced expression of miR-181a that could account for BCL2 gene down-regulation in this patient.
The PKD1 gene mutation identified in the patient, his father and his sister is a confounding factor. Deregulated apoptosis through alteration of BCL2 family member expression or function was implicated in several experimental PKD mouse and rat models (24), including juvenile cystic kidney mice (25), congenital polycystic kidney mice (26), PKC rats (27), and Han Sprague-Dawley rats (28). However, the most common gene variants associated with autosomal dominant polycystic kidney disease (ADPKD) in humans are PKD1 (85%) and PKD2 (15%) mutations (29). The pathogenic role of deregulated apoptosis is more controversial in Pkd1 and Pkd2 mouse models (30,31). The mechanisms leading to PKD in the absence of PKD1 differs from those leading to PKD in the absence of BCL2 (32) as kidney anatomical phenotypes differ, mouse survival differs, and the loss of Bim, a gene encoding a BH3-only pro-apoptotic protein that interplays with BCL2, prevents PKD in Bcl2−/− but not in Pkd1del34/del34 mice. Thus, even if an inherited PKD1 mutation was evidenced in our patient, BCL2 haplo-insufficiency could have played a complementary role in the development of his polycystic kidneys.
The same assumption could be made for the vascular phenotype observed in the patient. Vascular abnormalities, including intracranial aneurysms, thoracic aorta dissections, and aortic aneurysms, are commonly associated with ADPKD. The development or not of vascular complications could depend on modifier genes, probably involved in the structure or the function of the arterial wall (33), e.g. through increasing TGFβ signalling (34,35). Therefore, we cannot exclude that a modifying factor modulates the PKD1 phenotype in the studied patient. Deregulated apoptosis is also involved in the pathogenesis of cardiac and arterial diseases, e.g. a deregulated interplay between pro- and anti-apoptotic proteins of the BCL2 family can lead to the depletion of vascular smooth muscle cells that induces aortic aneurysms and dissections (36,37). BCL2 protein was involved in vascular development, i.e. in vitro sprouting assays identified a decreased angiogenesis in the absence of Bcl-2 (38,39) while Bcl2−/− mice demonstrated a significant decrease in vascular density (40,41) suggesting that the patient phenotype involved defective angiogenesis in addition to defective apoptosis.
As for Bcl2−/− lymphocytes, an accelerated spontaneous apoptosis was observed in the patient CD3-positive T cells, which was partially corrected by T-cell stimulation with and anti-CD3 antibody and associated with a decreased expression level of BCL2 mRNA and protein in these cells. Importantly, a similar decreased in BCL2 mRNA level was detected in cells of the aorta wall. We looked for mutations in the coding sequences of BCL2 gene and extended our investigations to the promoter and 3’UTR region of the gene, but we failed to detect any constitutive variant of the gene sequence. We looked also for an abnormal methylation pattern of BCL2 gene promoter without detecting any pathogenic variation methylation.
The 3'-UTR of BCL2 mRNA, which is 5.2 kb in length, contains multiple predicted miRNA-binding sites that allow miRNAs to play a central role in regulating BCL2 gene expression in normal and pathological conditions [for review, see (42)]. For example, miR-15a and miR-16-1 inhibit BCL2 expression in B-cells and their deregulation is responsible for BCL2-dependent resistance to apoptosis of chronic lymphocytic leukaemia cells (11). MicroRNA array analysis performed in patient and control T-cells identified an increased expression level of miR-181a in patient lymphocytes. This miRNA was shown recently to target BCL2 gene in different human cell types, including astrocytes (23), glioma (12), chronic lymphocytic leukaemia (13) and acute myeloid leukemia (43,44) cells, a deregulation often associated with drug resistance. In human, 6 mature miR-181 s are encoded by 3 independent sequences located on 3 separate chromosomes, namely miR-181a1 and miR-181b1 on chromosome 1, miR-181a2 and miR-181b2 on chromosome 9, miR-181c and miR-181d on chromosome 19. The corresponding premature miRNAs lead to the expression of 4 sets of mature miRNA, miR-181a, miR-181 b, miR-181c and miR-181d which share the same ‘seed’ sequence ‘ACAUUCA’ (45). MiR-181a was involved in normal T cell development (46,47) and its deregulated expression may play a role in the pathogenesis of cancer and autoimmune diseases (48–50). Our data indicate that deregulation of miR181a could account for the low level of BCL2 gene expression in the patient cells.
In conclusion, we report a unique case of a patient whose phenotype recapitulates most of the features of the bcl2−/− mice. A dramatic decrease in BCL2 gene and protein expression was identified in his T-cells and aorta wall cells, correlating with T-cell hypersensitivity to apoptosis. This decreased expression of BCL2 gene expression was associated with the abnormal accumulation of miR-181a in the patient cells. The abnormal expression of miR-181a is the only identified parameter that could explain BCL2 gene expression and behave as a modifying factor that alters the phenotype of a PKD1 constitutive variant.
Patients and Methods
Clinical case
A 40-year-old male, the first child of non-consanguineous parents, had a younger sister aged 37 years affected by isolated classical polycystic kidney disease (PKD) inherited from her father, who had received renal transplant at age 52, complicated by cerebral ruptured aneurysm. PKD of the sister remained uncomplicated. The patient had developed a rare T cell lymphoblastic lymphoma that had been cured when he was 2-year old. He had suffered from learning difficulties when he was a child, from scoliosis that appeared at adolescence, and from myopia diagnosed when he was 16 (5/10 right side, 6/10 left side). Finally, he had developed white hairs at the age of 20. He was referred initially to our hospital at 31 with abdominal and thoracic aortic dissection and aortic root aneurysm, and got surgery for the abdominal aortic dissection. He measured 173 cm, his OFC was 52 cm, had mild facial asymmetry, and limited extension of both thumbs. He suffered from atypical PKD with cysts smaller than usually seen in this disease, not complicated of renal insufficiency. He had hypertension treated by avlocardyl, tarka, mediatensyl and aldactone. He also had lymphopenia (0.51 10e3/mm3, N = 1–4) with normal immunophenotyping of hematopoietic cells. His cerebral MRI was normal. He had normal standard karyotype and 180 K array CGH. Hair histology was normal. Direct sequencing identified a PKD1 mutation NM_000296.3: p.(Asn1870Asp) in the patient, the father and sister. One year later, dilatation of the ascending aorta required new surgery.
CD3+ T cell sorting and apoptosis assay
Heparinized blood was collected from the patient, his parents, his sister, and healthy donors with informed consent. Peripheral blood mononucleated cells were isolated by Ficoll Hypaque (Eurobio), and lymphocytes were sorted using a CD3+ magnetic isolation kit and AutoMACS separator according to the manufacturer’s instructions (Miltenyi Biotec). Two-hundred thousand CD3+ T-lymphocytes were cultured for 72 and 96 h in 200 μl RMPI supplemented with 10% Fetal Calf Serum (FCS), in the presence or absence of an anti-CD3 antibody (clone OKT3, 1 µg per well, BioLegend). The nuclear chromatin was stained with Hoechst 33342 (10 μg/ml; Sigma) for 30 min at 37 °C to detect typical apoptotic modifications by fluorescence microscopy.
RNA extraction and real-time qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. RNA was reverse transcribed by M-MLV reverse transcriptase with random hexamer primers (Promega). Real-time PCR was performed with AmpliTaq Gold polymerase in a 7500 FAST thermocycler (Applied Biosystems) using the SYBR Green detection protocol as outlined by the manufacturer. Briefly, 15 ng of total complementary DNA, 50 nM of each primer, and 1× SYBR Green mix were used in a total volume of 20 μl. Human-specific forward and reverse primers were RPL32-F: TGTCCTGAATGTGGTCACCTGA and RPL32-R: CTGCAGTCTCCTTGCA CACCT; TUBULINA-F: TCCAGATTGGCAATGCCTG and TUBULINA-R: GGCCATCG GGCTGGAT used as a standardizing control; BCL2-F: AACTGTACGGCCCCAGCAT and BCL2-R: GCCAAACTGAGCAGAGTCTTCAG. BCLXL-F: GAACGGCGGCTGGGATA and BCLXL-R: GCTCTCGGCTGCTGCATT. MCL1-F: CGTTGTCTCGAGTGATGATCCA and MCL1-R: TCACAATCCTGCCCCAGTTT. Human RPL32 and TUBULINA were used as internal controls. BCL2L11-F: GCCCCACCTGCCAGC and BCL2L11-R: CAGCAGGGAGGATCTTCTCATAA.
Immunoblot analyses
Lymphocytes were lysed for 30 min in RIPA buffer containing 50 mM Tris, 150 mM NaCl, 1 mM NaF, 0.1% NP40, 0.25% DOC, 1 mM Na3VO4, 1 mM PMSF and protein inhibitor cocktail (Sigma). Cell lysates were centrifuged at 13 000 g for 20 min at 4 °C and protein concentrations were determined with the BCA protein assay (Sigma). Proteins were run on a 10% SDS-PAGE and transferred to nitrocellulose membrane (Millipore, Bedford, MA). The membrane was blotted with antibodies directed against BCL2 (Dako, Denmark) in PBS, 0.05% Tween 20, 5% non-fat dried milk, washed, and probed with the appropriate secondary antibody coupled to horseradish peroxidase (Santa Cruz biotechnologies) before analysis with a chemiluminescence detection kit (Santa Cruz Biotechnology). Quantification has been made using ImageJ software.
BCL2 exons, 3’-UTR and promoters sequencing
Genomic DNA from CD3+ sorted cells was extracted according to manufacturer protocol (Norgen Biotek, Thorold, ON, Canada). PCR was performed with AccuPrime Taq high fidelity DNA polymerase (Invitrogen) using the following primer sets. BCL2ex1-F: TTTCTGTGAAGCAGAAGTCTGGG, BCL2ex1-R: TTGTATTTTTTAAGTACAGCATGA TCC; BCL2ex2-F1: CATCACAGAGGAAGTAGACTG, BCL2ex2-R1: CTCGTAGC CCCTCTGCGACA; BCL2ex2-F2: TCCTCTGGGAAGGATGGCGC, BCL2ex2-R2: CCCAGCCTCCGTTATCCTGGATCC; BCL2ex3-F: ATGCC TTTGTGGAACTGTACGG, BCL2ex3-R: TCACTTGTGGCCCAGATAGG; BCL2-3’-F: CCTATCTGGGCCACAAGT GA, BCL2-3’-R: GGCAGTAAATAGCTGATTCGACG; BCL2int1-F: GTAAGTTCTCTGC ACAGG, BCL2int1-R: CAGTCTACTTCCTCTGTGATG; BCL2prom2-F: CACCTGTC TTCACAGCAGGGC; BCL2prom2-R: CCTGGACCC TTTCTGGC. All the PCR amplicons were purified and sequenced by the Sanger method at Cogenics (Meylan, France).
BCL2 promoter methylation
Two micrograms of total genomic DNA was modified by bisulphite treatment according to the manufacturer’s instructions (MethylDetector, Active Motif). 2 µg of bisulphite DNA were provided at DNAVision (Gosselies, Belgium) for DNA methylation study of the promoter of human BCL2 (NM_000633-910 bp). Briefly, 2 PCR amplicons were done using respectively BCL2(I)-F: GGGTGGTTTAGAGGAGGGTTT; BCL2(I)-R: ATTCTCCCCCCTAAACCCCCTCT and BCL2(II)-F: GGTTTAGGGGGGAG AATTT; BCL2(II)-R: ATTCCCAAACTTCTACTTCACAA. After purification of biotinylated amplicons, pyrosequencing runs were performed with BCL2(I)-PSQprobe: TTT. TTTTTTTTTTTGAATG; BCL2(II)-PSQprobeS1: TTAGGGGGGAGAATTT; BCL2(II)-PSQprobeS2: AAATGTATTTGTTGTT; BCL2(II)-PSQprobeS3: TATTATAGGATTTTTG.
Array-comparative genomic hybridization
Array-CGH was performed using the SurePrint G3 Human aCGH Microarray 1 M (Agilent Technologies, Santa Clara, CA). This platform is a high-resolution 60-mer oligonucleotide-based microarray representing 974 016 probes (1 M) that allow a genome-wide survey and molecular profiling of genomic aberrations with an average resolution of ∼4 kb. Array-CGH experiments were performed with the maximum amount of DNA available. Array-CGH analysis was performed according to the Agilent protocol and immediately scanned on a DNA Microarray Scanner (Agilent Technologies). Mapping data were analysed on the human genome sequence build hg19 using ensemble (www.ensembl.org). Copy number variations (CNV) were assessed in the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Whole exome sequencing
Genomic DNA was extracted from a peripheral blood sample. With the SureSelect Human All Exon V2 kit (Agilent) to capture the coding regions, we used 3 µg of DNA per subject (index case, parents and sister). The resulting libraries were sequenced on a HiSeq 2000 (Illumina) as paired-end 75 bp reads in accordance with the manufacturer's recommendations. BAM files were aligned to a human genome reference sequence (GRCh37/hg19) using BWA (Burrows-Wheeler Aligner; v0.7.6) and duplicate paired-end reads were removed by Picard 1.77 (www.picard.sourceforge.net). Indel realignment and base quality score recalibration were conducted with Genome Analysis Toolkit (GATK; v2.1-10). Variants were annotated with SeattleSeq SNP Annotation (snp.gs.washington.edu/SeattleSeqAnnotation138/). Rare variants present at a frequency above 1% in dbSNP 138 (http://www.ncbi.nlm.nih.gov/projects/SNP/), the NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) and ExAC Browser (http://exac.broadinstitute.org/) or present from 100 local exomes of unaffected individuals were excluded. Candidate variants and segregation analysis were confirmed by standard PCR and Sanger sequencing of DNA extracted from peripheral blood.
Micro-RNA analysis
microRNA expression analysis was performed with Agilent® Unrestricted Human miRNA V3 8x15K Microarray (Agilent Technologies, AMADID 21827). Each sample was hybridized on separate arrays. Agilent miRNA Microarray System with miRNA complete labelling and hybridization kit was used for Cy3 labelling. Briefly, isolated total RNA were dephosphorylated, labelled with pCp-Cy3 and hybridized on arrays for 20 h at 55 °C in a rotating oven (Robbins Scientific) at 20 rpm. Slides were washed and scanned by using an Agilent G4470C DNA microarray scanner using defaults parameters. Microarray images were analysed by using Feature Extraction software version (10.7.3.1) from Agilent technologies. Default settings were used.
Gene expression analysis
Gene expression was analysed with Agilent® SurePrint G3 Human GE 8x60K Microarray (Agilent Technologies, AMADID 28004) with a single-color design, labelled with Cy3 using the one-color Agilent labelling kit (Low Input Quick Amp Labeling Kit 5190-2306), adapted for small amount of total RNA (100 ng total RNA per reaction). Hybridization was then performed on microarray using 800 ng of linearly amplified cRNA labelled, following the manufacturer protocol (Agilent SureHyb Chamber; 800 ng of labelled extract; duration of hybridization of 17 h; 40 µl per array; Temperature of 65 °C). After washing in acetonitrile, slides were scanned by using an Agilent G2565 C DNA microarray scanner with default parameters (100° PMT, 3 µm resolution) at 20 °C in free-ozone concentration environment. Microarray images were analysed by using Feature Extraction software version (10.7.3.1) from Agilent technologies. Default settings were used.
Data processing and analysis
Raw data files from Feature Extraction were imported into R with LIMMA (Smyth, 2004, Statistical applications in Genetics and molecular biology, vol3, N°1, article3), an R package from the Bioconductor project, and processed as follows: gMedianSignal data were imported, control probes were systematically removed, and flagged probes (gIsSaturated, gIsFeatpopnOL, gIsFeatNonUnifOL) were set to NA. Inter-array normalization was performed by quantile normalization. To get a single value for each transcript, taking the mean of each replicated probe summarized data. Missing values were inferred using KNN algorithm from the package ‘impute’ from R bioconductor. Normalized data were then analysed. To assess differentially expressed genes between two groups, we start by fitting a linear model to the data. Then, we used an empirical Bayes method to moderate the standard errors of the estimated log-fold changes. The top-ranked genes were selected with the following criteria: an absolute fold-change > 2 and an adjusted P-value (FDR) < 0.05. PCA were computed using the prcomp function from R by setting the centring to TRUE.
Conflict of Interest statement. None declared.
Funding
‘Taxe d’apprentissage’ program Genomic Core Facilities - TA2010 and by the AOI2010 from the Regional Council of Burgundy and Dijon University Hospital.
URLs
References
Author notes
First two authors contributed equally to this work.
Last two authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}