




Abstract
Although genetic variations in several genes encoding for synaptic adhesion proteins have been found to be associated with autism spectrum disorders, one of the most consistently replicated genes has been CNTNAP2, encoding for contactin-associated protein-like 2 (CASPR2), a multidomain transmembrane protein of the neurexin superfamily. Using immunofluorescence confocal microscopy and complementary biochemical techniques, we compared wild-type CASPR2 to 12 point mutations identified in individuals with autism. In contrast to the wild-type protein, localized to the cell surface, some of the mutants show altered cellular disposition. In particular, CASPR2-D1129H is largely retained in the endoplasmic reticulum (ER) in HEK-293 cells and in hippocampal neurons. BiP/Grp78, Calnexin and ERp57, key ER chaperones, appear to be responsible for retention of this mutant and activation of one signaling pathway of the unfolded protein response (UPR). The presence of this mutation also lowers expression and activates proteosomal degradation. A frame-shift mutation that causes a form of syndromic epilepsy (CASPR2-1253*), results in a secreted protein with seemingly normal folding and oligomerization. Taken together, these data indicate that CASPR2-D1129H has severe trafficking abnormalities and CASPR2-1253* is a secreted soluble protein, suggesting that the structural or signaling functions of the membrane tethered form are lost. Our data support a complex genetic architecture in which multiple distinct risk factors interact with others to shape autism risk and presentation.
INTRODUCTION
The autism spectrum disorders (ASDs), commonly called autism, are neurodevelopmental conditions that may affect as much as 1% of the population (1,2). Despite an important role of hereditary factors (3), the genetics of the ASDs is complex (4), making the identification of susceptibility genes challenging. Based on recent molecular and genetic studies, one of the candidate genes most frequently linked to autism is contactin-associated protein-like 2 (CNTNAP2) (5). Rare variations at this locus have been implicated in both the ASDs (6–10) and intellectual disability with epilepsy (11–15), as well as Tourette syndrome (16), Schizophrenia (17) and ADHD (18). Similarly, a common variation in this gene has been associated with ASD diagnosis (19), language performance in both typically developing individuals (20) and others with language-related disorders (21,22), and aberrations of brain structure and function (23–25).
CASPR2 is a neuronal cell adhesion molecule known in rodents to be necessary in the clustering of the Kv1 potassium channels at juxtaparanodes (26). In myelinated nerves, CASPR2 is confined to the juxtaparanodal region of the axon where it associates with the immunoglobulin domains of TAG-1 to form a scaffold which clusters the potassium channels Kv1.1 and Kv1.2 (27–29). Data on the distribution of CASPR2 in pyramidal cells in the human temporal neocortex indicate that CASPR2 is localized at the axon initial segment, a region that is characterized by a high concentration of K+ channels (30,31). In rat hippocampal neurons, CASPR2 appears to be predominantly localized in post-synaptic compartments such as cell bodies and dendrites (26), and it was found in synaptic plasma membranes to be associated with TAG-1 (8). Yet, work in rat-dissociated hippocampal cultures (32) and Drosophila points to a presynaptic localization (12), a finding consistent with its association within the Neurexin superfamily. No information is thus far available on CASPR2 structure, the subtype of synapse to which it localizes or its synaptic function.
In ASD patients, Bakkaloglu et al. (8) found 11 rare variants, eight of which were predicted to be deleterious because they occurred in evolutionarily conserved regions of the gene (Fig. 1A). In particular, CASPR2-D1129H was present in monozygotic twins, both affected with autism. Their father, carrying the same mutation, did not have an autism diagnosis. Other studies (6,11) found that individuals homozygous for a rare frame-shift mutation (single-base G deletion at nucleotide 3709 in exon 22) presented cortical dysplasia, focal epilepsy and ASD with language regression. As above, this variant was inherited. A surgical biopsy of the anterior temporal lobe of two children carrying the mutation revealed cortical dysplasia including evidence of abnormalities of neuronal migration, structure and widespread astrogliosis. No information is currently available on the molecular and cellular defects resulting from any of these mutations.
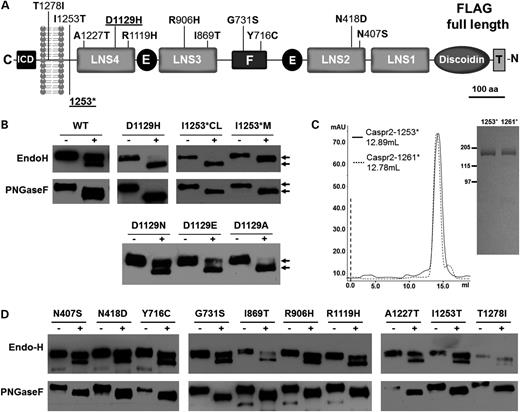
Endoglycosidase H digestion reveals improper cellular trafficking for multiple CASPR2 mutations. (A) Schematic drawing of the entire domain organization of CASPR2 is shown. Nonsynonymous mutations of the coding region are shown on their approximate location. Mutations are described by Bakkaloglu et al., 2008 (above) and by Strauss et al., 2006 (below) (6,8). Mutants shown underlined are presented in detail in this study. ICD, intracellular domain; LNS, Laminin–Neurexin–Sex hormone-binding globulin domains; E, EGF domains; F, Fibrinogen-like domain; Discoidin, discoidin domain; T, FLAG Tag. (B) Western blot analysis of a typical Endo-H and PNGase F experiment of CASPR2-WT and two mutants. Arrows on the right side of the image indicate the different migration. M, protein present in the medium; CL, protein extracted form cell lysate. (C) Gel filtration chromatography (left panel) and Coomassie blue staining (right panel) analysis of CASPR2-1253* compared with CASPR2-1261*. (D) Western blot analysis of the Endo-H and PNGase F experiments of the remaining 10 mutants.
To ascertain whether the reported autism-related mutations (6,8,11) are benign polymorphisms or may affect the structure and expression of the protein, we studied the glycosylation processing and subcellular localization of the wild type and mutants. We also measured their rates of degradation and ascertained that the unfolded protein response (UPR) was activated through ATF6. We found that CASPR2-D1129H is arrested in the endoplasmic reticulum (ER) and found associated with chaperons BiP, Calnexin and ERp57, leading to stimulation of the UPR and subsequent proteasomal degradation. In contrast, CASPR2-1253* folds properly, but is secreted in the extracellular space. Our results support a disease model in which multiple distinct factors, genetic and environmental, may be critical contributors to the disease outcome and as such must be considered in looking forward towards treatment.
RESULTS
Endoglycosidase-H (Endo-H) digestion reveals improper cellular trafficking for multiple CASPR2 mutations
Based on protein sequence analysis, CASPR2 contains 13 potential N-linked glycosylation sites in its extracellular domain. Glycoproteins acquire their N-linked carbohydrate moieties upon their entry into the ER as high mannose glycans. Subsequently, during the passage through the Golgi apparatus, most of the mannose residues are trimmed and rebuilt with a variety of glycan residues until a final composition is achieved (33). The enzyme Endo-H preferentially digests high mannose sugars, thus, specific alterations in the trafficking and biosynthetic processing of the mutants can be revealed by susceptibility to Endo-H digestion (34,35).
Using stably transfected cells, western blot analysis of CASPR2-WT subjected to Endo-H treatment reveals that the majority of the protein is not sensitive to the enzymatic treatment, indicating that it is fully mature and is presumably presented to the cell surface (Fig. 1B). The small fraction of the protein that is sensitive to Endo-H digestion represents the immature protein being processed in the ER. A control treatment with PNGase F showed a band shift consistent with the removal of several N-linked sugars, irrespective of their composition, suggesting that the conjugated oligosaccharides are similarly accessible under these experimental conditions. Due to the extreme phenotype displayed by CASPR2-D1129H and CASPR2-1253*, we further focused on these mutants, but a comparative analysis of all the remaining autism mutants (8) is reported in Figure 1D. Endo-H appears to fully digest CASPR2-D1129H, indicating that the majority of the protein resides in the ER and does not reach the Golgi apparatus (Fig. 1B), suggesting that trafficking of CASPR2-D1129H is severely impaired. These data indicate that D1129 is extremely important for the structural integrity of the LNS4 domain of CASPR2 (see also Fig. 6), as suggested by the absolute conservation of the Asp residue in this position across mammals. To gain further insights into this trafficking abnormality and ascertain whether the critical alteration was general substitution for Asp or its specific replacement with His, we analyzed three other mutants: CASPR2-D1129A (a small, hydrophobic and non-polar residue), CASPR2-D1129N (a polar residue similar in size and other chemical features to D) and CASPR2-D1129E (E is similar to D although it carries a net negative charge and its side chain is bulkier). Endo-H digestion shows that CASPR2-D1129N has a digestion pattern similar to the wild-type protein, whereas CASPR2-D1129A closely resembles CASPR2-D1129H and the presence of E at position 1129 has an intermediate digestion pattern (Fig. 1B). The remaining 10 mutants (Fig. 1D) display a level of ER retention ranging from the wild type to the D1129H extremes. Whereas most of them, including N407S, N418D, A1227T, I1253T and T1278I, resemble the wild type, the remaining mutants show various degrees of Endo-H sensitivity, culminating with the R1119H.
Because of the frame-shift mutation, a premature stop codon is introduced at position 1253 (6,11) (Fig. 1A). Thus, CASPR2-1253* is devoid of its transmembrane and intracellular domains, but has a C-terminal sequence of 16 residues that differ from the wild-type protein (wild-type sequence DHLDSASADFPYNPGQGQA …; mutant sequence DHLIQPVRIFHIIQDKAKL*, single letter amino acid code). Immunoblots of the conditioned medium of transfected HEK-293 cell show that CASPR2-1253* is secreted into the cell culture medium as a fully processed protein, whereas protein extracted from the cell lysate shows a molecular mass slightly smaller than that of the secreted protein (Fig. 1B). As in the case of other secreted forms of transmembrane glycoproteins (36), this difference is due to the complete glycosylation processing acquired in the Golgi and required for secretion. Moreover, consistent with the different processing of these two forms, only the cellular fraction of CASPR2-1253* is fully sensitive to the enzymatic treatment. Importantly, because the protein was found in the cell culture medium, we can rule out that the messenger RNA for CASPR2-1253* is targeted by the nonsense-mediated decay (37,38), or that the protein is prematurely degraded by the ER-associated protein degradation (ERAD) (39). To ascertain whether the premature stop or the extra sequence affects the general folding or oligomerization state of CASPR2-1253*, the protein was affinity purified from conditioned HEK-293 cell culture medium (Fig. 1C). For comparison, another truncation mutant, CASPR2-1261*, was also engineered from the wild-type construct and affinity purified. This position corresponds to the last residue before the putative transmembrane domain, but in this case the wild-type sequence in the C-terminal direction is conserved. The expression levels of both the proteins are comparable (>1 mg/l of conditioned medium) and Coomassie stained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) shows that gel electrophoretic migration of CASPR2-1253* is virtually identical to that in the wild-type (Fig. 1C). Gel filtration elution profiles of both the proteins, a rough measure of their molecular size and oligomerization state, is also virtually identical, consistent with the fact that CASPR2-1253* is only a few residues shorter than the wild-type protein truncated at position 1261. Although these measurements do not determine the absolute mass or oligomerization state of either protein, they indicate that the additional 16 residues found in this mutant do not compromise with the overall folding, oligomerization properties or carbohydrate composition.
Immunofluorescence in HEK-293 cells demonstrates different % of ER retention for some of the mutants
The Endo-H deglycosylation patterns observed are consistent with abnormal trafficking of several CASPR2 mutants. Thus, by using confocal immunofluorescence microscopy, the subcellular distribution of previously identified mutations could be determined. CASPR2 stably transfected HEK-293 cells were stained with anti-FLAG and anti-calreticulin, a marker of the ER. As expected, CASPR2-WT is detected predominantly on the cell surface of HEK-293 cells (Fig. 2A), whereas the fluorescent signal of CASPR2-D1129H and CASPR2-1253* largely overlapped with the calreticulin staining, indicating a strong ER retention for CASPR2-D1129H, as predicted by the Endo-H deglycosylation patterns. The three other D1129 mutants also display a subcellular localization consistent with the Endo-H results. To quantify the amount of ER retention, the fluorescent signal of the FLAG epitope of CASPR2 was overlaid with the fluorescent signal of calreticulin and their co-localization was estimated in the total cell volume. Three complete Z-stacks for each mutant were taken and analyzed with ImageJ software. With this approach, CASPR2-WT shows a 51 ± 2% of ER co-localization (Fig. 2B). A careful analysis of the distinct channels indicates that the overlap comes from the overexpression of the protein that is being processed in the ER, consistent with the Endo-H results. Interestingly, Golgi staining with the marker giantin shows negligible co-localization with CASPR2 (Fig. 2C), suggesting that Golgi transit is probably rapid. Consistent with the Endo-H digestions, CASPR2-D1129H shows >80% of ER co-localization and negligible surface staining, indicating that this mutation may disrupt the fold of LNS4 domain where D1129 is located (Fig. 1A). The immature form of CASPR2-1253* strongly co-localizes with calreticulin before secretion and because it is no longer membrane tethered, it lacks surface staining. Consistent with the Endo-H results, the other CASPR2 variants show variable degrees of ER co-localization with some variants showing co-localization values ranging 45% for T1278I to >70% for both Y716C and R1119H (Fig. 3A and B). Interestingly, I869T, the variant that, apart from CASPR2-1253*, is most strongly implicated in disease on the basis of genetic data, shows an intermediate level of ER retention. As such, it seems that different variants may perturb cellular disposition in distinct ways, compounding the enormous complexity underlying relationships between cellular studies and behavioral end-points.
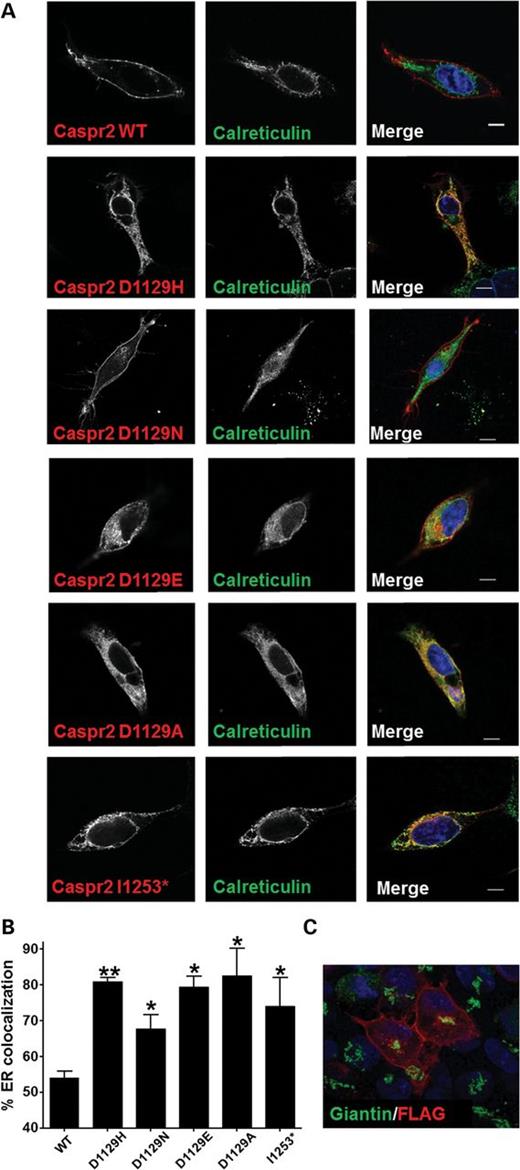
Immunofluorescence staining in HEK-293 cells demonstrates ER retention of mutants. (A) Immunofluorescence co-localization of CASPR2 and calreticulin in stable HEK-293 cell lines. Cells were stained with anti-calreticulin and anti-FLAG antibody. (B) FLAG and calreticulin pixel co-localization to estimate the amount of ER retention of each mutant. Data are presented as mean ± SEM values calculated from z-stacks from three separate cells. *P < 0.01; **P < 0.001. (C) Immunofluorescence co-localization of FLAG and Giantin, a Golgi marker.
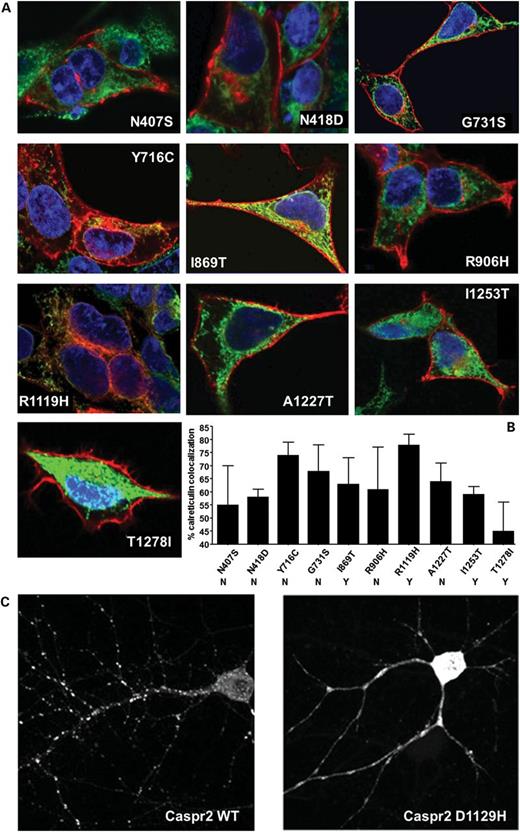
Immunofluorescence staining in HEK-293 cells and neurons demonstrates ER retention of the remaining mutants. (A) Immunofluorescence co-localization of CASPR2 and calreticulin in stable HEK-293 cell lines. Cells were stained with anti-calreticulin and anti-FLAG antibody. (B) FLAG and calreticulin pixel co-localization to estimate the amount of ER retention of each mutant. Data are presented as mean ± SEM values calculated from z-stacks from three separate cells. Y and N below the mutant name refer to the potential of the variant to be deleterious based on PolyPhen and SIFT algorithms as presented in Table 2 by Bakkaloglu et al. (8). (C) FLAG staining of rat hippocampal neurons transfected with CASPR2-WT and D1129H mutant. Note the different disposition of the mutant protein with respect to the wild type.
Immunofluorescence in hippocampal neurons confirms severe trafficking defects of CASPR2-D1129H
To ensure that the results obtained in HEK-293 cells were not an artifact of the undifferentiated cell system used, CASPR2-WT and mutants were cloned into a pEF-BOS expression vector and transfected in rat primary hippocampal neurons. Visualization at 14 days in vitro (DIV14) following transfection at DIV7 shows that CASPR2-WT is efficiently transported from the soma into the dendritic arborizations of the cultured neurons, in a punctate pattern typical of synaptic structures (Fig. 3C). Under the same experimental conditions, CASPR2-D1129H remains localized in the soma and inside the dendritic branches, confirming the severe trafficking deficiency of this variant.
Unexpectedly rapid turnover of CASPR2-WT is delayed in D1129H variant
Glycosylation processing and subcellular localization experiments indicate that the D1129H mutation causes severe trafficking abnormalities likely arising from impairment in protein folding. To test whether D1129H is affecting the protein expression, CASPR2-WT and CASPR2-D1129H mutant were transiently transfected into HEK-293 cells and expression of the protein was monitored by immunoblotting at 12, 24 and 48 h after transfection. CASPR2 WT and D1129H mutant were expressed using the same vector where the insert only differs in the D1129H substitutions. They were transfected under identical conditions using the same amount of cDNA. Despite the replication of conditions, the amount of expressed CASPR2-D1129H protein is significantly reduced compared with the WT protein. Moreover, it appears that the increased expression of WT protein correlated with the accumulation of mature protein (slower migrating band), which fails to appear in the D1129H mutant (Fig. 4A), and this is consistent with the incomplete glycosylation of the protein that remains in the ER. These results suggest that the mutant protein is either expressed significantly more slowly than the wild type, or that it is more rapidly degraded before entering the Golgi apparatus.
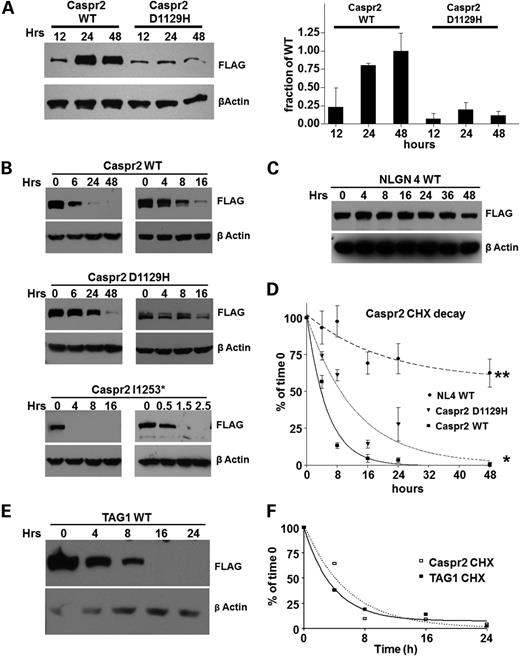
Unexpectedly rapid processing of CASPR2-WT delayed in D1129H mutant. (A) Left panel: representative immunoblot of the time course of expression of wild type and D1129H mutants CASPR2, using HEK-293 cells. Equal amounts of plasmids identical in sequence except for the point mutation in CASPR2 were transfected in identical HEK-293 monolayer lawns. The same membrane exposed to anti-βactin as a loading control is shown below. Right panel: quantification of expression levels in HEK-293 cells. Data are presented as mean ± SEM of the protein/β-actin ratio for four experiments. (B) Immunoblots representing the rate of degradation of CASPR2-WT, CASPR2-D1129H and CASPR2-1253*, after blocking protein synthesis with 0.35 mm cycloheximide. (C) The same CHX experiment used neuroligin-4 as an additional control. (D) Band intensity of (B) and (C) was quantified, and data are presented as mean ± SEM for three experiments. Solid line, wild type; dotted line, D1129H; dashed line, NLGN4. Asterisks: * indicates that P < 0.01; ** indicates that P < 0.001. (E) Immunoblots of CHX degradation of TAG1. (F) Band intensity of (E) was quantified and degradation rate calculated as described above. The degradation curve of CASPR2 obtained from (D) is shown for comparison.
To distinguish between these two possibilities, we monitored the degradation rates of CASPR2 by blocking the translation process at the ribosomal level using cycloheximide (CHX) and measured the decay of the protein over time. Identical plates of stably expressing HEK-293 cells were added with CHX for 6, 24, 48 h (Fig. 4B) and the amount of CASPR2 was subsequently measured by immunoblotting using β-actin as a reference protein to normalize the loading amount. The use of stably transfected lines enables starting the experiments at a steady-state rate of expression, thus bypassing the slower expression of CASPR2-D1129H. As the degradation rate of CASPR2-WT appears to be more rapid than anticipated, shorter incubation times (4, 8, 16 h) were analyzed to obtain better resolution of decay rates (Fig. 4B). Under these conditions, we observed a complete degradation of CASPR2-WT within 24 h (half-life of 3.7 h), whereas the CASPR2-D1129H shows a significantly longer half-life (8.6 h) (Fig. 4B and D). Because CASPR2-1253* mutant is a secreted protein, CHX does not allow monitoring intracellular degradation rates, but roughly indicates the time required for protein maturation and secretion. For CASPR2-1253*, the experiments were done at 30, 90, and 150 min. Under these conditions, CASPR2-1253* is processed to maturity and completely secreted in about 30 min (Fig. 4B). Similar results for all three CASPR2 variants were obtained using transient transfections (data not shown).
Because the half-life of a receptor can be enhanced by the presence of a cognate endogenous ligand, we tested whether contactin 2 (TAG-1) (27,28) had any influence on the rate of degradation of CASPR2-WT. Under the same experimental conditions, TAG-1 has a half-life that is comparable with CASPR2-WT (2.3 h) (Fig. 4E and F). Because TAG-1 is thought to bind CASPR2 in cis (27,28), we co-transfected both CASPR2 and TAG-1 in the same cells and followed their degradation after CHX treatment. Even under these experimental conditions, co-transfection of CASPR2 and TAG-1 did not change the stability of either protein (data not shown). Because of the unexpectedly short half-life of both CASPR2 and TAG1, we used neuroligin-4 (NLGN4), a well-studied cell-adhesion synaptic protein, as an additional control. Our data are consistent with the published results (40) regarding the degradation rates of NLGN4 measured with the same assay and show that NLGN4 is significantly more stable than CASPR2 under these conditions (half-life of 14 h and plateauing at 57% of the expressed protein) (Fig. 4C and D).
Taken together, these data indicate that despite the size, multidomain organization and glycosylation pattern, CASPR2-WT expresses and matures rapidly, but it is also rapidly degraded. The single point mutation D1129H dramatically delays both expression and degradation of CASPR2, indicating that the mutant remains in the ER likely bound to different chaperones attempting to complete folding before degradation is triggered. CASPR2-1253* also matures rapidly and is completely secreted in the extracellular environment. The results of the CASPR2-WT protein are consistent with a dynamic synaptic function, potentially involving synaptic structural reorganization, as observed for other neurexin superfamily members (41,42).
Caspr2-D1129H is retained by ER chaperones and preferentially degraded by the proteasome
To investigate whether the potential misfolding caused by the D1129H mutation leads to ER retention through common ER-resident chaperones, we have studied the association of the wild type and CASPR2-D1129H with specific chaperones. We used anti-FLAG antibody to immunoprecipitate CASPR2-WT and D1129H and subsequently searched for bound ER chaperones by immunoblotting. Our data show that CASPR2-D1129H uniquely precipitates BiP/Grp78 that typically recognizes hydrophobic regions exposed by the misfolded proteins (Fig. 5A). Calnexin, a chaperone that mainly assists in the initial folding of glycoproteins, is pulled down more avidly by the mutant protein. Also, as CASPR2 is rich in disulfide bonds, we tested whether the misfolding causes exposure of free cysteines, thus triggering a thiol-mediated retention mechanism. No association was detected between wild type and mutant protein with PDI. However, ERp57, another member of the PDI family of oxidoreductases, was found to slightly cross react with D1129H but not with the wild-type protein (Fig. 5A).
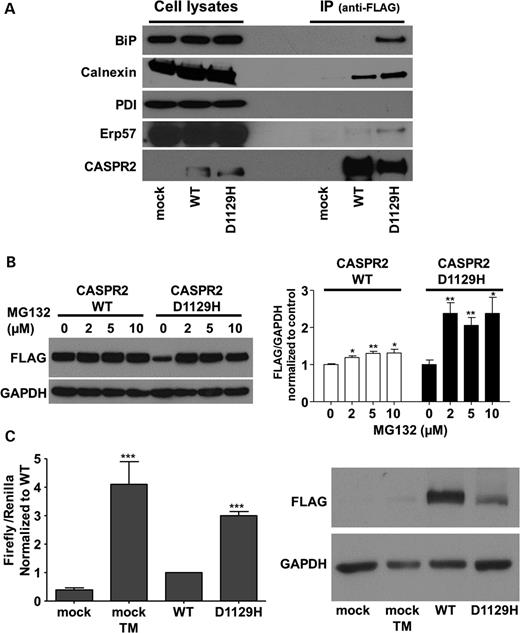
Interaction with ER chaperones, activation of ATF6 and ERAD. (A) Immunoprecipitation of ER chaperones with full-length CASPR2-WT and CASPR2-D1129H. Anti-FLAG antibody was used to detect immunoprecipitated CASPR2 as a control (bottom). Mock, untransfected cells. (B) Left panel: immunoblot of CASPR2-WT and CASPR2-D1129H after incubation of the cells with the proteasome inhibitor MG132. Right panel: quantification of the western blot densitometry of the left panel (three experiments with each concentration). (C) Left, the graph shows Firefly normalized to Renilla luciferase as average ± SEM of seven experiments each done in triplicate. The values are expressed in relative light units (RLU) and data normalized to wild type. Cells were mock-treated (negative control) or treated with 2 μg/ml tunicamycin (TM) as a positive control for 16 h prior to harvesting. T-test was used for statistical analyses relative to wild type. Differences between CASPR2-D1129H mutant and CASPR2-WT show a significance of P < 0.0001. Right, western blot of the expressed CASPR2-WT and CASPR2-D1129H in HEK-293 cells used to induce the expression of p(5x)ATF6-luc.
Finally, to examine the influence of the degradation pathway of the CASPR2-WT and D1129H mutant, the inhibitor MG132 was used to specifically block the proteasome pathway. After 24 h incubation, as little as 2 µm of MG132 causes a large increase in cellular levels for CASPR2-D1129H, much higher than what we observe for the wild-type protein, suggesting that the mutant protein is shuttled to the proteosome for degradation by ERAD (Fig. 5B), bypassing the secretory pathway.
CASPR2-D1129H activates the UPR
To evaluate whether ER retention of CASPR2-D1129H mutant was governing the activation of the UPR, we analyzed the ATF6 signaling pathway, one of the three ER stress sensors. The vector p(5x)ATF6-luc containing binding sites for ATF6 upstream of the luciferase reporter gene was used. Since those sequences are found in the promoters of most UPR target genes, the luciferase signal will indicate the activation of ATF6 in promoting an ER stress response. The p(5x)ATF6-luc vector was transfected in HEK-293 cells together with the plasmids encoding for CASPR2 either wild type or mutant, and with a third plasmid encoding for the Renilla luciferase gene to normalize for the transfection efficiency. Firefly luciferase activity 24 h after transfection (Fig. 5C) normalized to Renilla shows that ATF6 activation is significantly higher for the CASPR2-D1129H compared with the wild-type protein. This indicates that the accumulation of the mutant protein is activating at least one of three signaling pathways in an attempt to restore cellular homeostasis.
DISCUSSION
Homozygous mutations in CNTNAP2 give rise to severe neurodevelopmental disorders that include cortico-dysplasia-focal epilepsy (6) and Pitt–Hopkins Syndrome (12). Surprisingly, striking parallels exist between behavioral and neuropathological features in humans and mice (43) homozygous for disruptive mutations in CNTNAP2. We sought here to explore whether candidate human mutations might have distinctive phenotypes at the molecular and cellular levels. We characterized the subcellular localization of all of the point mutations observed in patients to date, and present a detailed analysis of two of these.
We find that D1129H causes significantly lower protein expression of CASPR2 and retention of the protein in the ER with an accompanying loss of mature glycosylation processing (Figs 1 and 2), indicating a folding impairment in the LNS4 domains of CASPR2. Three transmembrane ER proteins, PERK (PKR-like ER kinase), IRE1 (inositol-requiring kinase 1) and ATF6 (activating transcription factor 6), sense protein misfolding and initiate an ER-to-nucleus signaling cascade known as the UPR. This signaling pathway tries to reestablish folding homeostasis by inducing the expression of chaperones that enhance protein folding. At the same time, global protein translation is attenuated to reduce the folding load in the ER, while the proteosomal degradation rate is increased (44). We show here that CASPR2-D1129H, but not wild type, causes a significant activation of ATF6, indicating that overexpression of the mutant protein promotes activation of a protective cellular response (Fig. 5C) possibly through binding with BiP, Calnexin and ERp57 (Fig. 5A). In addition to its role as a folding chaperone, BiP also functions as an ER stress regulator: normally BiP is bound to PERK, ATF6 and IRE1, thus blocking their activation. However, in the presence of misfolded proteins, BiP remains associated with them allowing the three branches of UPR (PERK, TF6 and IRE1) to be activated (extensively reviewed in (44)). Importantly, prolonged activation of the UPR caused by unmitigated ER stress may lead to profound alterations of neuronal function or even cell death (45). Interestingly, a link between UPR and ASD has been recently described for mutations in the synaptic cell adhesion molecule-1, a membrane glycoprotein belonging to the immunoglobulin superfamily (46). Our data provide a first link between CASPR2 mutations and the UPR. Since only one signaling pathway of considerable complexity has been investigated, other studies warrant consideration. The surprisingly short half-life of CASPR2, alone or in combination with TAG1, suggests that these adhesion proteins are rapidly inserted in the cell membrane and internalized for degradation. Compared with the relatively long half-life of synaptic NLGN4 (Fig. 4C) or NLGN3 (35), CASPR2 and TAG1 may participate in more rapid turnover processes in the central nervous system. As hypothesized above, our results are consistent with the notion that the CASPR2-WT protein may be a modulator of structural reorganization of the synapse, as observed for other neurexin superfamily members (41,42).
Because a high-resolution crystal structure of CASPR2 is not available, we built homology models of the LNS4 domain, and to improve the robustness of the predictions, we used four different structural templates (see the materials and Methods section). These models enabled us to inspect the structure of LNS4 of CASPR2 and the three-dimensional environment where D1129 is located. In all models examined, D1129 is in a basic environment closely surrounded by two histidines (H1113 and H1130), R1105 and the backbone nitrogen of S1114 with which the terminal carboxylic group of D1129 side chain may form an electrostatic interaction or hydrogen-bonding network to tighten interactions between β-strands (Fig. 6). In particular, whereas H1130 assumes slightly different orientations depending on the modeling template (not shown), D1129 and H1113 are always in the same reciprocal orientation as shown in Figure 6. The in silico mutagenesis function in Pymol (47) enabled us to compare these structural models with our experimental results. In all models tested, and consistent with our experimental findings, Asp to His substitution causes a major clash with the H1113 ring, as exemplified by the red disks between the two side chains. Because maintaining this orientation seems to be forbidden, we predict that H1129 assumes other poses that may be incompatible with the local folding of this loop. The same holds true for D1129E that shows significant steric hindrance with H1113. The Asn substitution is the most benign (little to no steric clash, distances similar to Asp side chain and the possibility to form hydrogen bonds, are all conserved features). Although D1129A substitution does not seem to be sterically prohibited, the mutated protein shows severe trafficking abnormalities. As visible in the model (lower right panel), this variant probably lacks the ability to make an electrostatic interaction or form a hydrogen bond with its surroundings, eventually resulting in loop instability. Thus, substitution of the Asp with the introduction of a His or other amino acids may disrupt an established charge relationship or hydrogen bonding, likely affecting local protein folding, giving rise to the processing defects described here.
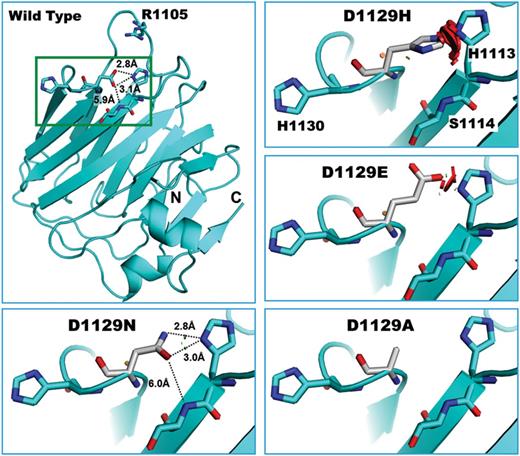
Homology model of LNS4 of CASPR2—Homology model shown here was built using the crystal structure of LNS2 of neurexin-1α (∼22% sequence identity) (50). The top left panel shows the position of D1129 with respect to R1105, H1130, H1113 and S1114, and potential hydrogen-bonding distances. N, N-terminus; C, C-terminus of LNS4. Remaining panels illustrate the potential rearrangement of the side chains of each D1129 mutant with respect to the surroundings. Small brown or green disks are shown when atoms are almost in contact or slightly overlapping (D1129N). Large red disks indicate significant van der Waals radii overlap (D1129H and D1129E).
Taken together, these results highlight the following overall mechanism underlying the fate of Caspr2-D1129H: the amino acid substitution causes a local misfolding in LNS4. BiP and other ER chaperones remain bound to the mutant (Fig. 5A) causing the protein to remain in the ER (Figs 1–3). As BiP is not available to suppress ATF6 and other UPR activators, the UPR is triggered (Fig. 5C) and the protein is eventually degraded by the proteosomal pathway (Fig. 5B).
A second mutation, a 3709delG in exon 22 of CNTNAP2 gene that results in a premature stop codon at position 1253, was described independently by Strauss et al. (6) and Jackman et al. (11) in the Old Amish population. Although we cannot test whether this mutation targets the mRNA for the nonsense-mediated decay in vivo (37,38), our experiments indicate that the disease-associated 3709delG mutant is not prematurely degraded by the ERAD (39) because it is rapidly secreted (Figs 1B, C and 3B). This also shows that the premature stop codon does not interfere with proper domain folding as is the case for other proteins such as neuroligin-4 (48). Biophysical characterization of α-neurexin 1 (αNRXN1) (49,50), another member of the neurexin superfamily to which CASPR2 belongs, indicates that the extracellular domain of αNRXN1 is monomeric in solution with an elongated shape. Gel filtration profiles of αNRXN1 (49) and the two CASPR2 constructs shown here, developed in the same laboratory, indicate that their extracellular domains are likely monomeric, despite the sequence of 16 C-terminal residues that differ in CASPR2-1253* from the wild-type protein. Furthermore, although the immunofluorescence data show that CASPR2-1253* is largely localized in the ER, the kinetic data indicate that the bulk of CASPR2-1253* is not retained in this compartment but is readily secreted. Thus, recycling and degradation machineries that would trigger the UPR are likely not impacted by this mutation.
Based on Endo-H and immunofluorescence data for an additional ten variants examined, we find that each mutation affects CASPR2 subcellular localization to different degrees. Analogous amino acid changes (I869T, I1253T and I1278T) do not result in a common biochemical phenotype, but this is not surprising, given that phenotypic consequences are dependent on the immediate three-dimensional surrounding of the amino acid side chain rather than on the chemical properties of the amino acid. For example, whereas I869 is in one of the predicted LNS domains, I1253 is in a predicted unstructured domain and I1278 is buried in the transmembrane domain. Moreover, because only a subset of the variants we analyzed appear to impact trafficking, it remains possible that some will influence association with protein partners like TAG1 or other ligands molecules yet to be discovered.
The CNTNAP2 gene contains an extensive number of mutations associated with ASD (6,8,11), and to our knowledge, these are the first mutants that have been linked to aberrations in protein trafficking. Compared with some of the known neuroligin mutations (40,35,51) CASPR2-D1129H seems to have several common features. These mutant proteins appear to have reduced glycosylation processing accompanied by ER retention and abnormal subcellular localization in both HEK-293 cells and neurons. Although a wealth of information is available on the phenotype with a complete lack of CASPR2 (CNTANP2−/− mouse) (27,43), it would be interesting to ascertain whether the presence of a soluble CASPR2 can affect neuronal maturation and migration by competitive antagonism of the membrane-associated orthologous protein. The processing and trafficking assays we have employed cannot speak to how individual variants impact behavior, but do serve as a means of assessing whether such candidate mutations have biochemical consequences. We show here that CNTNAP2 inherited variants perturb CASPR2 trafficking and activate an ER stress response. Incomplete penetrance was observed for all CASPR2 variants studied here, with presentation in heterozygous carriers ranging from severe to apparently unaffected (6,8). Although patients homozygous for the CASPR2-1253 all function at a very low level (6), individuals homozygous for the other variants we evaluated have yet to be identified. As such, the cellular readouts presented here provide important insights into the potential consequences of individual variants, information that cannot currently be inferred from work in humans. Our data highlight an imperfect relationship between genetic variation, cellular consequences, and clinical outcomes and require a model in which multiple distinct factors, genetic and environmental, converge to shape risk and presentation.
MATERIALS AND METHODS
Cloning of CASPR2
The full-length human CASPR2 was acquired from Open Biosystem (Thermo Fisher Scientific Inc.). The coding sequence of CASPR2 was fully sequenced and was cloned downstream of the FLAG octapeptide epitope and the pre-pro-trypsin leader peptide into pcDNA3.1 expression vector (36). This arrangement allows the mature protein to be physiologically inserted into the cell membrane as a type 1 membrane protein without interfering with its C-terminal PDZ-binding domain (Fig. 1A). The FLAG-CASPR2 protein starts after the leader peptide at position Ala28. For efficient expression in neuronal cells, FLAG-CASPR2 fragment full length was cloned into pEF-BOS (52) under control of the human polypeptide chain elongation factor 1α promoter. Mutations identified in the human population (6,8,11) were inserted into FLAG-CASPR2 expression plasmids using a Stratagene QuickChange site-directed mutagenesis kit (Agilent Technologies, Inc. Santa Clara, CA, USA). To introduce the CASPR2-1253* mutation, the same frame shift reported by Strauss et al. (6) was engineered, and all clones were verified by restriction digestion and DNA sequencing.
Cell culture and transfection
Human embryonic kidney 293 (HEK-293) cells were obtained from American Type Culture Collection and were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 2 mm glutamine, 10% FBS, maintained in a humidified incubator at 10% CO2 and 90% air and periodically tested to ensure the absence of mycoplasma contamination. Stable cell lines were made for each mutant and kept in DMEM/10% FBS supplemented with 500 µg/ml G418 (Geneticin, Sigma). For transient transfections, cells were transfected using FuGENE (Roche) and cell lysates were collected after 24–48 h as described below. Primary rat hippocampal neurons were dissected from E18 embryos and maintained in Neuobasal (Gibco) medium added with B27 (Gibco)+ glutamine and glutamic acid. Cells were transfected at DIV7 with Lipofectamine 2000 and analyzed at DIV14.
Antibodies
The following antibodies were commercially obtained: anti-FLAG mouse monoclonal (Sigma, St. Louis, MO, USA), secondary antibody anti-mouse HRP conjugated (Jackson Immunoresearch, West Grove, PA, USA), β-actin mouse monoclonal, anti calnexin, antiBiP and anti Erp57 (Abcam, Cambridge, MA, USA); anti-PDI (Assay Designs, Ann Harbor, MI, USA). Secondary antibody anti-rabbit HRP conjugated, calreticulin rabbit polyclonal (Assay Designs, Ann Harbor, MI, USA), CASPR2 rabbit polyclonal (Millipore, Temecula, CA, USA), secondary antibody anti-mouse conjugated with Cy5 and secondary antibody anti-rabbit conjugated with Fluorescein isothiocyanate (Jackson Immunoresearch, West Grove, PA, USA). All antibodies were used according to the manufacturers' recommendations.
Immunoprecipitation and western blot analysis
Cell lysates from HEK-293 cells transfected with FLAG-CASPR2-WT and mutants were probed by western blot for expression, protein degradation and deglycosylation experiments. Plates were washed and cells were scraped off the plate with cold phosphate buffer saline (PBS), sedimented and resuspended in 95 µl of lysis buffer (50 mm Tris buffer pH7.4, 140 mm NaCl, 3 mm MgCl2, 0.5% NP40) and 5 µl of the protease inhibitor Leupeptin at 5 µg/ml final dilution. After 20 min incubation on ice, the soluble proteins in the lysate were separated from the insoluble fraction by centrifugation at 20 000g for 10 min at 4°C. Protein content was quantified with the Bradford protein assay. For chaperone detection (Fig. 5A), cells lysates were prepared as described with the exception that 10 mmN-ethylmaleimide (Sigma) was added in the lysis buffer and in all the following incubation steps, to covalently block sulfhydryl groups. Cell lysate total protein (1.5 mg) was pre-cleaned with immobilized protein G-Sepharose and this was followed by incubation with the anti-FLAG antibody as previously described (51). SDS-PAGE loading buffer was added to the cell lysate, and the samples were heated at 95°C for 5 min. Fifty micrograms of protein cell extract were separated by 10% SDS-PAGE (Invitrogen) and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked in PBS 5% non-fat milk and incubated with a mouse monoclonal anti-FLAG or rabbit anti–β-actin antibodies followed by peroxidase-conjugated secondary antibodies. To quantify the intensity of the bands, films were scanned, and band intensity was quantified with the Molecular Analyst™ image analysis software (AlphaEase FC 6.0.0). All data are presented as means ± SEM for three independent experiments unless otherwise noted. Graphs are created using GraphPad Prism (version 4.0; GraphPad Software, Inc., San Diego, CA, USA).
Deglycosylation assay
Cell lysates from HEK-293 cells stably expressing CASPR2-WT and mutants were immunoprecipitated with anti-FLAG antibody. The antibody was then captured with protein A Sepharose beads that were washed twice with PBS and centrifuged at 6 000g for 1 min. The immunoprecipitated protein was then denatured by adding 1X denaturing buffer and heated to 95°C for 5 min. For Endo-H and PNGase F (NEB, Ipswich, MA), denatured proteins were incubated with the enzyme, or PBS as a control, for 2 h at 37°C following manufacturer instructions. To stop the reactions, 4X SDS-PAGE loading buffer was added to each sample and heated at 95°C for 5 min.
Protein degradation assays
The protein stability of CASPR2 wild type and mutants was measured by adding CHX (35 μm) to stably transfected HEK-293 cells. Cells were initially exposed to CHX for 6, 24, 48 h or for 4, 8, 16 h sequences. Rates of degradation were monitored by quantitative immunoblotting. Data are presented as means ± SEM for three experiments. The proteasome inhibitor, MG132 (2, 5, and 10 µm; Sigma), was applied to the cells 24 h after transient transfection, and the Caspr2 expression levels were assessed after treatment for 24 h by western blot (35).
Immunofluorescence confocal microscopy
HEK-293 stably expressing CASPR2-WT and mutants were grown on poly-D-lysine-coated coverslips in six-well plates and cultured in complete DMEM for 24 h. Cells were washed twice with Hank's balanced salt solution (HBSS) and fixed using methanol for 5 min. at room temperature. Fixed cells were washed four times with HBSS and then incubated with HBSS, 2% normal goat serum (NGS), 1% bovine serum albumin (BSA) for 30 min at room temperature. Cells were then incubated for 1 h at room temperature with anti-FLAG (1:800), anti-calreticulin (1:800), or anti-CASPR2 (1:100) antibodies, followed by another wash in HBSS. Subsequently, cells were incubated for 1 h at room temperature with Alexa Fluor 488 anti-mouse IgG (Invitrogen, Carlsbad, CA, USA) and Alexa Fluor 633 anti-rabbit IgG secondary antibodies (Invitrogen, Carlsbad, CA, USA) in the dark. Controls slides were treated with secondary antibodies alone to ascertain the level of non-specific staining. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole dilactate (DAPI) (Sigma). Images were captured using Olympus FluoViewTM FV1000 laser-scanning confocal microscope. Acquired images were analyzed using Image J Software. Confocal images are the representation of 0.3 µm sections of cells. Single planes and Z-stacks were used to determine the amount of surface versus ER co-localization of wild type and mutated proteins inside the region of interest (ROI) defined by entire image of the cell that excludes the black background. Co-localization data are the result of three independent experiments. For each sample the ROI was analyzed with the Image J Software using the Colocalization Threshold plugin (NIMH, Bethesda, MD, USA).
Luciferase assay
Transfections of HEK-293 cells were performed in six-well plates. Typically, 300 ng of CASPR2, either wild type or D1129H, were co-transfected with 490 ng of p(5x)ATF6-luc (Firefly) and 10 ng of pRL-TK (Renilla) for transfection efficiency (53). A total of 800 ng of DNA comprehensive of the three plasmids were mixed with 3 µl of FuGENE in serum-free DMEM culture medium following the manufacturer's protocol (Roche). The cells were lysed 24 h post-transfection using the dual-luciferase reporter assay recommended protocol (Promega), with the provided passive lysis buffer. Both Firefly and Renilla luciferase activity were measured using a Glomax MultiPlus (Promega), and luciferase activity was calculated normalizing the Firefly luciferase activity relative to the Renilla. All measurements were performed in triplicate for seven independent experiments. Mock condition is the transfection of the pcDNA3.1 empty vector.
Protein modeling
Models were created with ModWeb server (version SVN.r1368M) using the following template: rat neurexin 1β (PDB ID: 2R1B); the second LNS/LG domain of neurexin-1α (PDB ID: 2H0B), Laminin G-like domain 3 from human perlecan (PDB ID: 3SH4), and the LG1-3 region of laminin (PDB ID: 2WJS).
ACKNOWLEDGEMENTS
We are grateful to Dr Giudo Gaietta for helpful advice in the use of the confocal microscope and to Ms Eva N. Rubio-Marrero for the technical help during the resubmission process. We would like to thank the Robert Wood Johnson Foundation (grant # 67038) for their support of the Child Health Institute of New Jersey.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by National Institutes of Health grants, P42-ES10337-08 and RO1-GM18360-39 to P.T.; RO1-MH092906-01 and Autism Speaks #2617 to D.C.; Compagnia San Paolo Bando in Neuroscienze to ADJ; Confocal microscopy employed the facilities of the National Center for Imaging and Microscopy (NCMIR) at UCSD supported by NIH P41 RR004050 and P41GM103412 (MHE). BSA was supported by a New Investigator Development Award, a Human Genetics Pilot Award and a Rose F. Kennedy Intellectual and Developmental Disabilities Research Center (P30HD071593) Pilot Award from the Albert Einstein College of Medicine.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}