




Abstract
Obesity has always existed in human populations, but until very recently was comparatively rare. The availability of abundant, energy-rich processed foods in the last few decades has, however, resulted in a sharp rise in the prevalence of obesity in westernized countries. Although it is the obesogenic environment that has resulted in this major healthcare problem, it is acting by revealing a sub-population with a pre-existing genetic predisposition to excess adiposity. There is substantial evidence for the heritability of obesity, and research in both rare and common forms of obesity has identified genes with significant roles in its aetiology. Application of this understanding to patient care has been slower. Until very recently, the health risks of obesity were thought to be well understood, with a straightforward correlation between increasing obesity and increasing risk of health problems such as type 2 diabetes, coronary heart disease, hypertension, arthritis and cancer. It is becoming clear, however, that the location of fat deposition, variation in the secretion of adipokines and other factors govern whether a particular obese person develops such complications. Prediction of the health risks of obesity for individual patients is not straightforward, but continuing advances in understanding of genetic factors influencing obesity risk and improved diagnostic technologies mean that the future for such prediction is looking increasingly bright.
INTRODUCTION
According to the World Health Organization, the escalating international epidemic of obesity is now the most significant contributor to ill health (1). More than 30% of US adults are obese, i.e. body mass index (BMI) >30 kg/m2 (2), and it is feared that one in three children born in the early 21st century will develop diabetes with a consequent reduction in lifetime expectancy. Obesity accounts for 5–7% of national health expenditure in the US (3) and now outranks both smoking and drinking in its deleterious effects on health and health costs (4). Prevalence levels in the UK are following closely behind. (5). The paediatric picture is just as gloomy with a 2–2.8-fold increase over a 10-year period in childhood obesity (6).
Obesity is a major risk factor for cardiovascular diseases, pulmonary diseases (such as sleep apnoea), metabolic diseases (e.g. diabetes and dyslipidaemia), osteoarticular diseases, for several of the commonest forms of cancer (7) and for serious psychiatric illness. Furthermore, childhood obesity is associated with early onset type 2 diabetes (8) and with an increased mortality risk for coronary heart disease in adulthood (9).
If the escalating population prevalence of obesity and its serious implications for public health are generally accepted [with some notable exceptions (10)], its causes and physiological consequences at the individual level are still elusive. In only a few decades, the industrialized world has gone from a calorie-poor to a calorie-rich environment. Obviously, the recent unlimited availability of low-cost calorie-dense food, along with increasing sedentarity, has played a major role in the adult obesity pandemic.
In the 1960s, Neel (11) proposed the ‘thrifty gene’ hypothesis, whereby genes that predispose to obesity would have had a selective advantage in populations that frequently experienced starvation. People who possess these genes in today's obesogenic environment might be those that ‘overreact’— not just becoming slightly overweight, but extremely obese. This can be seen in certain high-risk groups, such as Pima Indians and Pacific Islanders (12), and recent studies in the USA have shown that there is a disproportionate level of obesity in African-Americans and Hispanic-Americans, compared with Caucasians (2). Recent support for the thrifty gene hypothesis was provided by Pritchard and co-workers (13), who found that many genes involved in the glucose and lipid metabolism have been subject to positive selection in the last 10 000 years, especially in Asian and African ethnic groups.
OBESITY IS A HIGHLY HERITABLE TRAIT
Heritability estimates for obesity are high (typically >0.70), comparing well with other complex, polygenic diseases such as schizophrenia (0.81) (14) and autism (0.90) (15), and are significantly higher than for other complex traits such as hypertension (0.29) (16) and depression (0.50) (17). In addition, the use of quantitative obesity sub-phenotypes that can be accurately measured has resulted in significant measures of heritability for skinfold thickness (18–20), waist circumference (21) and total and regional fat distribution (22).
RARE FAMILIAL OBESITY
For over a decade, obesity genetics has been predominantly driven by research into monogenic or syndromic obesity. The cloning of the mouse ob gene and its human homologue, leptin (23), proved to be a paradigm for the field that resulted in the identification of many genes involved in the regulation of appetite via the leptin–melanocortin pathway. These variants account for ∼5% of morbid human obesity and include leptin (24) and its receptor (25), the α-melanocortin-stimulating hormone receptor (MC4R) (26,27), pro-opiomelanocortin (POMC) (28) and prohormone convertase-1 (29).
Alongside these ‘pure’ forms of obesity, where the gene defect is in appetite regulation and the disease is characterized by severe early onset obesity because of hyperphagia, syndromic forms have provided additional insights into the mechanisms underlying obesity. There are around 30 different Mendelian disorders that have obesity as a significant clinical feature (reviewed in 30).
COMMON POLYGENIC OBESITY
The Human Obesity Gene Map summarizes the present situation in the field of common polygenic obesity (31). There are currently 253 quantitative-trait loci (QTLs) identified in 61 genome-wide scans, and 52 genomic regions contain QTLs supported by two or more studies. As in any complex genetic disease, there are many unconfirmed genetic associations: some of these may be due to inadequate sample sizes for association studies of genes of modest effect or due to inadequate examination of the genetic variation within these candidate genes. Researchers in the field of complex human genetics are increasingly aware of these problems and examples such as the TCF7L2 association with diabetes (32) demonstrate that sufficiently powerful studies can generate statistically strong results (P<10−8). There are currently 22 gene associations supported by at least five positive studies (summarized in Table 1). These genes include members of the leptin–melanocortin pathway, proinflammatory cytokines and uncoupling proteins. The largest numbers of studies have been carried out on ADRB2 and PPARG, but the fact that they also have reported associations with asthma and T2D, respectively, may account for this.
Genes with five or more positive associations of variants with obesity or obesity-related phenotypes in the published literature [adapted from (31)]
| Gene symbol | Full name | Chromosomal location | Number of studies | P-value |
|---|---|---|---|---|
| ACE | Angiotensin I-converting enzyme (peptidyl-dipeptidase A) 1 | 17q24.1 | 6 | 0.05–0.0023 |
| ADIPOQ | Adiponectin, C1Q and collagen domain containing | 3q27 | 11 | 0.05–0.001 |
| ADRB2 | Adrenergic, beta-2-, receptor, surface | 5q31–q32 | 20 | 0.05–0.0001 |
| ADRB3 | Adrenergic, beta-3-, receptor | 8p12–p11.2 | 29 | 0.05–0.001 |
| DRD2 | Dopamine receptor D2 | 11q23.2 | 5 | 0.03–0.002 |
| GNB3 | Guanine nucleotide binding protein (G protein), beta polypeptide 3 | 12p13.31 | 14 | 0.05–0.001 |
| HTR2C | 5-hydroxytryptamine (serotonin) receptor 2C | Xq24 | 10 | 0.05–0.0001 |
| IL6 | Interleukin 6 (interferon, beta 2) | 7p21 | 6 | 0.03–0.003 |
| INS | Insulin | 11p15.5 | 7 | 0.05–0.0002 |
| LDLR | Low density lipoprotein receptor (familial hypercholesterolaemia) | 19p13.2 | 5 | 0.04–0.001 |
| LEP | Leptin (obesity homologue, mouse) | 7q31.3 | 10 | 0.05–0.003 |
| LEPR | Leptin receptor | 1p31 | 16 | 0.04–0.0001 |
| LIPE | Lipase, hormone-sensitive | 19q13.2 | 5 | 0.05–0.002 |
| MC4R | Melanocortin 4 receptor | 18q22 | 8 | 0.04–0.002 |
| NR3C1 | Nuclear receptor sub-family 3, group C, member 1 (glucocorticoid receptor) | 5q31 | 10 | 0.05–0.001 |
| PLIN | Perilipin | 15q26 | 5 | 0.05–0.0008 |
| PPARG | Peroxisome proliferative activated receptor, gamma | 3p25 | 30 | 0.05–0.001 |
| RETN | Resistin | 19p13.2 | 5 | 0.048–0.001 |
| TNF | Tumor necrosis factor (TNF superfamily, member 2) | 6p21.3 | 9 | 0.05–0.004 |
| UCP1 | Uncoupling protein 1 (mitochondrial, proton carrier) | 4q28–q31 | 10 | 0.05–0.001 |
| UCP2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 11q13.3 | 11 | 0.05–0.001 |
| UCP3 | Uncoupling protein 3 (mitochondrial, proton carrier) | 11q13 | 12 | 0.049–0.0005 |
| Gene symbol | Full name | Chromosomal location | Number of studies | P-value |
|---|---|---|---|---|
| ACE | Angiotensin I-converting enzyme (peptidyl-dipeptidase A) 1 | 17q24.1 | 6 | 0.05–0.0023 |
| ADIPOQ | Adiponectin, C1Q and collagen domain containing | 3q27 | 11 | 0.05–0.001 |
| ADRB2 | Adrenergic, beta-2-, receptor, surface | 5q31–q32 | 20 | 0.05–0.0001 |
| ADRB3 | Adrenergic, beta-3-, receptor | 8p12–p11.2 | 29 | 0.05–0.001 |
| DRD2 | Dopamine receptor D2 | 11q23.2 | 5 | 0.03–0.002 |
| GNB3 | Guanine nucleotide binding protein (G protein), beta polypeptide 3 | 12p13.31 | 14 | 0.05–0.001 |
| HTR2C | 5-hydroxytryptamine (serotonin) receptor 2C | Xq24 | 10 | 0.05–0.0001 |
| IL6 | Interleukin 6 (interferon, beta 2) | 7p21 | 6 | 0.03–0.003 |
| INS | Insulin | 11p15.5 | 7 | 0.05–0.0002 |
| LDLR | Low density lipoprotein receptor (familial hypercholesterolaemia) | 19p13.2 | 5 | 0.04–0.001 |
| LEP | Leptin (obesity homologue, mouse) | 7q31.3 | 10 | 0.05–0.003 |
| LEPR | Leptin receptor | 1p31 | 16 | 0.04–0.0001 |
| LIPE | Lipase, hormone-sensitive | 19q13.2 | 5 | 0.05–0.002 |
| MC4R | Melanocortin 4 receptor | 18q22 | 8 | 0.04–0.002 |
| NR3C1 | Nuclear receptor sub-family 3, group C, member 1 (glucocorticoid receptor) | 5q31 | 10 | 0.05–0.001 |
| PLIN | Perilipin | 15q26 | 5 | 0.05–0.0008 |
| PPARG | Peroxisome proliferative activated receptor, gamma | 3p25 | 30 | 0.05–0.001 |
| RETN | Resistin | 19p13.2 | 5 | 0.048–0.001 |
| TNF | Tumor necrosis factor (TNF superfamily, member 2) | 6p21.3 | 9 | 0.05–0.004 |
| UCP1 | Uncoupling protein 1 (mitochondrial, proton carrier) | 4q28–q31 | 10 | 0.05–0.001 |
| UCP2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 11q13.3 | 11 | 0.05–0.001 |
| UCP3 | Uncoupling protein 3 (mitochondrial, proton carrier) | 11q13 | 12 | 0.049–0.0005 |
Genes with five or more positive associations of variants with obesity or obesity-related phenotypes in the published literature [adapted from (31)]
| Gene symbol | Full name | Chromosomal location | Number of studies | P-value |
|---|---|---|---|---|
| ACE | Angiotensin I-converting enzyme (peptidyl-dipeptidase A) 1 | 17q24.1 | 6 | 0.05–0.0023 |
| ADIPOQ | Adiponectin, C1Q and collagen domain containing | 3q27 | 11 | 0.05–0.001 |
| ADRB2 | Adrenergic, beta-2-, receptor, surface | 5q31–q32 | 20 | 0.05–0.0001 |
| ADRB3 | Adrenergic, beta-3-, receptor | 8p12–p11.2 | 29 | 0.05–0.001 |
| DRD2 | Dopamine receptor D2 | 11q23.2 | 5 | 0.03–0.002 |
| GNB3 | Guanine nucleotide binding protein (G protein), beta polypeptide 3 | 12p13.31 | 14 | 0.05–0.001 |
| HTR2C | 5-hydroxytryptamine (serotonin) receptor 2C | Xq24 | 10 | 0.05–0.0001 |
| IL6 | Interleukin 6 (interferon, beta 2) | 7p21 | 6 | 0.03–0.003 |
| INS | Insulin | 11p15.5 | 7 | 0.05–0.0002 |
| LDLR | Low density lipoprotein receptor (familial hypercholesterolaemia) | 19p13.2 | 5 | 0.04–0.001 |
| LEP | Leptin (obesity homologue, mouse) | 7q31.3 | 10 | 0.05–0.003 |
| LEPR | Leptin receptor | 1p31 | 16 | 0.04–0.0001 |
| LIPE | Lipase, hormone-sensitive | 19q13.2 | 5 | 0.05–0.002 |
| MC4R | Melanocortin 4 receptor | 18q22 | 8 | 0.04–0.002 |
| NR3C1 | Nuclear receptor sub-family 3, group C, member 1 (glucocorticoid receptor) | 5q31 | 10 | 0.05–0.001 |
| PLIN | Perilipin | 15q26 | 5 | 0.05–0.0008 |
| PPARG | Peroxisome proliferative activated receptor, gamma | 3p25 | 30 | 0.05–0.001 |
| RETN | Resistin | 19p13.2 | 5 | 0.048–0.001 |
| TNF | Tumor necrosis factor (TNF superfamily, member 2) | 6p21.3 | 9 | 0.05–0.004 |
| UCP1 | Uncoupling protein 1 (mitochondrial, proton carrier) | 4q28–q31 | 10 | 0.05–0.001 |
| UCP2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 11q13.3 | 11 | 0.05–0.001 |
| UCP3 | Uncoupling protein 3 (mitochondrial, proton carrier) | 11q13 | 12 | 0.049–0.0005 |
| Gene symbol | Full name | Chromosomal location | Number of studies | P-value |
|---|---|---|---|---|
| ACE | Angiotensin I-converting enzyme (peptidyl-dipeptidase A) 1 | 17q24.1 | 6 | 0.05–0.0023 |
| ADIPOQ | Adiponectin, C1Q and collagen domain containing | 3q27 | 11 | 0.05–0.001 |
| ADRB2 | Adrenergic, beta-2-, receptor, surface | 5q31–q32 | 20 | 0.05–0.0001 |
| ADRB3 | Adrenergic, beta-3-, receptor | 8p12–p11.2 | 29 | 0.05–0.001 |
| DRD2 | Dopamine receptor D2 | 11q23.2 | 5 | 0.03–0.002 |
| GNB3 | Guanine nucleotide binding protein (G protein), beta polypeptide 3 | 12p13.31 | 14 | 0.05–0.001 |
| HTR2C | 5-hydroxytryptamine (serotonin) receptor 2C | Xq24 | 10 | 0.05–0.0001 |
| IL6 | Interleukin 6 (interferon, beta 2) | 7p21 | 6 | 0.03–0.003 |
| INS | Insulin | 11p15.5 | 7 | 0.05–0.0002 |
| LDLR | Low density lipoprotein receptor (familial hypercholesterolaemia) | 19p13.2 | 5 | 0.04–0.001 |
| LEP | Leptin (obesity homologue, mouse) | 7q31.3 | 10 | 0.05–0.003 |
| LEPR | Leptin receptor | 1p31 | 16 | 0.04–0.0001 |
| LIPE | Lipase, hormone-sensitive | 19q13.2 | 5 | 0.05–0.002 |
| MC4R | Melanocortin 4 receptor | 18q22 | 8 | 0.04–0.002 |
| NR3C1 | Nuclear receptor sub-family 3, group C, member 1 (glucocorticoid receptor) | 5q31 | 10 | 0.05–0.001 |
| PLIN | Perilipin | 15q26 | 5 | 0.05–0.0008 |
| PPARG | Peroxisome proliferative activated receptor, gamma | 3p25 | 30 | 0.05–0.001 |
| RETN | Resistin | 19p13.2 | 5 | 0.048–0.001 |
| TNF | Tumor necrosis factor (TNF superfamily, member 2) | 6p21.3 | 9 | 0.05–0.004 |
| UCP1 | Uncoupling protein 1 (mitochondrial, proton carrier) | 4q28–q31 | 10 | 0.05–0.001 |
| UCP2 | Uncoupling protein 2 (mitochondrial, proton carrier) | 11q13.3 | 11 | 0.05–0.001 |
| UCP3 | Uncoupling protein 3 (mitochondrial, proton carrier) | 11q13 | 12 | 0.049–0.0005 |
It is not the intent of the current review to go into detail for all these candidate genes, but instead several genes that have recently generated discussion within the field will be highlighted for the reader.
GAD2
The glutamic acid decarboxylase gene (GAD2) was first reported to be associated with obesity and feeding behaviours in morbidly obese adults (33), and this result was subsequently replicated in obese children (34). However, an independent study failed to replicate these findings (35). Very recently, a study has reported associations with type 2 diabetes (36), but the effect of the single nucleotide polymorphisms (SNPs) was in the opposite direction. These findings illustrate the difficulty in determining whether a gene is truly associated with a complex genetic disease. In the current situation, failure to replicate the association, or the direction of its effect, does not invalidate the original findings, it just illustrates that small numbers of studies are not enough to prove or disprove that a gene is involved in a disease process. This is particularly true when it is likely that the aetiological variants have not been genotyped, as the most recent paper concludes.
Visfatin
Pre-B cell colony enhancing factor (PBEF1) was first identified as a protein secreted by lymphocytes over 10 years ago (37). It was re-named visfatin in a very recent paper (38), in which it was also shown to be expressed by adipocytes and expression was increased in visceral adipose tissue when compared with subcutaneous. This finding has now been replicated (39). The initial visfatin paper also reported that visfatin had insulin mimetic activity (38). This was supported by a report that visfatin levels were altered in T2D patients (40). Another study demonstrated a correlation between plasma visfatin concentration, or visceral visfatin mRNA expression, and obesity, although not specifically with visceral fat mass or waist-to-hip ratio (41). The first published genetic study of visfatin concluded that the variants tested did not play a major role in obesity or T2DM (42). It should be noted that although visfatin is often referred to as an adipokine, it is neither exclusively expressed by adipose tissue nor is it a typical cytokine, in fact, it is a type II phosphoribosyltransferase whose structure has recently been solved (43).
Ghrelin/ghrelin receptor/obestatin
The growth hormone secretagogue receptor (GHSR) was identified in 1996 (44) and its endogenous ligand ghrelin in 1999 (45). Subsequently, it has been demonstrated that ghrelin is a hormone predominantly secreted by the stomach and small intestine that has a significant role in appetite regulation and gastrointestinal function (reviewed in 46). Recently, GHSR variants have been reported to be associated with both common obesity (47) and a rare familial short stature/obesity syndrome (48). However, the evidence for ghrelin is not quite as clear with initial positive results (49–52), but latterly, some negative results (53,54). There is the possibility that the recent identification of another appetite-regulating hormone that is derived from proghrelin, named obestatin, that has opposing effects to those of ghrelin (55) may be one possible explanation for the differing results of different studies.
Bardet–Biedl syndrome genes
Given the evidence that common variants of genes involved in rare familial forms of obesity have been associated with common obesity, e.g. leptin and the leptin receptor [see Rankinen et al. (31) for details], we have examined BBS gene variants for this possibility. Twelve SNPs within the BBS1, 2, 4 and 6 genes were selected and genotyped in adult and childhood obesity sample sets, with a total of 3242 subjects. Somewhat surprisingly, no associations were observed for the BBS1 SNPs as this is the gene most frequently associated with BBS. However, SNPs in BBS2, 4 and 6 were associated with obesity, although not necessarily in both adults and children. The BBS6 variants also showed some association with quantitative traits associated with the metabolic syndrome such as dyslipidaemia and hyperglycaemia (56).
EPIGENETICS
It is also becoming clear, in both rare and common forms of obesity, that epigenetic influences, defined as any heritable influence on genes that occurs without a change in the DNA sequence, are also important. It has been known for sometime that the rare Prader-Willi and Angelman syndromes are due to variations in genomic imprinting at the proximal long arm of chromosome 15 (reviewed in 57). There is also an initial report of genomic imprinting, playing a role in common obesity at three different genomic loci (58). If we consider the combination of DNA methylation and histone modification patterns, with the possibility of paramutation as well (59), it would be highly surprising if epigenetics was not a significant contributor to the complexity of the genetics of common obesity.
GENES AND OBESITY-ASSOCIATED HEALTH RISKS
Some individuals are more predisposed than others to obesity-associated diseases, but it is presently difficult to identify the ‘at risk’ individuals who would benefit the most from individualized monitoring and care. All fat is not equal and, in particular, the site of fat accumulation can have important implications. Physiologically, metabolically deleterious and life-threatening forms of obesity associate with a preferential accumulation of fat in the visceral adipose tissue and with ectopic fat deposition in insulin-sensitive tissues, such as muscle, liver and pancreas. This aberrant fat content strongly correlates with severe generalized insulin resistance and the development of a chronic inflammatory state, partly due to the infiltration of the adipose tissue by macrophages (reviewed in 60).
Convincing evidence has recently been put forward that several fat-derived cytokines, including the anti-inflammatory adiponectin, strongly modulate the risk of the metabolic syndrome and T2D associated with obesity (diabesity) (61). Variation within the adiponectin gene is reported to modulate plasma levels of adiponectin and also to predict risk for diabesity and associated coronary heart diseases (62). Paradoxically, the adiponectin variant alleles that protect against the development of diabesity by maintaining high adiponectin concentrations also associate with obesity risk in both adults and obese children (63). Individuals with high adiponectin levels can be severely obese, but seem to enjoy metabolic protection (64). In the general population, the same alleles, together with the T2D protective PPAR-g 12Ala allele associates with a CHD protective risk factor pattern, elevated adiponectin and insulin sensitivity, but also with a dramatic increase of 3 units of BMI (65).
Altogether, these data suggest that obesity can be metabolically ‘neutral’ if there is an effectively unlimited availability of small insulin-sensitive subcutaneous adipocytes for fat storage. This appears to protect against the progressive development of fat-related chronic inflammatory disease. This protective pattern is, at least in part, genetically driven. In contrast, it has also been shown that in the context of obesity, carrying gene variants that worsen obesity-associated insulin resistance dramatically increases risk for diabesity and also for CHD (66).
BIOMARKERS FOR ASSESSING OBESITY-ASSOCIATED HEALTH RISKS
If well-characterized biomarkers were available, therapeutic intervention to prevent or delay the onset of obesity or its complications in susceptible individuals might become possible (67). In addition, biomarkers might help us to separate different sub-types of obesity, including those most associated with diabetes, cardiovascular disease and cancer (68–71). Some prospective biomarkers for these different states of health and disease are illustrated in Figure 1.
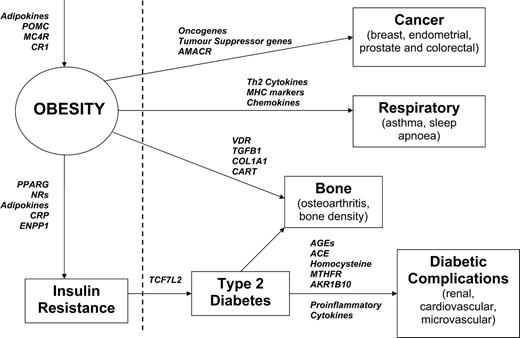
Examples of clinically relevant biomarkers for assessing the development of obesity and its sequelae. The dotted line represents the division between clinical health and disease. Key (in alphabetical order): ACE, angiotensin I-converting enzyme (peptidyl-dipeptidase A) 1; AGEs, advanced glycation end-products; AKR1B10, aldo-keto reductase family 1, member B10 (aldose reductase); AMACR, alpha-methylacyl-CoA racemase; CART, cocaine- and amphetamine-regulated transcript; COL1A1, collagen, type I, alpha 1; CR1, cannabinoid receptor 1; ENPP1, ectonucleotide pyrophosphatase/phosphodiesterase 1; MC4R, melanocortin-4 receptor; MTHFR, 5,10-methylenetetrahydrofolate reductase (NADPH); NRs, nuclear receptors; POMC, pro-opiomelanocortin; PPARG, peroxisome proliferative activated receptor, gamma; TCF7L2, transcription factor 7-like 2 (T-cell specific, HMG-box); TGFB1, transforming growth factor, beta 1; VDR, vitamin D (1,25-dihydroxyvitamin D3) receptor.
In addition to proteins in tissues and body fluids, gene expression profiles or circulating metabolites (72,73), suitable biomarkers might include genetic variants. As discussed earlier, genetic analysis of common forms of obesity has revealed associations with a range of individual genes, each of modest effect, but although individually these markers have modest predictive value, typically conferring a relative risk of 1.2–1.5, they might be much more informative when used in combination (74–77). Despite replicated associations, no systematic analysis of the utility of these key genes, and their protein products as biomarkers for obesity-related health risk, has yet been carried out. The challenge will be to understand how the data can be combined, incorporating new information from proteomics and metabonomics, to define a minimal effective set of biomarkers for risk prediction.
It may be that because several potentially important biomarkers are susceptible to environmental influences and/or measurement error (78–80), and it is well established that some biomarkers fluctuate with the menstrual cycle (81–84), gene variants could give a firmer indication of background susceptibility, although repeated measurements of circulating proteins and metabolites may be necessary to judge whether a genetically vulnerable individual is being adversely affected by environmental conditions. Ultimately, this could allow design of a two-stage monitoring system for severe obesity: initially, subjects would be genotyped for important genetic predictors and serum biomarkers could then be monitored on a regular basis to track their condition and alert their doctor to any adverse changes. In the future, this monitoring could be carried out with minimally invasive technology using micro-fluidics-based lab-on-a-chip systems (85,86), sending information directly through a PDA-like device to care providers. Such an integrated preventative system could reduce obesity-related morbidity and mortality and have profound implications for healthcare economics.
OBESITY, HEALTH AND GENETICS
The controversy about the consequences of obesity on health was recently fuelled by two papers: a follow-up study of former obese subjects ‘cured’ by bariatric surgery found no beneficial effect on mortality (87), suggesting that losing weight, even for very obese people, does not always improve health. Furthermore, a large Scandinavian epidemiological study showed that overweight subjects with no associated co-morbidities (i.e. with no metabolic syndrome) who intended to lose weight, and succeeded in doing so, died earlier than those who maintained or increased their weight (88).
Thus, it seems that obesity is more phenotypically and genetically heterogeneous and more complex than previously thought. Genetics (and probably epigenetics) plays an important role in the energetic imbalance leading to fat accumulation, but being obese does not necessarily mean being ill and, indeed, it is likely that very good health is required to establish and to maintain extreme obesity. As demonstrated by the data on adiponectin, genetics also determines, at least in part, the extent to which the inflation of fat depots predisposes to dysmetabolism, eventually dramatically increasing morbidity and mortality risk. Unravelling the genetic background associated with every stage of obesity and its consequences for health is of paramount importance, as it may help to suggest less emotive and more efficient ways to manage the obesity phenomenon.
Acknowledgements
AJW wishes to thank Miss Prabhjeet Phalora for her assistance in preparing Table 1.
Conflict of Interest statement. None declared.

{kind=link}