




Abstract
Mutations in the leucine-rich repeat kinase 2 gene (LRRK2) have been recently identified in families with autosomal dominant late-onset Parkinson disease (PD). The LRRK2 protein consists of multiple domains and belongs to the Roco family, a novel group of the Ras/GTPase superfamily. Besides the GTPase (Roc) domain, it contains a predicted kinase domain, with homology to MAP kinase kinase kinases. Using cell fractionation and immunofluorescence microscopy, we show that LRRK2 is localized in the cytoplasm and is associated with cellular membrane structures. The purified LRRK2 protein demonstrates autokinase activity. The disease-associated I2020T mutant shows a significant increase in autophosphorylation of ∼40% in comparison to wild-type protein in vitro. This suggests that the pathology of PD caused by the I2020T mutation is associated with an increase rather than a loss in LRRK2 kinase activity.
INTRODUCTION
Parkinson disease (PD) is the second most common neurodegenerative disorder affecting 1–2% of the population aged 65 and older. Recent studies have identified mutations in the leucine-rich repeat kinase 2 (LRRK2, PARK8) gene, located on the chromosomal region 12q11.2–q13.1 (OMIM 607060), as the cause of an autosomal dominant inherited form of familial PD (1,2). LRRK2 mutations are found in 5% of persons having a first-degree relative with PD and in 0.4–1.6% of sporadic PD cases (3–6). The LRRK2 gene has been predicted to encode a 285 kDa protein, belonging to the Roco protein family, a novel group of the Ras/GTPase superfamily (1,2). These proteins consist of multiple domains with varying composition. All family members have two domains in common: the GTPase domain Ras of complex proteins (Roc) and the C-terminal of Roc (COR) domain (7). Information on the biological function of Roco proteins is sparse. The best studied Roco protein so far is the death domain bearing metazoan DAP-kinase, which is involved in apoptotic pathways (7–10).
In addition to the Roc and COR domains, LRRK2 contains N-terminal leucine-rich repeats (LRRs), a MAP kinase kinase kinase (MAPKKK) domain and C-terminal WD40 repeats (2). The fusion of a Ras-like domain with a MAPKKK domain makes LRRK2 an ideal candidate for intra-molecular signal transduction. In addition to participating in active signalling, LRRK2 could also function as a scaffolding protein similar to kinase suppressor of Ras (Ksr), a multiple domain protein with similarity to MAPKKK but lacking kinase activity. Ksr participates in MAPK signalling as a scaffolding factor by binding Raf, MEK and ERK (reviewed in 11). The predicted LRRs, as well as the predicted C-terminal WD40 repeats within the LRRK2 sequence, are found in a large variety of proteins with different functions. Both domains are thought to serve as assembly points for larger protein complexes (12,13). Phylogenetic analysis of the LRRK2 kinase domain has demonstrated a similarity to both RIP and mixed lineage kinases, which are part of the tyrosine kinase-like (TKL) branch of the human kinome (14). Both kinase families are involved in stress-induced cell signalling and mediate apoptosis (15,16). However, a functional role for LRRK2 itself in these pathways remains to be determined.
As the biological function of wild-type LRRK2 and the nature of disease-associated LRRK2 mutations are completely unknown, we undertook a biochemical analysis of LRRK2 and its PD-associated variant I2020T that bears a mutation in the kinase domain. Our findings indicate that LRRK2 acts as a true protein kinase. Furthermore, the I2020T mutation in LRRK2 causes a significant increase in kinase activity.
RESULTS
LRRK2 is a 280 kDa membrane-associated protein
For functional and biochemical studies, we cloned LRRK2 from human cDNA and generated a series of constructs for the expression of haemagglutinin (HA)-, Strep/Flag- and green fluorescent protein (GFP)-tagged LRRK2 fusion proteins. Besides the disease-associated mutant I2020T, a kinase-dead variant of LRRK2 was generated, serving as negative control in the kinase assays described later (Fig. 1A). HEK293 cells, transiently transfected with C-terminal Strep/Flag-tagged wild-type or mutant human LRRK2, express a ∼280 kDa protein recognized by anti-Flag antibody (Fig. 1B). In addition to the predicted 280 kDa band, a weaker signal at ∼180 kDa could also be detected in some cases, probably representing an N-terminal degradation product of LRRK2.
To determine the subcellular localization of LRRK2, we used two approaches: subcellular fractionation and fluorescence microscopy. For the biochemical detection of the subcellular distribution of LRRK2 in vitro, transfected cells were fractionated by differential centrifugation. The distribution of subcellular organelles in the obtained fractions was then analysed by western blotting, using specific antibodies for mitochondria (TOM20), cytoskeleton (β-tubulin), peroxisomes (PMP70), microsomes (BiP/GRP78, Sec61α) and soluble cytosolic proteins (p50cdc37). The Strep/Flag-tagged LRRK2 fusion protein was found exclusively in membranous fractions, i.e. fractions enriched in mitochondria (10K pellet) and microsomal membranes (160K pellet), but was absent from the cytosol (Fig. 2A). This indicates that LRRK2 is attached to particulate structures within the cytoplasm of these cells.
In order to investigate whether LRRK2 is a membrane-associated or an integral membrane protein, the 160K pellet was treated with sodium carbonate, pH 11.5 (17). LRRK2, together with two other known membrane-associated proteins—the luminal ER marker BiP/GRP78 (78 kDa glucose regulated protein) and the peripheral cytosolic ER-associated marker valosin-containing protein (VCP)—was extracted from microsomal membranes, whereas the integral membrane protein Sec61α was recovered in the membrane pellet (Fig. 2B). This suggests that LRRK2 is a membrane-associated protein rather than integrated into membranes.
Furthermore, HEK293 cells that expressed the Strep/Flag-tagged LRRK2 kinase domain were subjected to subcellular fractionation. In contrast to full-length LRRK2, the kinase domain construct was found in the cytosol, whereas little or no fusion protein was detected in the particulate fractions (both 10K and 160K pellets, Fig. 2A). Thus, the kinase domain is not implicated in the association of LRRK2 to membranous structures.
LRRK2 co-localizes with discrete cytoplasmic structures
Immunofluorescence microscopy was used to determine the subcellular localization of GFP-tagged LRRK2 transiently expressed in HEK293 cells. After fixation, cells were permeabilized and co-immunolabelled with antibodies specific for distinct subcellular structures. In order to minimize potential artefacts due to overexpression, only cells where GFP-tagged LRRK2 was expressed at low level were chosen for imaging analysis. The LRRK2 fusion protein demonstrated a diffuse cytoplasmic distribution and co-localization with particulate structures and organelles (Fig. 3, column 2). Partial co-localization was observed with inner cellular structures, i.e. mitochondria (TOM20), ER (PDI) and Golgi (58K Golgi), confirming results from subcellular fractionation studies. In contrast, no overlap was observed with peroxisomes (PMP70) and with the actin cytoskeleton (phalloidin–TRITC) or intermediate filaments (Vimentin). The strongest co-localization found was an overlap with β-tubulin. Similar results were obtained with COS7 cells (data not shown). Together, within the resolution limits of light microscopy and biochemical fractionation, these data suggest that LRRK2 is a cytoplasmic protein associated with a subset of organelles and inner cellular membranes, i.e. mitochondria, ER and Golgi and with the microtubules.
LRRK2 forms homodimers
The kinase domain of LRRK2 is predicted to belong to the class of MAPKKK. One feature of such kinases is the formation of dimers. Moreover, for Raf-1 and MLK-3—the latter being one of the closest relatives of LRRK2 in vertebrates—homodimerization is required for activity (18,19).
In order to address the question of dimerization, we performed a co-purification experiment with differentially tagged LRRK2 fusion proteins co-expressed in HEK293 cells: a Strep/Flag-tagged LRRK2 bait and a HA-tagged LRRK2 prey. In addition to the full-length LRRK2, we used a bait construct bearing only the kinase domain. These constructs are summarized in Figure 1A. As shown in Figure 4A (lower panel), both the full-length and the LRRK2 kinase domain baits were precipitated with the same efficiency by streptactin resin demonstrated by western blotting with an anti-Flag antibody. Analysis of the precipitated proteins with the anti-HA antibody showed that only the full-length LRRK2 bait could pull out HA-tagged LRRK2, whereas the kinase domain only did not display any interaction with full-length LRRK2 (Fig. 4A, upper left panel). Thus, full-length LRRK2 interacts with itself, but the interaction is not mediated by the kinase domain.
As dimerization can serve as a mechanism to activate the catalytic activities of kinases, we next tested whether a disease-associated mutation in the kinase domain would affect the observed dimerization of LRRK2. By site-directed mutagenesis, we introduced the I2020T mutation into the full-length LRRK2 cDNA construct. Western blot analysis showed that mutated LRRK2 is expressed at a similar level as the wild-type protein (Fig. 1B). A Strep/Flag-tagged bait of the LRRK2-I2020T variant was compared to the wild-type in its ability of co-purifying the LRRK2-HA prey. As shown in Figure 4B, no difference in the amounts of co-purified LRRK2-HA protein was observed. Thus, oligomerization of LRRK2 is not altered by the PD-associated mutation I2020T.
The kinase domain of LRRK2 interacts with HSP90 and its co-chaperone p50cdc37
As no physiological substrate of LRRK2 has been reported to date, we started a search for proteins interacting with the LRRK2 kinase domain. The tandem affinity purification (TAP) tag technique has been successfully used to identify protein complexes in yeast under non-denaturing conditions (20). We performed TAP using two bait constructs containing a combination of a tandem StrepII and Flag-epitope (Strep/Flag-tag) fused to full-length LRRK2 and the kinase domain only. Transiently transfected cells were collected after 2 days and subjected to affinity purification starting with streptactin purification followed by Flag immunoprecipitation (IP). The purified protein complexes were then resolved by SDS–PAGE and stained with colloidal Coomassie blue (Fig. 4C). In addition to the purified bait (tagged kinase domain), two further bands were stained, which were identified as HSP90 and its co-chaperone p50cdc37 by tryptic in-gel proteolysis, followed by mass spectrometry (Fig. 4C). Performing a streptactin purification of the full-length protein, we co-isolated the same proteins, demonstrated by western blot analysis (Fig. 4D). In comparison to the kinase domain only, HSP90 and p50cdc37 were bound to the full-length protein to a significantly lower extent (data not shown). The interaction with the HSP90/p50cdc37 chaperone system was shown for several kinases, including the MAPKKK Raf-1 and MLK-3 (21–24). In both instances, they do not serve as substrates but associate as chaperones participating in maintenance of proper folding of the kinase. With our approach, we so far did not identify a substrate of LRRK2 but accumulated evidence that LRRK2 possesses kinase activity and may be active in transfected cells.
The I2020T mutation increases LRRK2 kinase activity
For other MAPKKK, like Raf-1 or the leucine zipper bearing MLK-3, it is known that dimerization and autophosphorylation of serine and threonine residues occur upon activation (18,19,25,26). The observation that recombinantly expressed LRRK2 can form dimers, its association with the HSP90/p50cdc37 chaperone system and its high grade of homology with MAPKKK prompted us to question whether LRRK2 shows kinase activity. With no physiological candidate substrate for LRRK2 available and autophosphorylation being a common feature of active kinases (25–27), we focused our analysis on the autophosphorylation activity of LRKK2. As a negative control, we generated a predicted LRRK2 ‘kinase-dead’ mutant by exchanging a conserved lysine residue within the ATP-binding site of the kinase to a methionine. Corresponding mutations in Raf-1 (K375W or K375M), for example, have been shown to be unable to catalyze the phosphotransfer reaction (Fig. 1C) (28,29).
The full-length Flag-tagged LRRK2 was precipitated from HEK293 cell lysates by using anti-Flag M2 agarose resin. LRRK2 was directly assayed for autophosphorylation by offering radioactive labelled ATP and performing subsequent autoradiography of the SDS–PAGE separated and blotted proteins. The assay was performed under conditions allowing autophosphorylation of Raf-1 in vitro (Fig. 5B) (28). As shown in Figure 5A, affinity purified LRRK2 exerted kinase activity and was able to phosphorylate itself, whereas only residual phosphorylation was observed when the K1906M mutant was used.
We further questioned whether a disease-associated mutation in the kinase domain would bear functional consequences on kinase activity. Thus far, two mutations, I2020T and G2019S, have been identified in the kinase domain of LRRK2, which are both predicted to be positioned in the kinase activation loop (1,30,31). Here, we analysed the I2020T mutation. This LRRK2 mutant was affinity purified in parallel with wild-type LRRK2. The autophosphorylation assay revealed that mutant LRRK2 also shows kinase activity (Fig. 5A). Quantification of autophosphorylation rates normalized to LRRK2 protein levels revealed a significant increase of ∼40% of the I2020T mutant in comparison to wild-type (Fig. 5C).
Taken together, the data reported here indicate that LRRK2 indeed shows kinase activity, a function which was predicted on the basis of sequence similarity. Furthermore, the I2020T mutation increases the kinase activity.
DISCUSSION
Until now, little is known on the function of LRRK2 and what has been speculated so far is based on the predicted domain structure and the mutations therein. In this study, we addressed the question of subcellular LRRK2 localization and whether we could establish a functional assay for LRRK2 protein activity. As it is predicted to be a protein kinase, we analysed the kinase activity of wild-type LRRK2 and compared it with the kinase mutant I2020T. Furthermore, to describe interacting proteins with the possibility of identifying a substrate of LRRK2, we purified the protein using the TAP technique.
The challenge for any functional analysis of LRRK2 is its large size and complex domain structure. In silico analysis of the LRRK2 protein did not reveal any targeting signal for specific subcellular destinations, suggesting a cytosolic localization of LRRK2. Surprisingly, our data obtained by subcellular fractionation excluded a cytosolic localization. We found LRRK2 in membranous fractions but not in the cytosol. Moreover, immunofluorescence analyses suggest that LRRK2 is partially localized to the microtubular cytoskeleton and inner cellular membranes, i.e. mitochondria, ER and Golgi. Although these results need to be confirmed for endogenous LRRK2, once suitable, specific antibodies are at hand; both localizations are attractive for a potential function of LRRK2 with respect to the pathogenesis of PD. Specifically, mitochondria are the focus of Parkinson's research based on (1) the recent identification of parkin, DJ-1 and PINK1 mutations, (2) indications that these proteins may have protective effects on the mitochondria and (3) the hypothesis that mitochondrial dysfunction may play a central role in the aetiology of sporadic PD (32–35).
The observed partial localization with the cytoskeleton is a feature already shown for another member of the Roco-protein family: the human death-associated protein kinase (DAP-kinase) (7). In contrast to DAP-kinase which localizes to the actin cytoskeleton (9), our analysis suggests that LRRK2 is associated with microtubules but not with actin or intermediate filaments. Thus, LRRK2 could be involved in cytoskeletal regulation but in a way mechanistically different from that of the DAP-kinase.
The predicted LRRK2 structure includes a GTPase and a kinase domain. Both domains might confer measurable biochemical activities potentially affected by mutations in Parkinson patients. Here, we focused on the kinase activity and a mutation within the kinase domain. We established an assay to monitor autophosphorylation. Indeed, LRRK2 is able to autophosphorylate and to dimerize; both features are shared by many MAPKKK, including the Roco-protein DAP-kinase, MLK-3 or Raf-1 (9,25,26). Moreover, we found that the HSP90/p50cdc37 chaperone complex binds to LRRK2, which is known to assist both folding and activation of other kinases (22). In summary, this suggests that LRRK2 exerts its function, at least in part, as an active protein kinase.
The I2020T mutation confers an isoleucine to threonine exchange next to the DFG motif, (DYG in LRRK2) at the beginning of the activation loop of the kinase domain, which prompts to be highly conserved in almost all MAPKKK (36). Conformational changes in the activation loop, which in many kinases are induced by phosphorylation, are needed to switch between the inactive and the active state of a kinase (37). We found an increase of ∼40% in the kinase activity of the LRRK2 mutant I2020T, which is consistent with mutations in homologous positions of other kinases like B-Raf that is associated with cancer (38). Our results point to a gain-of-function of the PD-associated I2020T mutation. The gain-of-function is also in line with the dominant feature of known LRRK2 mutations. In addition to an overall increase in kinase activity, the mutation could also alter substrate specificity. As with oncogenic kinase variants, kinase inhibitors could then be considered as a treatment option. The effectiveness of such therapeutic strategy has been proved with respect to specifically inhibiting the bcr-abl protein kinase within chronic myelogenous leukaemia (CML) through the kinase inhibitor 2-phenylaminopyrimimidine STI571 (Gleevec), a small-molecule tyrosine kinase inhibitor for the treatment of CML (39).
In summary, we could show that LRRK2 shares common biochemical features with other MAPKKK, such as autophosphorylation, dimerization and interaction with kinase-specific chaperones. Furthermore, the autokinase activity of the LRRK2 mutant I2020T was found to be increased when compared with the wild-type. The latter finding has to be interpreted with caution, as long as it lacks confirmation on physiologically relevant substrates. These physiological substrates may reside and co-localize with LRRK2 at inner cellular membranes and microtubules. As can be speculated from its multimodular structure, LRRK2 could be involved in functions as diverse as maintenance of microtubular structure and dynamics, vesicular trafficking (ER, Golgi compartment) and/or cytoskeletal rearrangements. Further studies focusing on the biochemical and functional properties of LRRK2 domains are underway. They will shed light on the physiological function of LRRK2 and how disease-associated mutations in LRRK2 interfere with these functions.
MATERIALS AND METHODS
Plasmids and cloning
Human LRRK2 was cloned via PCR from cDNA that had been generated from lymphoblast mRNA. LRRK2 was cloned domain-wise in six fragments, with each fragment cloned into pcDNA3.0 (Invitrogen) and verified by sequencing. The full-length sequence was generated by subsequent fusion of the subconstructs. The HA and Strep/Flag tag were introduced in frame at the 3′ end (C-terminus) of the constructs. The disease-associated I2020T and the kinase-impaired K1906M mutation were introduced into LRRK2 by site-directed mutagenesis using the QuikChange® II mutagenesis kit (Stratagene). For fluorescence microscopy, humanized GFP cDNA, derived from pFRED143 (40), was cloned in frame at the 3′ end (C-terminus) of LRRK2.
Cell culture
HEK293 cells were cultured in DMEM supplemented with 10% FBS at 37°C and 5% CO2. For IP, TAP or cell fractionation, experiments cells were transfected with Effectene (Qiagen) according to the manufacturer's protocol and kept under full medium for an additional 48 h.
Electrophoresis and western blot
For western blot analysis, protein samples were separated by SDS–PAGE and transferred onto Hybond-P PVDF membranes (GE Healthcare). After blocking non-specific binding sites with 5% non-fat dry milk in TBST (1 h, RT) (25 mm Tris, pH 7.4, 150 mm NaCl, 0.1% Tween-20), membranes were incubated overnight at 4°C with primary antibodies in blocking buffer [mouse anti-Bip/GRP78 (BD), 1:1000; mouse anti-p50cdc37 (BD), 1:1000; rat anti-HA 5F10, 1.3 µg/ml; mouse anti-p97/VCP (Progen), 1:1000; rabbit anti-PMP70 (kindly provided by Professor Dr A. Völkl, University of Heidelberg, Germany), 1:1000; rabbit anti-Sec61α (Acris), 1:1000; mouse anti-TOM20 (BD), 1:1000; mouse anti-β-tubulin (Sigma), 1:2000], washed with TBST and incubated for 1 h with horse radish peroxidase (HRP)-coupled secondary antibodies. For detection of the Flag-epitope, membranes were incubated with HRP-coupled monoclonal anti-Flag M2 antibody (Sigma), 1:1000. Membranes were washed and antibody–antigen complexes were visualized using the ECL+chemiluminescence detection system (GE Healthcare) on Hyperfilms (GE Healthcare).
Cell fractionation
Cells were harvested via trypsinization, washed once with cold PBS, resuspended in cold homogenization buffer (20 mm HEPES pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 250 mm sucrose, protease inhibitors, Roche) and homogenized. Homogenates were centrifuged at 700g for 10 min to pellet nuclei, debris and non-disrupted cells (cell pellet). The supernatant was centrifuged at 10 000g for 20 min to obtain the 10K pellet. Cytosol and 160K pellet were prepared by ultracentrifugation of the 10K supernatant (160 000g for 1 h).
Carbonate extraction
160K fractions were diluted with sodium carbonate (final concentration 100 mm), pH 11.5, and incubated for 30 min on ice. The suspensions were centrifuged for 1 h at 160 000g at 4°C. The supernatants were recovered and proteins precipitated with 10% trichloracetic acid. Membrane pellets and precipitated proteins were subjected to SDS–PAGE and western blotting analysis.
Tandem affinity purification
The TAP was done with a C-terminal TAP tag consisting of a tandem StrepII tag and a Flag epitope (Strep/Flag-tag). HEK293 cells transiently expressing the Strep/Flag-tagged constructs were lysed in 50 mm Tris–HCl pH 7.4, 150 mm NaCl, 0.5% Nonidet P-40, protease inhibitors and 1 mm orthovanadate for 1 h at 4°C. Following sedimentation of nuclei, the cleared supernatant was incubated for 2 h at 4°C with streptactin superflow (IBA). Prior to washing, the lysates with suspended resin were transferred to microspin columns (GE Healthcare). Washing (once with lysis buffer and twice with TBS) was done in the microspin columns. Washing solution was removed from the columns by centrifugation (10 s, 2000g) after each washing step. Protein baits were eluted with desthiobiotin (2 mm in TBS). The eluates were used for LRRK2 co-precipitation experiments of LRRK2-Strep/Flag constructs versus LRRK2-HA.
For MS analysis, a second purification step was added. For this step, the eluates were transferred to anti-Flag M2 agarose (Sigma) and incubated for 2 h at 4°C. The beads were washed three times with TBS in microspin columns. Proteins were eluted with Flag peptide (Sigma) in PBS at 200 µg/ml peptide. After purification, samples were separated by SDS–PAGE and stained with colloidal Coomassie blue according to standard protocols prior to MS identification (41).
Mass spectrometry
Proteins were identified by MALDI-MS and MSMS on an AB4700 (Applied Biosystems) instrument. Tryptic in-gel proteolysis was done according to standard protocols (42). Peptides were spotted on steal targets with the dried droplet method using α-cyano-4-hydroxycinnamic acid (Sigma) as matrix (42). Obtained MS and MS/MS spectra were analysed by GPS explorer software suite (Applied Biosystems).
Kinase activity assay (autophosphorylation assay)
For kinase assays (autophosphorylation assays), Strep/Flag-tagged full-length wild-type LRRK2, LRRK2-I2020T or LRRK2-K1906M were transiently expressed in HEK293 cells (4×14 cm culture dishes per construct, 2×14 cm dishes for the vector control). After cell lysis and removal of the nuclei, the purification of LRRK2 variants was done by IP with anti-Flag M2 agarose. The resin was washed three times in lysis buffer. The tagged proteins were not eluted because the kinase assays were directly performed on the resin. Each sample was divided into four aliquots and stored in TBS+10% glycerol at −80°C until use.
For the kinase assay, one aliquot of each condition (wild-type LRRK2, LRRK2-I2020T and LRRK2-K1906M) was divided into three sub-aliquots (half, one-third, one-sixth). Each sub-aliquot, as well as one aliquot of the vector control, was incubated with 50 µm ATP, 3 µCi [γ-32P] ATP in 30 µl assay buffer (25 mm Tris–HCl pH 7.5, 5 mm β-glycerophosphate, 2 mm DTT, 0.1 mm orthovanadate; 10 mm MgCl2; Cell Signaling) for 1 h at 30°C. Reaction was stopped with Laemmli buffer. Protein samples were resolved by SDS–PAGE and transferred onto low fluorescence Hybond-LFP PVDF membranes (GE Healthcare). Imaging was done either by exposition of a film or on a phosphorimager plate (Fuji) scanned on a FLA3000 reader (Fuji). Loading was determined by western blot analysis. The used antibody was Cy3-labelled (anti-Flag M2). Fluorescence imaging was performed on an FLA3000 reader. Alternatively, loading was measured by the Coomassie stain of the PVDF membranes.
As a positive control for autophosphorylation, GST-Raf-1 and a kinase-deficient mutant of GST-Raf301 (K375W) were expressed in COS7 cells. The cells were starved overnight and stimulated with tetradecanoyl phorbol-ester acetate (TPA) for 20 min prior to lysis. GST-Raf-1 and GST-Raf301 were purified by glutathione–Sepharose beads (GE Healthcare) and subjected to kinase assay as described in (28). The kinase assays were separated by SDS–PAGE and blotted. Blots were exposed to X-ray film and subsequently probed with Raf-1-specific antiserum to assure that similar amounts of Raf-1 proteins were loaded.
Statistics
Quantification of the autoradiograms and the loading controls was done with the Image analysis software ImageJ (Rasband, W.S., US National Institutes of Health, Bethesda, MD, USA; http://rsb.info.nih.gov/ij/). To probe for significance of observed differences, a paired t-test was performed with 19 individual data-points (n=19) for wild-type LRRK2 and LRRK2-I2020T and 10 data-points (n=10) for the kinase-dead K1906M mutant originating from at least five independent experiments.
Immunofluorescence
HEK293 cells were grown on glass cover slips prior to transfection with GFP-tagged wild-type LRRK2. To avoid cell detachment, cover slips were pre-treated with poly-d-lysine (Sigma) and laminin (Sigma). Forty-eight hours post-transfection, cells were fixed for 15 min with 4% paraformaldehyde at RT. Fixed cells were permeabilized with PBS containing 0.1% Triton X-100 for 5 min, blocked with PBS containing 0.1% Tween-20 and 1% BSA and incubated for 3 h at RT with primary antibodies in blocking solution [mouse anti-58K Golgi, 1:100 (Abcam); mouse anti-PDI, 1:100 (Abcam); rabbit anti-PMP70, 1:200; mouse anti-TOM20, 1:500 (BD); mouse anti-β-tubulin, 1:500 (Sigma); mouse anti-Vimentin, 1:200 (Sigma)]. Cover slips were rinsed six times with PBS and labelled for 1 h with Alexa 568-conjugated goat anti-mouse, goat anti-rabbit IgG (Invitrogen) or phalloidin–TRITC (1:10 000, Sigma). For nuclear staining, the solution also contained 1 µg/ml 4,6-diaminodiphenyl-2-phenylindole (DAPI, Sigma). Cover slips were washed six times with PBS, mounted with FluorSave (Calbiochem) and evaluated by fluorescence microscopy using a Zeiss Apotome equipped with Cy3, FITC and DAPI optical filter sets. The obtained images provide an axial resolution comparable to confocal microscopy (43).
ACKNOWLEDGEMENTS
The authors would like thank Dr Ursula Olazabal and Gabriele Duetsch for critical reading of the manuscript, Utz Linzner for primer synthesis and Evelyn Botz for the preparation of the human lymphocyte cDNA. The authors are grateful to Qiagen for providing the transfection reagent Effectene. This project is funded by the German Federal Ministry for Education and Research (BMBF NGFN-SMP Proteomics, ‘Human Brain’) and NGFN‐SMP DNA, “Mitochondria”), BFAM (Bioinformatics for the Functional Analysis of Mammalian Genomes) and the European Union by EU grant LSHG-CT-2003-50520 (INTERACTION PROTEOME).
Conflict of Interest statement. None declared.
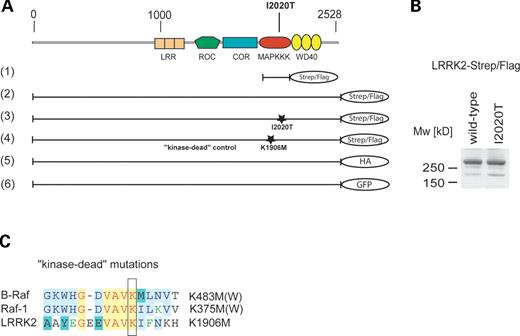
Figure 1. (A) Overview of LRRK2-domain structure and constructs used in this study. Human LRRK2 consists of five predicted domains: N-terminal LRRs, followed by a GTPase (Roc), a COR, an MAPKKK and a WD40 repeat domain. The kinase domain of human LRRK2 (1), the full-length LRRK2 (2), a disease-associated LRRK2 mutant I2020T (3) and a kinase-dead mutant LRRK2-K1906M (4) were cloned in frame into a modified pcDNA3.0, containing a C-terminal Strep/Flag tandem affinity tag. The mutations are localized, as marked, in the kinase domain of LRRK2. Additionally, wild-type LRRK2 was C-terminally tagged with an HA epitope (5) and a GFP tag (6). (B) LRRK2-Strep/Flag and LRRK2 I2020T-Strep/Flag constructs express an ∼280 kDa protein in HEK293 cells, visualized by western blotting (anti-Flag M2) after SDS–PAGE. (C) Sequence alignment of B-Raf, Raf-1 and LRRK2. For the kinase-dead mutation of LRRK2, a conserved lysine within the active centre of the kinase domain was exchanged by methionine. Homologous mutations in Raf-1 and B-Raf are known to disrupt their kinase activity.
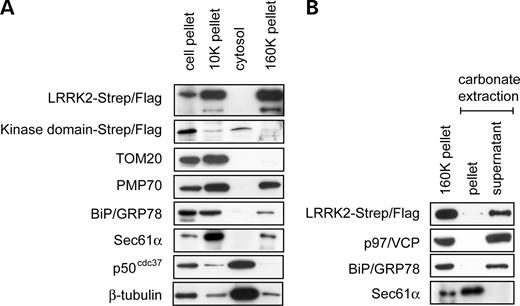
Figure 2. LRRK2 appears in the particulate fractions upon subcellular fractionation and is associated with membranes. (A) LRRK2 co-sediments with membranes. HEK293 cells were fractionated into a cell pellet (700g) an organelle pellet (10K pellet), a soluble cytosolic fraction (cytosol) and a microsomal fraction (160K pellet). The fractions were analysed by SDS–PAGE and western blotting with antibodies against the Flag-tag, TOM20 (mitochondria), PMP70 (peroxisomes), BiP/GRP78 (ER lumen), Sec61α (ER membrane), p50cdc37 (cytosol) and β-tubulin (microtubules). (B) Alkaline extraction of LRRK2. The 160K pellet was treated with 100 mm sodium carbonate. Membrane and soluble fraction (pellet and supernatant, respectively) were separated by centrifugation and analysed as in (A) using antibodies against the integral ER membrane protein Sec61α, the cytosolic ER-associated protein p97/VCP and the luminal ER protein BiP/GRP78.
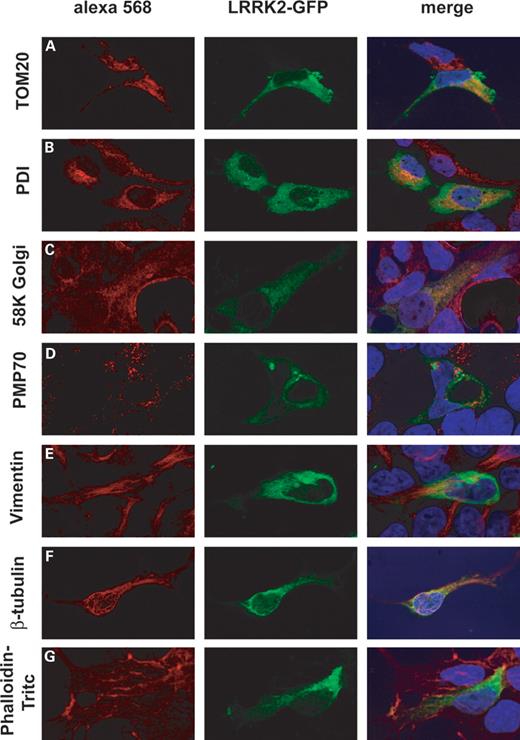
Figure 3. LRRK2-GFP localizes to mitochondria, endoplasmic reticulum, Golgi and the microtubular cytoskeleton. HEK293 expressing LRRK2-GFP (GFP fluorescence shown in the middle panel) were immunostained for (A) mitochondria (TOM20), (B) endoplasmic reticulum (PDI), (C) Golgi (58K Golgi), (D) peroxisomes (PMP70), (E) intermediate filaments (Vimentin), (F) microtubular cytoskeleton (β-tubulin) and (G) phalloidin–TRITC (actin cytoskeleton). The right panel depicts digitally merged images taken from the same micrograph section and merges red Alexa 568 staining (specific markers), green fluorescence GFP and nuclear staining with DAPI.
Figure 4. LRRK2 dimerizes and interacts with HSP90 and p50cdc37 (A) Co-purification of differently tagged LRRK2-constructs: HA-tagged full-length LRRK2 was tested for its ability to interact with two different Strep/Flag-tagged LRRK2 baits (a full-length and a kinase domain only construct). The constructs were co-expressed transiently in HEK293 cells prior to cell lysis and purification. The result of the co-purification of HA-tagged LRRK2 with the Strep/Flag-tagged baits is shown in the upper left panel (pellet). The co-precipitated HA-tagged LRRK2 was visualized by western blotting (3F10 anti-HA). Controls: In order to demonstrate equal expression of LRRK2-HA, a western blot (anti-HA) of the supernatants is shown (upper right panel). Equal loading of purified bait proteins was ensured by western blotting (anti-Flag, lower left panel). Purification efficiency of Strep/Flag-tagged baits was determined by Western blotting of the depleted supernatants: after their affinity binding to the beads, no detectable bait protein remained in the supernatants (lower right panel). (B) Both, Strep/Flag-tagged wild-type LRRK2 and I2020T mutant co-precipitate wild-type LRRK2-HA (upper panel). Equal expression of the Strep/Flag-tagged baits was confirmed by western blotting (lower panel). (C) TAP of Strep/Flag-tagged LRRK2 kinase domain and a vector control (expression of the Strep/Flag-tagged only) from transiently transfected HEK293 cells. After SDS–PAGE separation of the purified protein complexes and colloidal Coomassie staining, three protein bands were visible, identified by mass-spectrometry as bait (LRRK2 kinase domain, 39 kDa), p50cdc37 (50 kDa) and HSP90 (90 kDa). (D) Interaction of full-length LRRK2 with HSP90 and p50cdc37 was identified by western blotting.
![Figure 5. LRRK2 wild-type and the disease-associated mutant I2020T reveal kinase activity by autophosphorylation. (A) The Flag-tagged full-length wild-type LRRK2, a LRRK2-I2020T mutant or a kinase impaired (kinase-dead control) K1906M mutant were transiently expressed in HEK293 cells. LRRK2 variants affinity purified by IP with anti-Flag M2 agarose were directly subjected to the kinase assays. The purified protein samples were incubated with [γ-32P]-ATP for 1 h, subjected to SDS–PAGE and blotted onto PVDF membranes. Autoradiography from these blots was performed using a phosphoimaging system (upper panel): [γ-32P]-ATP incorporation can be observed for both the wild-type LRRK2 protein (first lane) and the I2020T mutant protein (second lane). The kinase-dead and the vector control are shown in the third and fourth lane, respectively. Equal protein input was controlled by staining the same blot with an anti-Flag antibody (lower panel). (B) Autophosphorylation of the MAPKKK Raf-1. Recombinant GST-Raf-1, a kinase-dead GST-Raf301 (K375W) and the empty vector were expressed in COS7 cells. Cells were starved overnight and stimulated with TPA (100 ng/ml, 20 min) prior to lysis. The purified GST-fusion proteins were subjected to the kinase assay. The autoradiogram is shown in the upper panel and the corresponding loading control, a western blot with an anti-Raf1 antibody, in the lower panel. (C) Quantification of phosphorylation levels of wild-type LRRK2, I2020T mutant and the K1906M kinase-dead control. Phosphorylation values were normalized to wild-type LRRK2 (100%). Differences were proved for their significance by a paired t-test. A 40% increase in phosphorylation level of I2020T mutant when compared with the wild-type LRRK2 was obtained with P<0.05. The observed phosphorylation of the kinase-dead control was <30% of the wild-type level (**P<0.005).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/hmg/15/2/10.1093/hmg/ddi439/2/m_ddi43905.jpeg?Expires=1716638846&Signature=Lo8WwvWwCow2ikh~CJrJki3KhbhaferI7EC4k2sd81BUBUOAKfs9zxZYwGnoFFiAZ5bqp~6AN~oZPDpWCL1zT4hd15WZvsXDRnryfgmYuWJSHycXPyaJzCQjVuGAIz7oQaCx-D3ylkd8kj8ME38R4SBsbY~d6kZ51JQLEOYqBWiWdbwDJ~jlU3370pE5ylbaIdRNFOhxPOTQWLzhnb6wCOqUt63GoLQwxUa6YNOQTulEbizoKtcqV~PjRXokKFUM4qgr2krMJtsu7OhLlieoI8zrlkscBbbwoWwAu5jAuPTKjYkeljLM~1bJscpNQbQEZa3ciGaOopiC12S1vjnVpA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Figure 5. LRRK2 wild-type and the disease-associated mutant I2020T reveal kinase activity by autophosphorylation. (A) The Flag-tagged full-length wild-type LRRK2, a LRRK2-I2020T mutant or a kinase impaired (kinase-dead control) K1906M mutant were transiently expressed in HEK293 cells. LRRK2 variants affinity purified by IP with anti-Flag M2 agarose were directly subjected to the kinase assays. The purified protein samples were incubated with [γ-32P]-ATP for 1 h, subjected to SDS–PAGE and blotted onto PVDF membranes. Autoradiography from these blots was performed using a phosphoimaging system (upper panel): [γ-32P]-ATP incorporation can be observed for both the wild-type LRRK2 protein (first lane) and the I2020T mutant protein (second lane). The kinase-dead and the vector control are shown in the third and fourth lane, respectively. Equal protein input was controlled by staining the same blot with an anti-Flag antibody (lower panel). (B) Autophosphorylation of the MAPKKK Raf-1. Recombinant GST-Raf-1, a kinase-dead GST-Raf301 (K375W) and the empty vector were expressed in COS7 cells. Cells were starved overnight and stimulated with TPA (100 ng/ml, 20 min) prior to lysis. The purified GST-fusion proteins were subjected to the kinase assay. The autoradiogram is shown in the upper panel and the corresponding loading control, a western blot with an anti-Raf1 antibody, in the lower panel. (C) Quantification of phosphorylation levels of wild-type LRRK2, I2020T mutant and the K1906M kinase-dead control. Phosphorylation values were normalized to wild-type LRRK2 (100%). Differences were proved for their significance by a paired t-test. A 40% increase in phosphorylation level of I2020T mutant when compared with the wild-type LRRK2 was obtained with P<0.05. The observed phosphorylation of the kinase-dead control was <30% of the wild-type level (**P<0.005).
References
Paisan-Ruiz, C., Jain, S., Evans, E.W., Gilks, W.P., Simon, J., van der Brug, M., de Munain, A.L., Aparicio, S., Gil, A.M., Khan, N. et al. (
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R.J., Calne, D.B. et al. (
Farrer, M., Stone, J., Mata, I.F., Lincoln, S., Kachergus, J., Hulihan, M., Strain, K.J. and Maraganore, D.M. (
Gilks, W.P., Abou-Sleiman, P.M., Gandhi, S., Jain, S., Singleton, A., Lees, A.J., Shaw, K., Bhatia, K.P., Bonifati, V., Quinn, N.P. et al. (
Toft, M., Mata, I.F., Kachergus, J.M., Ross, O.A. and Farrer, M.J. (
Zabetian, C.P., Samii, A., Mosley, A.D., Roberts, J.W., Leis, B.C., Yearout, D., Raskind, W.H. and Griffith, A. (
Bosgraaf, L. and Van Haastert, P.J. (
Aravind, L., Dixit, V.M. and Koonin, E.V. (
Bialik, S., Bresnick, A.R. and Kimchi, A. (
Cohen, O. and Kimchi, A. (
Kolch, W. (
Kobe, B. and Kajava, A.V. (
Smith, T.F., Gaitatzes, C., Saxena, K. and Neer, E.J. (
Manning, G., Whyte, D.B., Martinez, R., Hunter, T. and Sudarsanam, S. (
Meylan, E. and Tschopp, J. (
Xu, Z., Maroney, A.C., Dobrzanski, P., Kukekov, N.V. and Greene, L.A. (
Fujiki, Y., Hubbard, A.L., Fowler, S. and Lazarow, P.B. (
Farrar, M.A., Alberol, I. and Perlmutter, R.M. (
Leung, I.W. and Lassam, N. (
Gavin, A.C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M. et al. (
Grammatikakis, N., Lin, J.H., Grammatikakis, A., Tsichlis, P.N. and Cochran, B.H. (
Pearl, L.H. (
Zhang, H., Wu, W., Du, Y., Santos, S.J., Conrad, S.E., Watson, J.T., Grammatikakis, N. and Gallo, K.A. (
Zhang, W., Hirshberg, M., McLaughlin, S.H., Lazar, G.A., Grossmann, J.G., Nielsen, P.R., Sobott, F., Robinson, C.V., Jackson, S.E. and Laue, E.D. (
Leung, I.W. and Lassam, N. (
Morrison, D.K., Heidecker, G., Rapp, U.R. and Copeland, T.D. (
Pawson, T. (
Hafner, S., Adler, H.S., Mishak, H., Janosch P., Heidecker G., Wolfman, A., Ueffing, M., Kolch, W. (
Dent, P., Reardon, D.B., Morrison, D.K. and Sturgill, T.W. (
Taira, T., Saito, Y., Niki, T., Iguchi-Ariga, S.M., Takahashi, K. and Ariga, H. (
Zhang, L., Shimoji, M., Thomas, B., Moore, D.J., Yu, S.W., Marupudi, N.I., Torp, R., Torgner, I.A., Ottersen, O.P., Dawson, T.M. et al. (
Moore, D.J., Zhang, L., Troncoso, J., Lee, M.K., Hattori, N., Mizuno, Y., Dawson, T.M. and Dawson, V.L. (
Valente, E.M., Abou-Sleiman, P.M., Caputo, V., Muqit, M.M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A.R., Healy, D.G. et al. (
Ross, O.A. and Farrer, M.J. (
Nolen, B., Taylor, S. and Ghosh, G. (
Dibb, N.J., Dilworth, S.M. and Mol, C.D. (
Chalandon, Y. and Schwaller, J. (
Ludwig, E., Silberstein, F.C., van Empel, J., Erfle, V., Neumann, M. and Brack-Werner, R. (
Neuhoff, V., Arold, N., Taube, D. and Ehrhardt, W. (
Shevchenko, A., Wilm, M., Vorm, O. and Mann, M. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}