





Case report
A 35-year-old man was diagnosed with dilated cardiomyopathy (DCM) and was subsequently admitted for the pre-transplant evaluation. Optimal medical management had failed to improve his impaired left ventricular function, and heart failure became medically intractable. There was no family history of congenital anomalies. Transthoracic echocardiogram revealed dilated left ventricle and severe left ventricular dysfunction (ejection fraction; 14.2%) (Panel A). Cardiopulmonary exercise testing displayed a significantly reduced VO2 peak of 13.3 mL/kg/min. No anomalies in cardiac chamber and vessel were observed using cardiac magnetic resonance imaging. Right ventricular biopsy indicated myocardial injury derived from cardiomyopathy, characterized by myocardial replacement fibrosis and fat infiltration (Panel B). Also, further evaluation by computed tomography demonstrated associated extracardiac abnormalities, polysplenia (Panels C and D), pancreatic tail hypoplasia (Panel E), inversion of superior mesenteric vessels (Panel F), and intestinal malrotation.
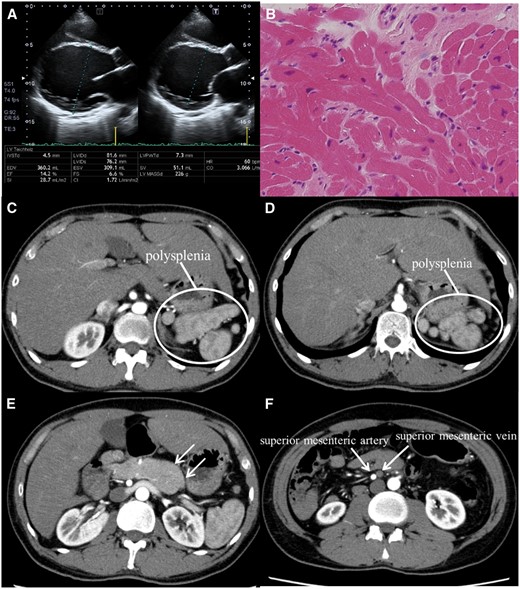
Polysplenia, one of the prominent features of heterotaxia, which is characterized by the features of left-sidedness including bilateral morphological left lung and interruption of the inferior vena cava with azygos continuation to the vein and cardiac anomaly. Although several associated genetic aberrations in ZIC3, EGF-CFC, and ACVR2B have been reported, the specific aetiology of heterotaxia remains unclear in most cases. Nevertheless, the high probability of cardiac anomalies with heterotaxia suggested the potential association of genes involved in body orientation and congenital cardiac abnormalities. To the best of our knowledge, this is the first report of the association of DCM with polysplenia and visceral heterotaxy. The DCM may also be one that these factors are similarly associated with.


{kind=link}