





Abstract
Although surfactant protein-D (SP-D) is a pneumoprotein that is predominantly synthesized by type II epithelial cells in the lung, individuals with increased circulating levels of SP-D are at an elevated risk of mortality from ischemic heart disease. Whether SP-D contributes directly to atherosclerosis is unknown. We determined the effects of SP-D gene deletion in a mouse model of atherosclerosis.
SP-D knockout (KO) mice were crossed with hyperlipidemic and atherosclerosis-prone apolipoprotein E (ApoE) KO mice to generate SP-D/ApoE double knockout (DKO) mice. Mice were placed on a high-fat diet for 12 weeks beginning at 8 weeks of age. Compared with ApoE KO mice, SP-D/ApoE DKO mice had significantly less atherosclerosis with reduced macrophage accumulation, decreased local macrophage proliferation, and increased smooth muscle cell coverage in plaques. Interestingly, SP-D deficiency worsened hypercholesterolemia and induced obesity and insulin resistance but suppressed plasma interleukin-6 (IL-6) levels. SP-D deficiency also reduced blood monocytes and neutrophils counts in ApoE KO mice.
SP-D deficiency reduces atherosclerosis in part by decreasing the accumulation and proliferation of macrophages and by reducing IL-6 levels systemically. SP-D is a promising therapeutic target for cachectic COPD patients with elevated circulating SP-D levels who are at increased risk of cardiovascular morbidity and mortality.
1. Introduction
Chronic obstructive pulmonary disease (COPD) is a strong independent risk factor for ischemic heart disease and cardiovascular (CV) mortality, independent of the effects of cigarette smoking.1 However, the mechanisms by which this occurs are largely unknown. One very interesting systemic biomarker of CV mortality is surfactant protein-D (SP-D). SP-D is detectable in plasma and circulating levels of SP-D are associated with increased risk of CV mortality, independent of other well-established risk factors such as age, sex, diabetes, plasma lipids, and C-reactive protein (CRP) in individuals with pre-existing heart disease and even those with COPD without overt heart disease.2 SP-D is secreted predominantly by alveolar type II epithelial cells3 and plays a critical role in pulmonary innate host defenses and in the regulation of surfactant homeostasis.4 Circulating levels of SP-D are increased in patients with COPD and genetic variations of SP-D have been associated with increased risk of COPD in various populations.5,6 More importantly, elevated SP-D levels in plasma have been associated with increased risk of exacerbations,7 rapid decline in lung density,8 and higher rates of mortality in patients with COPD.9 CV mortality is a leading cause of death in patients with COPD, especially those with mild to moderate grades of severity.10 It is not known, however, whether SP-D directly contributes to CV mortality (in COPD) or is just an epiphenomenon of smoking. To ascertain whether or not SP-D directly contributes to atherosclerosis, we generated SP-D/ApoE double knockout (DKO) mice by crossing SP-D knockout (KO) mice with apolipoprotein E (ApoE) KO mice and determined the effects of SP-D deficiency on plaque formation and the metabolic phenotypes related to obesity and atherosclerosis.
2. Methods
Further details are presented in the online Supplementary material.
2.1 Animals
The study utilized four strains of male mice including wild-type (WT, C57BL6/J; ApoE+/+, SP-D+/+; n = 16), SP-D KO (ApoE+/+, SP-D−/−; n = 16), ApoE KO (ApoE−/−, SP-D+/+; n = 18), and SP-D/ApoE DKO mice (ApoE−/−, SP-D−/−; n = 18). All mice were on a C57BL/6/J background. The WT and ApoE KO mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) while the SP-D KO mice were obtained from Dr. Sam Hawgood, who originally developed these mice.11 These animals were backcrossed at least 10 times to the C57BL6/J background and then bred to homozygosity. The SP-D KO mice were then crossed with the ApoE KO mice to generate DKO mice. All animal experiments were performed in conformity with NIH guidelines. This study was approved by the Animal Care Committee of the University of British Columbia (A10-0257 and A10-0310).
2.2 Study design, animal diet feeding, and preparation of tissues
The experimental schema is illustrated in Supplementary material online, Figure S1. Mice were divided into two batches and fed a high-fat diet (TD88137, Harlan, Supplementary material online, Table S1) for 12 weeks starting at 8 weeks of age. Mice were then sacrificed under anesthesia (isoflurane 2.5% inhalation), and the tissues were harvested carefully. Refer to the online Supplementary material for details.
2.3 Atherosclerotic lesion characterization
The brachiocephalic arteries were cross-sectioned in 5 µm thick slices starting from the proximal end of brachiocephalic arteries to the distal end of the bifurcation using Cryostat (Leica, UK). Five sections from each brachiocephalic artery (200 μm intervals from 0 to 800 μm distal to the aortic arch) were used for staining, and the results of each staining from these slides were averaged for each animal. H&E staining was performed for morphometric analysis. The necrotic core was defined as the component of the plaque that did not contain any matrix material (either collagen or elastin) or cells in H&E staining slide.12 Picrosirius red staining was performed for analysis of collagen content by measuring birefringence to plane-polarized light. Immunofluorescence staining was carried out using antibodies to CD68 (MCA1957, AbD Serotec), Ki67 (SP6, ab16667, Abcam), and alpha-smooth muscle actin (ab5694, Abcam). Immunoenzyme staining was carried out using antibodies to endothelial nitric oxide synthase (eNOS) (NB300-500, Novus Biologicals) and phospho-eNOS (SAB4300128, Sigma). Dihydroethidium (DHE) staining was performed for analysis of oxidative stress. See the online Supplementary material for details.
Aortic roots were sectioned into 10 μm slices, generating 20 sections that spanned the entire aortic root. Oil Red O staining was performed and the sections that captured the three maximal lesion area were used for quantitation. To determine atherosclerotic plaque coverage in the aortic intima, the entire aorta was dissected, pinned and then stained with Sudan IV.
2.4 Blood count and plasma analysis
Blood cell counts were determined using the Cell-Dyn 3700 hematology analyser (Abbott Park, USA). Plasma lipoproteins were analysed by high-performance liquid chromatography (HPLC) using molecular sieve columns (Skylight Biotech, Japan). Milliplex MAP Mouse Cytokine/Chemokine Magnetic Bead Panel (Millipore, USA) was used to analyse the levels of cytokines in plasma. This panel consisted of interleukin (IL)-1alpha, IL-1beta, IL-2, IL-6, IL-10, IL-12 (p40/p70), interferon gamma (IFN-gamma), interferon gamma-induced protein 10 (IP-10), tumor necrosis factor alpha (TNF-alpha), keratinocyte derived cytokine (KC), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1alpha), MIP-1beta, eotaxin, and regulated on activation, normal T-cell expressed and secreted (RANTES). Total cholesterol and glucose levels were quantified enzymatically. Plasma insulin, leptin, and adiponectin were measured with commercially available ELISA kits. High-sensitivity (hs)CRP and IL-9 levels were also measured in plasma from additional batch of mice. See the online Supplementary material for details.
2.5 BAL fluid analysis and morphometric measurements of airspace size
Milliplex MAP Mouse Cytokine/Chemokine Magnetic Bead Panel (Millipore) was used to analyse the levels of cytokines in cell-free BAL fluid. Macrophage count on cytospin prepared slides were recorded. For morphometric analysis, measurements of mean linear intercept (Lm) were performed using H&E stained lung sections. See the online Supplementary material for details.
2.6 Analysis of gene expression by real-time quantitative PCR
Gene expression in liver, spleen, aortic root, and retroperitoneal adipose tissues were evaluated using real-time quantitative PCR (CFX384 Touch Real-Time PCR Detection System, Bio-Rad). The relative amount of mRNA was calculated using ACTB (for aortic arch and spleen) or B2M (for adipose tissue and liver) mRNA as the invariant control. See the online Supplementary material and Supplementary material online, Table S2 for details.
2.7 Statistical analyses
All values were presented as mean ± SEM. Between-group comparisons were evaluated with a non-parametric test (Mann–Whitney U test for two group comparisons and Kruskal–Wallis test followed by Dunn’s test for multiple group comparisons). A two way ANOVA with Bonferroni’s multiple comparison test was used for the body weight analysis. Relations between parameters were evaluated by least-squares linear regression analysis. In all cytokine analyses, values below the limit of detection (3.2 pg/ml) were substituted by the lowest detectable value. A P value < 0.05 (two-tailed) was considered statistically significant. All statistical analyses were carried out using GraphPad Prism 6.
3. Results
3.1 SP-D deficiency in ApoE KO mice reduces atherosclerotic plaque size
Atherosclerotic plaque size was measured in the brachiocephalic arteries using H&E staining (Figure 1A and B). SP-D/ApoE DKO mice exhibited significantly reduced plaque size compared with ApoE KO mice (37% reduction, P = 0.043). Area within the internal elastic lamina was similar between the groups (Figure 1C). We also assessed plaque size at the aortic root and plaque coverage of the whole aorta. We found that SP-D deficiency in ApoE KO mice reduced plaque size in the aortic root (25% reduction, P = 0.037, Supplementary material online, Figure S2A) and coverage of the entire aorta though the latter was not statistically significant (23% reduction, P = 0.068, Supplementary material online, Figure S2B). WT and SP-D KO mice had few atherosclerotic plaques at the brachiocephalic artery, the aortic root, and the whole aorta (data not shown).
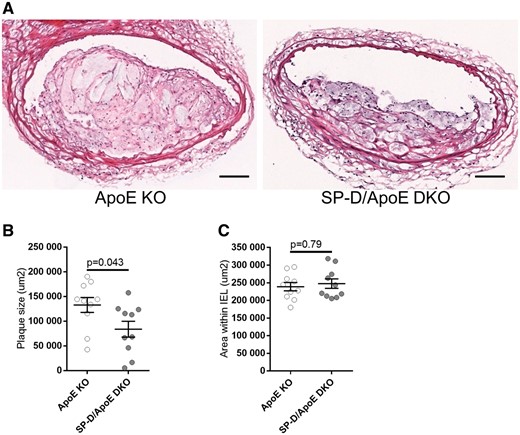
SP-D deficiency in ApoE KO mice reduces atherosclerotic plaque size in brachiocephalic arteries. (A) H&E staining of representative brachiocephalic arteries of ApoE KO and SP-D/ApoE DKO mice after 12 weeks of high-fat diet. Quantification of (B) atherosclerotic plaque size within the brachiocephalic artery, and (C) area within the internal elastic lamina (IEL) of the brachiocephalic artery. Five sections through the brachiocephalic artery (equally spaced at 200 um intervals) were evaluated, and the results were averaged for each animal. n = 10 for each group. Data represent mean ± SEM. P values were calculated using the Mann–Whitney U test. Scale bars: 100 μm.
3.2 SP-D deficiency reduces plaque macrophage accumulation and proliferation
We found that SP-D/ApoE DKO mice harboured significantly fewer macrophages in each plaque (46% reduction in CD68 positive area, P = 0.023, Figure 2A and C) compared with ApoE KO mice. Because there was good correlation between the area occupied by macrophages in the plaque and the total plaque size (R2 = 0.75, data not shown), there was no significant difference in the area of macrophages in the plaques between the two groups when total plaque size was taken into account (Figure 2D). More than 90% of Ki67 positive cells within plaques were CD68 positive (Figure 2B). SP-D/ApoE DKO mice had less CD68 cell proliferation in plaques compared with ApoE KO mice (68% reduction in the number of Ki67 positive cells per CD68 positive area, P = 0.011, Figure 2A and E). There was no significant difference in the number of proliferating cells within vessel media between groups (Figure 2F).
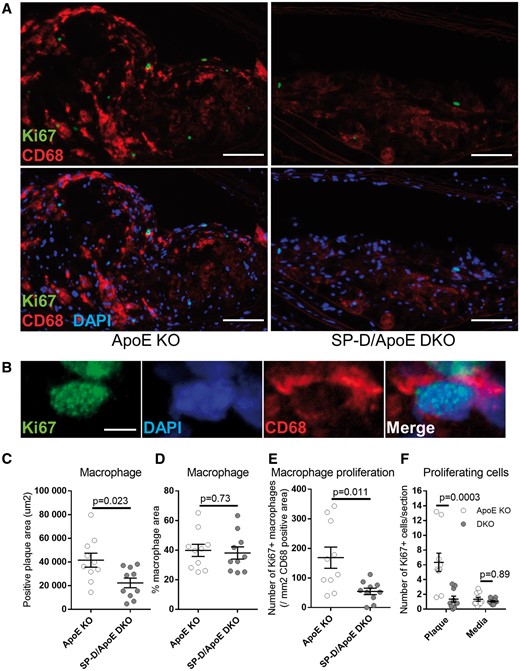
SP-D deficiency in ApoE KO mice reduces plaque macrophage accumulation and proliferation. (A) Representative images from brachiocephalic artery lesions of ApoE KO and SP-D/ApoE DKO mice with CD68 (red) and Ki67 (green) double immunostaining for detection of plaque macrophages and macrophage proliferation. Nuclei were stained with DAPI (blue). (B) Representative confocal images of proliferating macrophage stained with CD68 (red) and Ki67 (green) in ApoE KO mice brachiocephalic artery lesion. Quantification of (C) plaque macrophage area and (D) % macrophage area were based on CD68 staining, (E) proliferating macrophages was based on CD68 and Ki67 staining, and (F) proliferating cells within the plaque and vessel media was based on Ki67. n = 10 for each group. Data represent mean ± SEM. P values; Mann–Whitney U test. Scale bars: 100 μm (A); 4 μm (B).
3.3 SP-D deficiency increases plaque surface smooth muscle cell coverage
Although there was no significant difference in plaque smooth muscle cell (SMC) content based on alpha-smooth muscle actin staining, SP-D/ApoE DKO mice had more SMC coverage of the luminal surface of the plaque (Figure 3A, C and D), which is associated with a stable plaque phenotype in humans.13–15
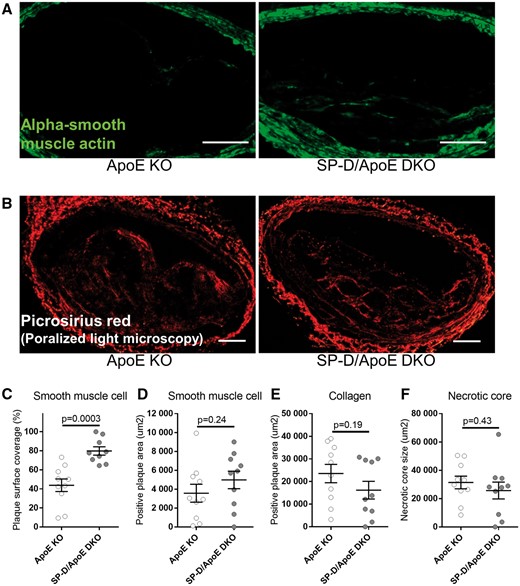
SP-D deficiency in ApoE KO mice increases plaque surface smooth muscle cell coverage. Representative images from brachiocephalic artery lesions of ApoE KO and SP-D/ApoE DKO mice with (A) alpha-smooth muscle actin (green) immunostaining for detecting smooth muscle cell (SMC) and (B) picrosirius red staining (polarized light microscopy) for collagen detection. Quantification of (C) plaque surface SMC coverage and (D) plaque SMC area were based on alpha-smooth muscle actin staining, (E) plaque collagen content was based on picrosirius red staining, and (F) necrotic core size was based on the component of the plaque that did not contain any matrix material or cells in H&E staining. n = 10 for each group. Data represent mean ± SEM. P values; Mann–Whitney U test. Scale bars: 100 μm.
Intraplaque collagen content and necrotic core size were similar between the groups (Figure 3B, E and F). eNOS production and reactive oxygen species production (DHE staining) were also similar between the groups (Supplementary material online, Figures S3 and S4). Phospho-eNOS was not visibly detectable in both ApoE KO and SP-D/ApoE DKO mice.
3.4 SP-D deficiency worsens hypercholesterolemia and induces obesity and insulin resistance in ApoE KO mice
To determine the effect of SP-D deficiency on lipoprotein profile of ApoE KO mice, we measured fasting plasma levels of total cholesterol and triglyceride by HPLC (Figure 4A and B). Total cholesterol levels were 39% higher in SP-D/ApoE DKO mice compared with ApoE KO mice (P < 0.0001). We confirmed these findings using an enzymatic colorimetric method (total cholesterol: 1114 ± 69 mg/dl in ApoE KO mice vs. 1518 ± 68 mg/dl in SP-D/ApoE DKO mice, P = 0.0005). SP-D/ApoE DKO mice also had elevated chylomicron cholesterol (CM-C) and very-low-density lipoprotein cholesterol (VLDL-C), and high-density lipoprotein cholesterol (HDL-C) levels. Low-density lipoprotein cholesterol (LDL-C) levels were unaffected. Triglyceride levels were also higher in SP-D/ApoE DKO mice (Figure 4B), reflecting higher levels of CM triglyceride, VLDL triglyceride, and LDL triglycerides (data not shown). In line with this unfavourable lipoprotein profile, SP-D/ApoE DKO mice had elevated body weight and fat tissue (gonadal, retroperitoneal, subcutaneous inguinal, and liver fat) compared with ApoE KO mice (Figure 5A and B). SP-D/ApoE DKO mice also had elevated fasting plasma insulin, glucose, leptin, and adiponectin levels (Figure 5C). We measured food intake in ApoE KO and SP-D/ApoE DKO mice at age 20 weeks. SP-D/ApoE DKO mice showed 47% decrease in food intake compared with ApoE KO mice (85.8 kcal/five mice/day in ApoE KO mice vs. 45.3 kcal/five mice/day in SP-D/ApoE DKO mice).
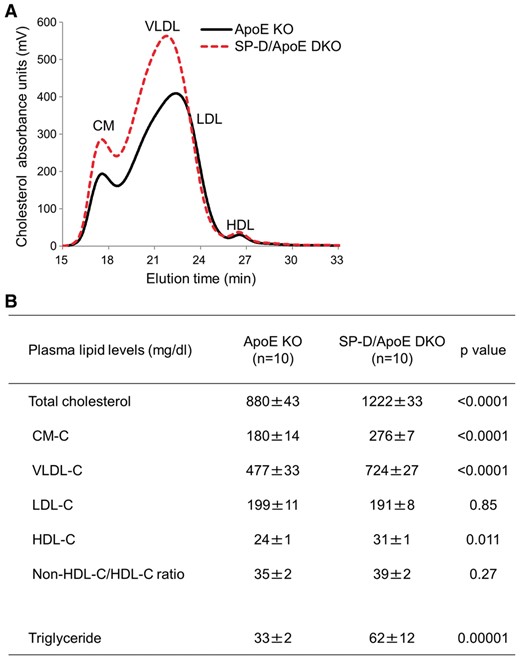
SP-D deficiency in ApoE KO mice increases fasting plasma total cholesterol. (A and B) Fasting plasma cholesterol and triglycerides analysis by HPLC. n = 10 for each group. Data represent mean ± SEM. P values; Mann–Whitney U test.
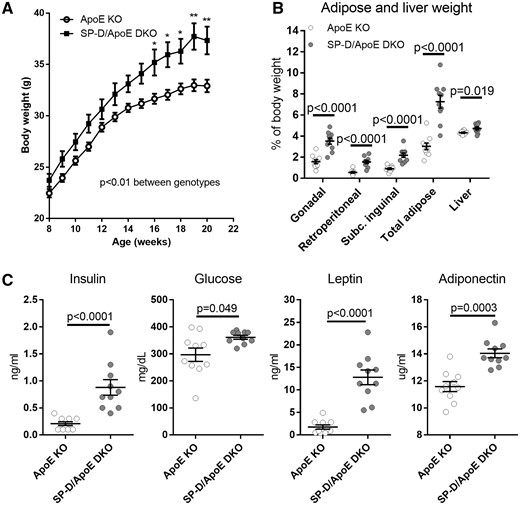
SP-D deficiency in ApoE KO mice increases body weight, adipose tissue weight, insulin, and leptin levels. (A) Body weight (n = 18 for each group, two way ANOVA with Bonferroni's multiple comparison test; *P < 0.05, **P < 0.01 vs. ApoE KO mice), (B) gonadal, retroperitoneal, subcutaneous inguinal, total adipose tissue, and liver weight (% body weight), and (C) insulin, leptin, glucose, and adiponectin levels in fasting plasma. n = 10 for each group, unless otherwise noted. Data represent mean ± SEM. P values; Mann–Whitney U test.
We also evaluated the effect of SP-D deficiency on lipoprotein profile of WT mice. SP-D KO mice had an unfavorable metabolic profile with an elevated body weight, increased retroperitoneal adipose tissue, and increased circulating levels of non-HDL-C (CM-C, VLDL-C, and LDL-C), fasting plasma insulin and leptin compared with WT mice (data not shown).
3.5 SP-D deficiency reduces plasma IL-6 in ApoE KO mice
We next evaluated the level of cytokines in plasma. Compared with WT mice, ApoE KO mice showed significantly elevated plasma IL-6 levels (Figure 6A), which is a pro-atherosclerotic cytokine, while other cytokine levels (IL-1alpha, IL-1beta, IL-2, IL-6, IL-10, IL-12 (p40/p70), IFN-gamma, IP-10, TNF-alpha, KC, MCP-1, MIP-1alpha, MIP-1beta, eotaxin, and RANTES) were similar (data not shown). Surprisingly, SP-D deficiency in ApoE KO mice suppressed plasma IL-6 levels to basal levels (95% reduction, P = 0.0013). We also measured IL-9 and hsCRP levels in plasma and the levels were not significantly different between the groups (Supplementary material online, Figure S5).
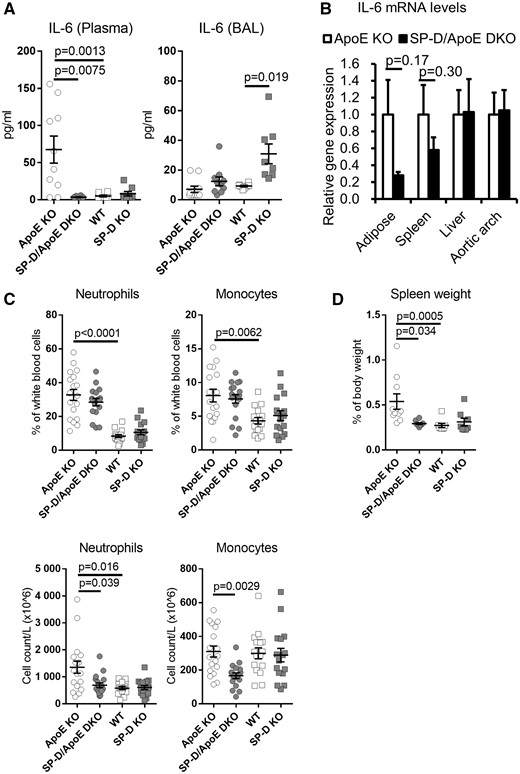
SP-D deficiency in ApoE KO mice decreases plasma IL-6 levels, blood monocyte and neutrophil counts, and spleen weight. (A) IL-6 levels in plasma and BAL. n = 8–10 for each group. (B) IL-6 mRNA expression in adipose tissue, spleen, liver, and aortic arch were quantified using real-time quantitative PCR. The expression of mRNA in SP-D/ApoE DKO mice was calculated relative to the housekeeping gene (B2M for adipose and liver, ACTB for spleen and aortic arch) and further normalized to the relative expression level in ApoE KO mice. n = 5 for each group. (C) Blood monocyte and neutrophil counts (% and absolute) by hematology analyser. n = 16–18 for each group. (D) Spleen weight. n = 8–10 for each group. Data represent mean ± SEM. P values were calculated using Mann–Whitney U test for two group comparisons, and Kruskal–Wallis test with Dunn’s multiple comparisons test for selected pairs (ApoE KO vs. WT, ApoE KO vs. SP-D/ApoE DKO, and WT vs. SP-D KO).
We then evaluated IL-6 mRNA expression levels in multiple organs (Figure 6B). SP-D/ApoE DKO mice showed reduced IL-6 expression levels in adipose tissue and spleen, but the relationship was not statistically significant. IL-6 expression levels in liver and aortic root were also similar between the groups. Similar to plasma levels, we observed no significant differences in MCP-1 and TNF-alpha gene expression levels in adipose tissue between ApoE KO mice and SP-D/ApoE DKO mice (data not shown).
3.6 SP-D deficiency leads to increased BAL fluid cytokine activation and accumulation of giant foamy macrophages in the lung
BAL cytokine levels were similar between ApoE KO mice and WT mice. Consistent with a previous study,16 SP-D deficient mice demonstrated increased BAL levels of IL-6, eotaxin, IP-10, MCP-1, MIP-1alpha, and MIP-1beta compared with WT mice and higher BAL levels of KC, eotaxin, IP-10, MCP-1, MIP-1alpha, and MIP-1beta compared with ApoE KO mice (Figure 6A and Supplementary material online, Figure S6). In contrast, SP-D deficient mice demonstrated reduced BAL levels of IL-2 compared with WT and ApoE KO mice. There were no cell count differences in BAL fluid between ApoE KO and SP-D/ApoE DKO mice (data not shown), but in keeping with previous studies in WT mice,11,17 SP-D/ApoE DKO mice demonstrated giant foamy alveolar macrophages in lung tissue (Supplementary material online, Figure S7A–C). There was no significant difference in Lm between ApoE KO and SP-D/ApoE DKO mice (Supplementary material online, Figure S7D).
3.7 SP-D deficiency reduces blood neutrophil and monocyte counts in ApoE KO mice
We evaluated the effect of SP-D deficiency on blood cell counts (Figure 6C). In line with previous studies,18,19 ApoE KO mice fed with a high-fat diet showed neutrophilia (% neutrophil: 33 ± 3% in ApoE KO mice vs. 8.3 ± 0.9% in WT mice, P < 0.0001) and monocytosis (% monocyte: 9.4 ± 1.7% in ApoE KO mice vs. 4.3 ± 0.5% in WT mice, P = 0.0062). Although there were no significant differences in % neutrophils or % monocytes, SP-D deficiency in ApoE KO mice showed significantly reduced absolute blood neutrophil counts (49% reduction, P = 0.039) and monocyte counts (46% reduction, P = 0.0029). Absolute blood lymphocyte counts were similar between ApoE KO mice and SP-D/ApoE DKO mice (data not shown).
We next evaluated the weight of the spleen (Figure 6D), which is a major reservoir for neutrophils and monocytes.20 Consistent with previous studies,21,22 ApoE KO mice showed splenomegaly (spleen per body weight: 0.54 ± 0.09% in ApoE KO mice vs. 0.29 ± 0.01% in WT mice, P = 0.0005). SP-D deficiency in ApoE KO mice significantly reduced spleen weight (% of body weight) to basal levels (46% reduction, P = 0.034).
3.8 SP-D deficiency does not affect expression of genes associated with endothelial activation, inflammation, or apoptosis in aortic arch
To determine whether or not SP-D deficiency modifies the expression levels of genes relevant to atherosclerosis, we measured the mRNA levels of a select number of genes in the aortic arch (Supplementary material online, Figure S8). There were no significant differences in the expression of genes related to endothelial activation (eNOS, ICAM1, and VCAM1), inflammation (IL-6, iNOS, TNF-α, MCP-1, and IL-10), apoptosis (Bax, Bcl2), cholesterol efflux (ABCA1), or scavenger receptors (Lox1, SRA1, and CD36) between the groups.
4. Discussion
This study describes the atherosclerotic, metabolic, and inflammatory phenotype of SP-D/ApoE DKO mice. SP-D deficiency in ApoE KO mice attenuates atherosclerosis, reduces macrophage accumulation and proliferation and increases SMC coverage in plaques, promoting their stability. Paradoxically, however, SP-D deficiency induces obesity and insulin resistance in ApoE KO mice, but suppresses systemic levels of plasma IL-6, a major adipokine and a pro-atherosclerotic cytokine. The reduction in systemic IL-6 levels, in turn, was associated with a significant decrease in circulating monocytes and neutrophils, which are independent risk factors for coronary artery disease in humans.23–25
In a large epidemiological study, we previously found that circulating SP-D levels were significantly related to CV mortality, independent of established risk factors such as obesity, diabetes, hyperlipidemia and CRP.2 To determine whether SP-D was directly involved in atherosclerosis and to resolve the role of SP-D in COPD-related CV comorbidities, we created a new murine model, SP-D/ApoE DKO mice, and evaluated plaque progression. SP-D deficiency reduced atherosclerotic plaque size in brachiocephalic artery and aortic root in ApoE KO mice. Because a previous study showed that in ApoE KO mice, brachiocephalic arterial plaques demonstrated features most similar to vulnerable plaques found in human atherosclerosis,26 we used this vessel for our primary plaque assessment.
4.1 SP-D deficiency and metabolic profile
Consistent with previous studies in humans,27,28 we found that SP-D deficiency in ApoE KO mice induced obesity and insulin resistance. SP-D deficiency in ApoE KO mice was also associated with increased total cholesterol levels, especially VLDL-C levels. The mechanism by which SP-D deficiency induces this phenotype is largely unknown. A recent study demonstrated that SP-D deficiency caused hyperphagia in young mice (7–17 weeks), but in the more aged mice (18–25 weeks), the energy intake was nearly identical.29 We measured food intake at 20 weeks of age in ApoE KO and SP-D/ApoE DKO. Surprisingly, food intake was lower in SP-D/ApoE DKO mice compared with ApoE KO mice. Although the mechanism for this observation is unclear, we found that SP-D/ApoE DKO mice had plasma leptin levels that were on average 7.3-fold higher than that in WT mice. It is well established that leptin inhibits food intake by suppressing the appetite control centres of the brain.30 SP-D KO mice also had significantly higher plasma leptin compared with WT mice, but the difference was small (1.2-fold increase, P = 0.010). It has also been shown that human adipose tissue and adipocytes express SP-D, and that this expression is decreased in obese subjects compared with non-obese subjects.31 SP-D is known to bind fatty acids and may modify the cellular uptake and catabolism of lipids.32 We also showed that SP-D KO mice had elevated LDL-C and VLDL-C levels, while HDL-C levels were similar. These data are similar to those previously published by Sorensen et al., who showed SP-D deficiency increases HDL-C in WT female mice, but not in male mice.33 It is possible that female sex hormones modify the role of SP-D in cholesterol metabolism.34
4.2 SP-D deficiency and IL-6
Despite the unfavourable metabolic profile of SP-D deficient mice, these mice harboured smaller atherosclerotic lesions most likely because of reduced inflammation. Inflammation is recognized to play a major role in all stages of atherogenesis.35 We showed that SP-D deficiency suppresses plasma IL-6 levels in ApoE KO mice. IL-6 is an upstream inflammatory cytokine that plays a critical role in orchestrating the downstream inflammatory response related to atherosclerosis.36 Not surprisingly given this central function of IL-6, circulating plasma IL-6 levels are strong predictors of both total and CV mortality, independent of the traditional risk factors for atherosclerosis.37 In mice, administration of exogenous IL-6 significantly accelerates atherosclerosis; whereas deficiency reduces it.38 How SP-D deficiency in ApoE KO mice reduced plasma IL-6 remains a mystery. IL-6 is secreted from a wide variety of cell types and is strongly induced in multiple organs and tissue (heart, aorta, lung, liver, kidney, muscle, gastrointestinal tract spleen, bone marrow derived inflammatory cells, and adipose tissue) under pro-inflammatory conditions.39 We evaluated IL-6 mRNA expression levels in multiple organs in our mice. SP-D/ApoE DKO mice showed reduced IL-6 expression levels in adipose tissue and spleen, albeit not significantly, compared with ApoE KO mice. Adipose tissue can be considered the largest organ in the body and is one of the major source of plasma IL-6 in obese and healthy subjects.40,41 We also found that SP-D deficiency in ApoE KO mice reduced both splenic IL-6 expression (though not significantly) and spleen weight. It is thus possible that SP-D deficiency may have reduced IL-6 derived from spleen. Other potential sources of IL-6 include muscle, lung, liver, and kidneys. Additional experiments will be needed to determine how (and if) SP-D regulates IL-6 expression and activation in these organs and its effect on atherogenesis.
4.3 SP-D deficiency and macrophage/monocyte/neutrophil
We also found that SP-D deficiency in ApoE KO mice modulated macrophages and monocytes, which are another major source of IL-6. In the double KO mice, there was reduced macrophage accumulation and proliferation in plaques, in addition to decreased circulating monocytes in the systemic circulation. We also found that these mice had reduced circulating levels of neutrophils in these mice, which may have contributed to their lower atherosclerotic burden.19,25,42 The mechanism by which this occurred is uncertain. However, since IL-6 regulates the maturation and release of hematopoietic cells, it is tempting to speculate that the reduced IL-6 expression related to the SP-D deficiency may have been responsible for this phenotype. Additional studies will be needed to validate this hypothesis.
4.4 SP-D deficiency and SMC
Recent paper showed that about 80% of SMCs in ApoE KO mouse lesions have no SMC marker expression like alpha-smooth muscle actin.43 Cholesterol loading causes loss of SMC markers and increases macrophage marker expression by vascular SMCs.44 We showed that SP-D/ApoE DKO mice have more SMCs, which retain alpha-smooth muscle actin staining, in the fibrous cap. This result suggests increased plaque stability and less de-differentiation of the SMCs due to less lipid-loading in SP-D/ApoE DKO mice.
4.5 Structure and function of SP-D
SP-D acts as dual function surveillance molecule to either suppress or enhance inflammation depending on the environmental milieu and its structure (Figure 7). SP-D is composed of two regions, a globular head and a collagenous tail domain.45,46 Previous studies have shown that SP-D’s pro-inflammatory functions are mediated by the tail domain which binds to CD91 and calreticulin and causes the activation of NF-kB. Its anti-inflammatory actions depend on the head domain, which binds to SIRP-1a and blocks NF-kB activation. The globular head also binds directly to pathogen-associated molecular patterns and induces opsonization and clearance of pathogens from lungs.45,46 In steady state, SP-D exists largely as a dodecamer, which exposes the globular head to the external environment and hides the tail domain. However, with oxidative stress (which occurs with smoking, or microbial insult or air pollution in lungs), the dodecameric form of SP-D breaks apart into trimers and even monomers, which causes the tails to become exposed to the external milieu. In healthy humans, the dodecamer:trimer ratio in the circulation is 1:1.6 but this ratio can decrease several-fold with an inflammatory insult.47
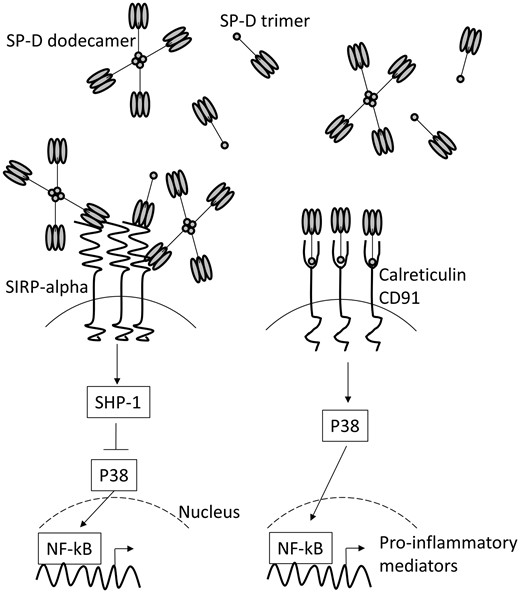
SP-D acts as dual function surveillance molecule to suppress or enhance inflammation. SP-D is composed of two regions, a globular head domain and a collagenous tail domain. It has been suggested that pro-inflammatory functions are mediated by the tail domains, via binding to CD91 and calreticulin and anti-inflammatory by the head domains, via binding to SIRP-alpha. The tail domains of SP-D dodecamers are hidden in the cruciform structure. Once the dodecamers break apart into the trimers, the tail domains become exposed.
4.6 SP-D deficiency and lung
As noted above, in the lungs, SP-D exists predominantly as a dodecamer and functions as an anti-inflammatory and anti-microbial molecule.45,46 It also acts to efficiently process all the other surfactant proteins and lipids in the lungs. Consistent with this concept, previous studies have shown that lungs of SP-D deficient mice exhibit a pro-inflammatory phenotype and demonstrate abnormal accumulation of apoptotic macrophages and giant foamy macrophages.11,16,17 Although these mice survive into adulthood, they spontaneously develop emphysematous changes in lungs. In keeping with these studies, we found that SP-D deficiency in ApoE KO mice was associated with increased levels of pro-inflammatory cytokines in BAL fluid and accumulation of giant foamy alveolar macrophages in their lungs. We did not find significant changes in Lm, which is a surrogate for emphysematous destruction, between SP-D deficient and non-deficient mice at 20 weeks of age. It is possible that significant emphysema may not be visually apparent until much later in life.
4.7 Preliminary in vitro study
To investigate whether SP-D directly modified macrophage proliferation, we conducted a preliminary in vitro study using RAW264.7 macrophages (Supplementary material online, File and Supplementary material online, Figure S9). These macrophages were treated for 24 h with recombinant human (rh)SP-D or nitrosylated SP-D (S-nitrosothiol (SNO)-rhSP-D). The latter was done because S-nitrosylation is one of the principal mechanism by which dodecameric SP-D becomes trimers in vivo.46 In our preliminary analysis, we did not find any significant impact of rhSP-D or SNO-rhSP-D on RAW264.7 macrophage proliferation. Additional work will be needed to determine the pathways by which SP-D modulates macrophage proliferation in plaques.
4.8 Limitations of the study
There were certain important limitations to this study. Firstly, although we found that SP-D deficiency was associated with reduced atherosclerosis in ApoE KO mice, we could not elucidate the exact mechanism by which SP-D deficiency mitigated atherosclerosis. These mice demonstrated strikingly lower circulating levels of IL-6 and reduced levels of blood neutrophils and monocytes compared with ApoE KO mice raising the possibility that the downregulation of the IL-6 and neutrophil/monocyte pathways may be responsible for the reduced atherosclerosis in these mice. Additional studies will be needed to reveal the precise mechanism involved in this process. Another limitation was that we could not determine which isoform of SP-D was responsible for the atherosclerotic phenotype. As discussed previously, SP-D in vivo can exist as monomer, trimer, dodecamer, or even a more complex structure. Its structure in the peripheral circulation remains mostly a mystery. It is plausible that post-translational modification may modify the atherogenic potential of SP-D in extrapulmonary vasculature. Additional work will be needed to sort this out. Thirdly, while SP-D is predominantly synthesized by type II alveolar cells and non-ciliated bronchiolar cells in lungs, SP-D expression can be found in extrapulmonary tissues and organs (salivary gland, lacrimal gland, ovary, uterus, oesophagus, stomach, testes, thyroid, and heart).48 Thus, it is possible that extrapulmonary production of SP-D may have contributed to the pro-atherogenic phenotype observed in the single ApoE KO mice. However, SP-D production (at the protein level) from extrapulmonary sources is very small (in both mice and humans),48–50 reducing the likelihood of this possibility.
4.9 Summary
We showed that SP-D deficiency in ApoE KO mice decreases atherosclerosis progression despite inducing obesity and insulin resistance. Although the exact mechanisms for this observation remain elusive, we showed that SP-D deficiency reduces macrophage accumulation and proliferation in plaques, suppresses plasma IL-6 levels and prevents monocytosis and neutrophilia in ApoE KO mice. These results suggest that SP-D plays a direct role in atherosclerosis possibly by modulating IL-6 related pathways. Thus, SP-D may be an important therapeutic target in cachectic COPD patients who have increased circulating levels of SP-D and are at increased risk of CV morbidity and mortality.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Acknowledgements
We thank Claire Smits and her team for assistance in animal experiments, Amrit Samra for assistance in histological staining, and Horacio Bach for preparing SNO-rhSP-D (University of British Columbia).
Conflict of interest: none declared.
Funding
This work was supported by grants from Canada Research Chair and the Canadian Institutes for Health Research.
References
Author notes
Time for primary review: 35 days

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}