





Abstract
Glutathione S-transferases (GSTs) play an essential role in the elimination of xenobiotic-derived electrophilic metabolites and also catalyze certain steps in the conversion of endogenous molecules. Their expression is controlled by different transcription factors, such as the antioxidant-activated Nrf2 or the constitutive androstane receptor. Here, we show that the Wnt/β-catenin pathway is also involved in the transcriptional regulation of GSTs: GSTm2, GSTm3, and GSTm6 are overexpressed in mouse hepatomas with activating Ctnnb1 (encoding β-catenin) mutations and in transgenic hepatocytes expressing activated β-catenin. Inversely, GSTm expression is reduced in mice with hepatocyte-specific knock out of Ctnnb1. Activation of β-catenin–dependent signaling stimulates GSTm expression in vitro. Activation of β-catenin in mouse hepatoma cells activates GSTm3 promoter–driven reporter activity, independently of β-catenin/T-cell factor sites, via a retinoid X receptor–binding site. By contrast, GSTm expression is inhibited upon Ras activation in mouse liver tumors and transgenic hepatocytes. Recent studies by different groups have shown that β-catenin–dependent signaling is involved in the transcriptional control of “perivenous” expression of various cytochrome P450s in mouse liver, whereas Ras signaling was hypothesized to antagonize the perivenous hepatocyte phenotype. In synopsis with our present results, it now appears that the Wnt/β-catenin pathway functions as a master regulator of the expression of both phase I and phase II drug-metabolizing enzymes in perivenous hepatocytes from mouse liver.
In hepatic biotransformation, enzymes of the so-called phase II of xenobiotic metabolism prepare metabolites of xenobiotic compounds for excretion via catalyzing their conjugation to endogenous substrates. Among phase II, enzymes of the glutathione S-transferase (GST) family play an essential role in the elimination of electrophilic xenobiotic metabolites. Members of the GST family are also involved in the conversion of endogenous molecules, e.g., in the degradation of aromatic amino acids, in steroid hormone biosynthesis, and in eicosanoid metabolism; for review, see Hayes et al., (2005).
As GSTs play a decisive role in the defense against electrophilic compounds and oxidative stress, it is not surprising that basal and inducible transcription of various GSTs, particularly of members of the GSTa and GSTm subfamilies, is under control of the antioxidant-activated transcription factor Nrf2 (nuclear factor erythroid 2–related factor 2) (Chanas et al., 2002; Hayes et al., 2000; Itoh et al., 1997). In account of their role in the detoxification of possible carcinogens, polymorphisms of different GST genes have been suspected not only to influence cancer susceptibility and prognosis but also to affect the response to anticancer drugs; for review, see McIlwain et al., (2006). In addition to antioxidants, several xenobiotic compounds have been identified to act on GST expression, such as the model inducers 3-methylcholanthrene and phenobarbital (PB), indicating a regulation of the respective GST isoforms by the nuclear receptors aryl hydrocarbon receptor (AhR) and constitutive androstane receptor (CAR) (Dwivedi et al., 1993; Langouet et al., 1996). Other nuclear receptors, i.e., the retinoid X receptor (RXR) α, the pregnane X receptor, and the peroxisome proliferator–activated receptor γ, have also been linked to the regulation of GST expression in liver (Dai et al., 2005; Park et al., 2004; Wu et al., 2004).
Altered expression of several GSTs has been observed in human as well as in rodent hepatocellular carcinoma (Grasl-Kraupp et al., 1993; Murray et al., 1993; Stahl et al., 2005). Interestingly, the same GSTm isoforms that were upregulated in a microarray analysis of mouse hepatomas carrying activating mutations in the Ctnnb1 (encoding β-catenin) proto-oncogene also exhibit significant differences in zone-specific expression in liver, being preferentially expressed in the “perivenous” areas of the liver lobule (Braeuning et al., 2006, 2007a; Hailfinger et al., 2006; Stahl et al., 2005). As several lines of evidence have demonstrated an essential role of β-catenin–dependent signal transduction in the control of the perivenous hepatocyte gene expression profile (Benhamouche et al., 2006, 2007a; Hailfinger et al., 2006; Sekine et al., 2006), these GSTm enzymes are therefore likely to be subject to a direct or indirect regulation by the β-catenin signaling pathway.
In the absence of appropriate agonists, cytosolic β-catenin is rapidly degraded in the proteasome after phosphorylation of several serine and threonine residues in its N-terminal domain by a cytosolic multiprotein complex containing, among others, glycogen synthase kinase (GSK) 3β. The β-catenin signaling pathway is physiologically stimulated by Wnt molecules binding to so-called Frizzled receptors on the cell surface. Upon Wnt binding, the β-catenin destruction complex is destabilized; β-catenin is no more degraded and can enter the nucleus, where it interacts with T-cell factor (TCF) family transcription factors (Behrens et al., 1996) and a variety of nuclear receptors (Mulholland et al., 2005). For review of this signaling pathway, see Huelsken and Behrens (2000).
According to a hypothesis developed in our laboratory, signaling through the Ha-ras/Raf/ extracellular signal-regulated kinase (ERK) cascade antagonizes the perivenous gene expression profile mediated by β-catenin (Braeuning et al., 2007a,b; Hailfinger et al., 2006). In support of this hypothesis, recently performed microarray analyses revealed a downregulation of GSTm isoforms in mouse liver tumors with activating mutations in Ha-ras or B-Raf (Jaworski et al., 2007; Stahl et al., 2005).
In this work, the role of β-catenin and Ha-ras in the regulation of GSTm expression in mouse liver is analyzed using both in vivo and in vitro approaches.
MATERIALS AND METHODS
Tissue samples.
Chemically induced mouse liver tumors were available to us from an earlier experiment conducted in our laboratory. In brief, male C3H/HeN mice received a single injection of 90 μg/g body weight N-nitrosodiethylamine. Thereafter, one group of animals were kept on a diet containing the tumor promoter PB, while the second group received control diet. Tumors had been previously characterized in terms of mutations in the proto-oncogenes Ha-ras and Ctnnb1 (Aydinlik et al., 2001). Additionally, paraffin-embedded liver samples were available to us from an analogous tumor promotion experiment in which PB treatment had been stopped 3 weeks before killing the animals (Bursch et al., 2005).
Transgenic mice (Tg(lox(pA)βCatS33Y)) with liver-specific expression of a point-mutated (S33Y) constitutively activated version of β-catenin were used (Braeuning and Schwarz, 2009; Schreiber et al., unpublished data). Mice were sacrificed at 8 weeks of age.
Transgenic Tglox(pA)H-ras* mice expressing a mutated Ha-ras gene were generated as previously described (Harada et al., 2004). Livers were isolated at 11 weeks of age and 4 weeks after adenoviral cre infection (1 × 108 plaque-forming units/mouse).
Mice with hepatocyte-specific knock out of β-catenin were obtained by interbreeding transgenic mice with loxP sites inserted in the introns flanking exons 3 and 6 of the Ctnnb1 gene (Huelsken et al., 2001), with Alb-Cre mice expressing Cre recombinase under control of the albumin promoter (Jackson laboratory, Bar Harbor, ME). These mice are also referred to as “Ctnnb1 knockout mice” in the following text. Mice were sacrificed at 8 weeks of age.
All animals received humane care and protocols compiled with institutional guidelines.
Immunohistochemical staining.
Immunostaining was performed using standard protocols as previously described (Hailfinger et al., 2006). In brief, 5-μm thick sections of paraffin-embedded tissue samples or 10-μm frozen sections were stained using antibodies against glutamine synthetase (GS; 1:1000 dilution; Sigma, Taufkirchen, Germany), E-cadherin (1:25; Cell Signaling, Danvers, MA), GSTm (1:500; gift of Dr R. Wolf, Biochemical Research Centre, University of Dundee, Dundee, UK), and a horseradish peroxidase–conjugated anti-rabbit secondary antibody (1:20; Dako, Glostrup, Denmark) with 3-amino-9-ethylcarbazole/H2O2 as substrates. Phosphorylated ERK was detected using a phospho-ERK1/2–specific antibody (1:1000; Cell Signaling) in combination with a biotinylated secondary antibody (1:200; Spa, Milan, Italy) and streptavidin-conjugated alkaline phosphatase with FastRed (Kementec, Copenhagen, Denmark) as substrate. Nuclei were counterstained by hematoxylin.
Isolation and cultivation of hepatocytes.
Hepatocytes were isolated by collagenase perfusion as previously described (Hailfinger et al., 2006) and cultivated in Dulbecco's Modified Eagle Medium (DMEM)/F-12 medium containing 100 U/ml penicillin and 100 μg/ml streptomycin. To induce β-catenin signaling, cells were incubated in medium conditioned by a stably Wnt3a-transfected 3T3 fibroblast cell line (gift of Dr R. Kemler, Max-Planck-Institute of Immunobiology, Freiburg, Germany) for 72 h. Medium conditioned by untransfected 3T3 cells was used as a control. Culture medium was changed daily. Perivenous and “periportal” hepatocyte fractions were isolated by combined digitonin/collagenase perfusion as recently described (Braeuning et al., 2006; Hailfinger et al., 2006).
Western blotting.
Whole cellular extracts were separated by SDS-PAGE (50 μg/lane) and transferred to PVDF membranes. Proteins were detected using antibodies described above against β-catenin (1:500; BD, Heidelberg, Germany), GS (1:5000) and GSTm (1:1000), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000; Chemicon, Chandler’s Ford, Hampshire, UK), and an alkaline phosphatase–conjugated secondary antibody (1:10,000; Tropix, Darmstadt, Germany) with CDP-star as a substrate. Chemoluminescence signals were monitored by use of a charged-coupled device camera system. GAPDH was used as a loading control.
Immunoprecipitation.
Whole cellular extracts were adjusted to 5 mg/ml protein concentration and incubated overnight with protein G–conjugated agarose beads (Roche, Mannheim, Germany) at 4°C to eliminate unspecific binding. Three hundred microliters of supernatant was incubated with 2 μg anti-RXR antibody (Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C for 3 h; afterward, 30 μl of protein G-agarose beads was added, followed by overnight incubation at 4°C. Beads were washed thrice with buffer, resuspended in 30 μl Lämmli buffer, and heated to 95°C for 10 min to release bound protein. Supernatant was used for Western blotting as described above.
Quantitative determination of messenger RNAs by RT-PCR.
Total RNA was isolated using Trizol (Invitrogen, Karlsruhe, Germany) followed by removal of residual DNA by DNAse I digestion (Roche). RNA was reverse transcribed by avian myeloblastosis virus reverse transcriptase (Peqlab, Erlangen, Germany). Expression analysis was performed using the LightCycler real-time PCR system (Roche). The 18s ribosomal RNA (rRNA) expression was used for normalization. Relative expression ratios of the target genes in the samples were computed based on the crossing point difference of a sample versus a control and its real-time PCR efficiency according to Pfaffl (2001). PCR efficiencies were as follows: GSTm2, 1.89; GSTm3, 1.92; GSTm6, 1.91; Axin2, 1.80; CAR, 1.96; Gpr49, 1.74; RXRα, 1.81; and 18s rRNA, 1.95. The primer pairs used for PCR amplification are given in Table 1.
Real-Time RT-PCR Primers Used in This Study
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
| GSTm2 | TGGAACCCAAAGTAGGATTACAAA | TGAGGACCAAGGCAGCACAC |
| GSTm3 | TGCACTGTGGCTCCCGGT | AGGCCTGGGGCAGCTCC |
| GSTm6 | TGCATGGAGTGCCGGTGT | CTGACATTGGAGGCTTGAGTGA |
| Axin2 | CGACGCACTGACCGACGATT | TCCAGACTATGGCGGCTTTCC |
| CAR | AACAACAGTCTCGGCTCCAAA | AGCATTTCATTGCCACTCCC |
| Gpr49 | AATCGCGGTAGTGGACATTC | GATTCGGAAGCAAAAATGGA |
| RXRα | ACATTGGGCTTCGGGACTGG | CTTGTTGTCTCGGCAGGTGTAGGT |
| 18s rRNA | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
| GSTm2 | TGGAACCCAAAGTAGGATTACAAA | TGAGGACCAAGGCAGCACAC |
| GSTm3 | TGCACTGTGGCTCCCGGT | AGGCCTGGGGCAGCTCC |
| GSTm6 | TGCATGGAGTGCCGGTGT | CTGACATTGGAGGCTTGAGTGA |
| Axin2 | CGACGCACTGACCGACGATT | TCCAGACTATGGCGGCTTTCC |
| CAR | AACAACAGTCTCGGCTCCAAA | AGCATTTCATTGCCACTCCC |
| Gpr49 | AATCGCGGTAGTGGACATTC | GATTCGGAAGCAAAAATGGA |
| RXRα | ACATTGGGCTTCGGGACTGG | CTTGTTGTCTCGGCAGGTGTAGGT |
| 18s rRNA | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
Real-Time RT-PCR Primers Used in This Study
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
| GSTm2 | TGGAACCCAAAGTAGGATTACAAA | TGAGGACCAAGGCAGCACAC |
| GSTm3 | TGCACTGTGGCTCCCGGT | AGGCCTGGGGCAGCTCC |
| GSTm6 | TGCATGGAGTGCCGGTGT | CTGACATTGGAGGCTTGAGTGA |
| Axin2 | CGACGCACTGACCGACGATT | TCCAGACTATGGCGGCTTTCC |
| CAR | AACAACAGTCTCGGCTCCAAA | AGCATTTCATTGCCACTCCC |
| Gpr49 | AATCGCGGTAGTGGACATTC | GATTCGGAAGCAAAAATGGA |
| RXRα | ACATTGGGCTTCGGGACTGG | CTTGTTGTCTCGGCAGGTGTAGGT |
| 18s rRNA | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
| GSTm2 | TGGAACCCAAAGTAGGATTACAAA | TGAGGACCAAGGCAGCACAC |
| GSTm3 | TGCACTGTGGCTCCCGGT | AGGCCTGGGGCAGCTCC |
| GSTm6 | TGCATGGAGTGCCGGTGT | CTGACATTGGAGGCTTGAGTGA |
| Axin2 | CGACGCACTGACCGACGATT | TCCAGACTATGGCGGCTTTCC |
| CAR | AACAACAGTCTCGGCTCCAAA | AGCATTTCATTGCCACTCCC |
| Gpr49 | AATCGCGGTAGTGGACATTC | GATTCGGAAGCAAAAATGGA |
| RXRα | ACATTGGGCTTCGGGACTGG | CTTGTTGTCTCGGCAGGTGTAGGT |
| 18s rRNA | CGGCTACCACATCCAAGGAA | GCTGGAATTACCGCGGCT |
Vector construction.
Two fragments of the 5′-upstream promoter region of mouse GSTm3, 1.0 and 4.9 kb in size, were amplified from genomic DNA using AccuTaq polymerase (Sigma) and the primer pairs 5′-CACCCTTGTGAAGGTTGGACTACAGGGCATA-3′ (1 kb_sense) and 5′-GCCTCGAGTTCTCAGACTGGCTTCAGGTAGGTTGAGC-3′ (antisense) and 5′-CACCGATTGGACAAATTGTGCAGGATCGCT-3′ (5 kb_sense) in combination with the reverse primer mentioned above. Amplicons were cloned into the luciferase reporter vector pT81luc by use of the Gateway conversion system (Invitrogen) following the manufacturer’s instructions to give pT81luc/GSTm3_1kb and pT81luc/GSTm3_5kb, respectively. Orientation and integrity of the inserts were controlled by dideoxy sequencing.
Deletion constructs.
Deletion mutants of the pT81luc/GSTm3_1kb construct (1) containing 0.2 kb of the 3′-terminus, (2) lacking the middle 0.6 kb, and (3) containing 0.3 kb of the 5′-terminus of the mGSTm3 promoter insert were created by BamHI and KpnI digestion followed by subsequent re-ligation of the truncated plasmids. Plasmids were denominated (1) pT81luc/GSTm3_1kb_3′, (2) pT81luc/GSTm3_1kb_mid, and (3) pT81luc/GSTm3_1kb_5′, respectively. Integrity of the inserts was verified by dideoxy sequencing. For an illustration of mGSTm3 promoter constructs used in this study, see Figure 6A.
Site-directed mutagenesis.
In silico analysis (MatInspector software; Genomatix, Munich, Germany) of the 1.0-kb GSTm3 promoter region contained in the plasmid pT81luc/GSTm3_1kb indicated the presence of three putative β-catenin/TCF-binding sites, termed A, B, and C. These sites were disabled by site-directed mutagenesis using the QuikChange Multi site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions, resulting in the plasmid pT81luc/GSTm3_1kb_ABC. Within the pT81luc/GSTm3_1kb_3′ deletion construct, additional mutations were introduced by use of the QuikChange II site-directed mutagenesis kit (Stratagene). Resulting plasmids were termed pT81luc/GSTm3_1kb_3′_A41G, pT81luc/GSTm3_1kb_3′_G114A, and pT81luc/GSTm3_1kb_3′_C192A according to the position denotation of the mutated base pairs. Mutagenic primers are listed in Table 2. For an illustration of mutant mGSTm3 promoter constructs used in this study, see Figure 6A. Correct insertion of all mutations was controlled by dideoxy sequencing.
Mutagenic Primers Used in This Study
| Name (mutation) | Sequence (5′–3′) |
| A(t274g)_se | CACACACCAGCTGTGCATTCTGTGCTCCCTTGC |
| B(a891g)_se | CCAAAGGGCTTCGGGTTCAGAGTCTCTGAGGAC |
| C(t986g)_se | CCCACCCACCTTTCCCTGTGCACCTCC |
| A41G_se | GCTGTGGGTGAAGCCAGAGGGCTTCGG |
| A41G_as | CCGAAGCCCTCTGGCTTCACCCACAGC |
| G114A_se | TGGGGTTAGGGCATTGAAGGCGGGACC |
| G114A_as | GGTCCCGCCTTCAATGCCCTAACCCCA |
| C192A_se | GCTCAACCTACCTGAAGACAGTCTGAGAACTCGAG |
| C192A_as | CTCGAGTTCTCAGACTGTCTTCAGGTAGGTTGAGC |
| Name (mutation) | Sequence (5′–3′) |
| A(t274g)_se | CACACACCAGCTGTGCATTCTGTGCTCCCTTGC |
| B(a891g)_se | CCAAAGGGCTTCGGGTTCAGAGTCTCTGAGGAC |
| C(t986g)_se | CCCACCCACCTTTCCCTGTGCACCTCC |
| A41G_se | GCTGTGGGTGAAGCCAGAGGGCTTCGG |
| A41G_as | CCGAAGCCCTCTGGCTTCACCCACAGC |
| G114A_se | TGGGGTTAGGGCATTGAAGGCGGGACC |
| G114A_as | GGTCCCGCCTTCAATGCCCTAACCCCA |
| C192A_se | GCTCAACCTACCTGAAGACAGTCTGAGAACTCGAG |
| C192A_as | CTCGAGTTCTCAGACTGTCTTCAGGTAGGTTGAGC |
Note. Mutagenic bases are printed in bold type.
Mutagenic Primers Used in This Study
| Name (mutation) | Sequence (5′–3′) |
| A(t274g)_se | CACACACCAGCTGTGCATTCTGTGCTCCCTTGC |
| B(a891g)_se | CCAAAGGGCTTCGGGTTCAGAGTCTCTGAGGAC |
| C(t986g)_se | CCCACCCACCTTTCCCTGTGCACCTCC |
| A41G_se | GCTGTGGGTGAAGCCAGAGGGCTTCGG |
| A41G_as | CCGAAGCCCTCTGGCTTCACCCACAGC |
| G114A_se | TGGGGTTAGGGCATTGAAGGCGGGACC |
| G114A_as | GGTCCCGCCTTCAATGCCCTAACCCCA |
| C192A_se | GCTCAACCTACCTGAAGACAGTCTGAGAACTCGAG |
| C192A_as | CTCGAGTTCTCAGACTGTCTTCAGGTAGGTTGAGC |
| Name (mutation) | Sequence (5′–3′) |
| A(t274g)_se | CACACACCAGCTGTGCATTCTGTGCTCCCTTGC |
| B(a891g)_se | CCAAAGGGCTTCGGGTTCAGAGTCTCTGAGGAC |
| C(t986g)_se | CCCACCCACCTTTCCCTGTGCACCTCC |
| A41G_se | GCTGTGGGTGAAGCCAGAGGGCTTCGG |
| A41G_as | CCGAAGCCCTCTGGCTTCACCCACAGC |
| G114A_se | TGGGGTTAGGGCATTGAAGGCGGGACC |
| G114A_as | GGTCCCGCCTTCAATGCCCTAACCCCA |
| C192A_se | GCTCAACCTACCTGAAGACAGTCTGAGAACTCGAG |
| C192A_as | CTCGAGTTCTCAGACTGTCTTCAGGTAGGTTGAGC |
Note. Mutagenic bases are printed in bold type.
Transfection and luciferase assay.
Mouse hepatoma cells from cell line 55.1c were transfected with vectors containing fragments of the mGSTm3 promoter as described above or with the empty pT81luc plasmid using Lipofectamine 2000 (Invitrogen). Plasmid pRL-CMV (Promega, Madison, WI) expressing Renilla luciferase under control of a constitutively active CMV promoter was cotransfected. After 24 h transfection, cells were treated with 15mM of the GSK3β inhibitor LiCl (or 15mM NaCl as a control) for additional 24 h. Afterward, luciferase activity was determined using the Dual Luciferase Kit (Promega) according to the manufacturer’s instructions and normalized to Renilla luciferase activity.
Statistical analysis.
For statistical analysis, Student’s t-test was used. Differences were considered significant when p < 0.05.
RESULTS
Expression of GSTm Isoforms in Ctnnb1- and Ha-ras–Mutated Mouse Liver Tumors
Recently, we have conducted a microarray analysis to analyze aberrant gene expression in mouse liver tumors with activating mutations in either the Ha-ras or the Ctnnb1 (encoding β-catenin) proto-oncogene. In this study, an upregulation of messenger RNAs (mRNAs) encoding the GST isoenzymes GSTm2, GSTm3, and GSTm6 had been observed in Ctnnb1-mutated hepatomas, suggesting that expression of these phase II enzymes is affected by β-catenin signaling. Tumors with activated Ha-ras, by contrast, exhibited a significant downregulation of the aforementioned GSTs (Stahl et al., 2005).
To extend the results of the microarray study, mRNA levels of the three GSTm isoforms were quantified in a larger number of tumors harboring mutations in either the Ctnnb1 or the Ha-ras proto-oncogene and the results were compared to normal liver tissue (Figs. 1A–C). The tumors with Ctnnb1 mutations were, as in the microarray study, taken from an experiment in which mice were treated with the tumor promoter PB until sacrifice, while Ha-ras–mutated tumors were isolated from livers of non-PB–treated mice. Since PB is known to induce expression of GSTm mRNA levels in mouse liver, expression in Ctnnb1-mutated tumors was compared with that of normal liver from PB-treated mice, while GSTm expression in Ha-ras–mutated tumors was compared with that of livers from non-PB–exposed mice to adjust for the PB effect. Expression of GSTm2 and GSTm3 in normal liver tissue was significantly elevated by PB treatment (Figs. 1A and 1B), while GSTm6 mRNA levels were not significantly altered (Fig. 1C). Mutational activation of β-catenin signaling in the tumor cells resulted in strongly elevated mRNA levels of all three GSTm isoforms, and mRNA levels were significantly higher in tumors with Ctnnb1 mutations than in the corresponding normal liver samples from the PB-treated mice. On the other hand, expression of the GSTm isoenzymes was significantly decreased in Ha-ras-mutated tumor cells when compared to the corresponding control samples (Figs. 1A–C). CAR mRNA levels were upregulated in Ctnnb1-mutated hepatomas, whereas a significant repression of CAR was observed in tumors with mutations in the Ha-ras proto-oncogene (Fig. 1D).
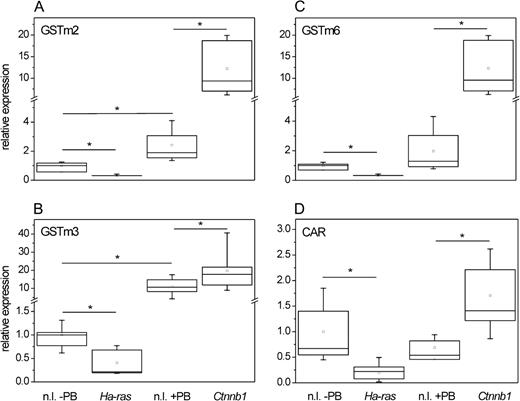
Expression of GSTm isoforms in mouse liver tumors with Ctnnb1 and Ha-ras mutations. RNA was isolated from normal liver samples (±PB treatment) and from mouse liver tumors harboring activating mutations in the indicated proto-oncogenes. Levels of different GSTm isoenzyme mRNAs were determined by real-time RT-PCR and normalized to 18s rRNA expression. Compared to the respective nontumorous tissue samples, Ctnnb1-mutated hepatomas expressed significantly elevated amounts of all three GSTm isoforms analyzed, GSTm2 (A), GSTm3 (B), and GSTm6 (C). Contrastingly, these mRNAs were downregulated in hepatomas carrying Ha-ras mutations. Comparable downregulation and upregulation were observed for CAR mRNA (D). Ctnnb1, Ctnnb1-mutated tumor; Ha-ras, Ha-ras-mutated tumor; n.l., normal liver. Number of samples analyzed: n = 11 (n.l. − PB); n = 12 (n.l. + PB); n = 6 (Ha-ras); n = 13 (Ctnnb1). Significant differences (p < 0.05, Student’s t-test) are indicated by asterisk.
The mRNA data were confirmed at the protein level by immunohistochemical staining of mouse liver tumors for GSTm. In addition, immunostaining was performed for GS and E-cadherin to demonstrate activation of either the β-catenin (GS) or the Ras/Raf/ERK signaling pathway (E-cadherin). GS is a model target of β-catenin signaling, which exhibits almost 100% correlation with β-catenin activation in hepatocytes with activated β-catenin and in mouse liver adenomas (Cadoret et al., 2002; Loeppen et al., 2002), whereas Ha-ras– and B-raf–mutated tumors lack GS expression. On the other hand, E-cadherin is consistently expressed in mouse liver tumors with activated Ha-ras or B-raf, but not in the Ctnnb1-mutated counterparts (Hailfinger et al., 2006), and was thus used to visualize tumors with an activated Ras/Raf/ERK pathway. Tumor-containing liver samples examined in this experiment were taken from a recent carcinogenesis study in which administration of PB had been stopped 3 weeks before killing of the animals, assuring that the effects on GSTm expression were not influenced by the presence of the tumor promoter at the time point of the analysis (Bursch et al., 2005). As evident from Figures 2A and 2B, GSTm was abundantly expressed in GS-positive tumors. Inversely, GSTm protein expression was undetectable in GS-negative (non-Ctnnb1–mutated) hepatomas with high expression of E-cadherin (Fig. 2C), which is indicative of Ras/Raf/ERK signaling.
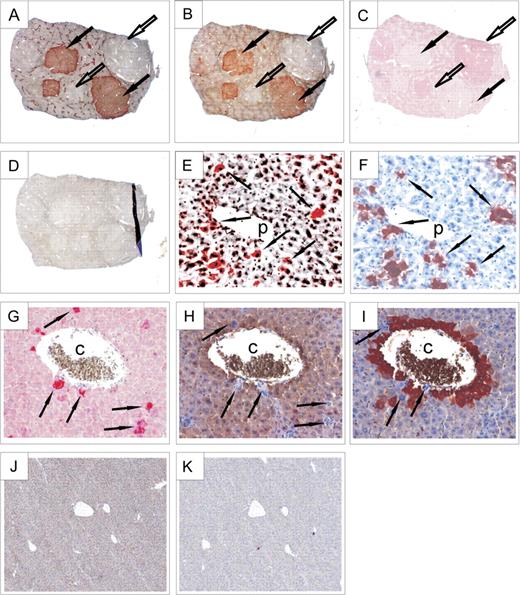
Expression of GSTm and other marker proteins in mouse liver as determined by immunohistochemical staining. (A) Staining for GS was used to identify tumors with Ctnnb1 mutations (GS positive); GS-negative tumors contained Ha-ras or B-Raf mutations. (B) GSTm protein was detected at high levels in GS-positive (Ctnnb1-mutated) tumors (designated by black arrows), whereas it was largely absent in GS-negative hepatomas (indicated by white arrows). Please also note zonation of GS and GSTm expression in neighboring normal liver tissue. (C) By contrast, E-cadherin expression, used to identify Ha-ras/B-raf–mutated tumors, was inversely correlated to GSTm and GS. (D) Control staining w/o primary antibody demonstrated the specificity of the detected signals. (E) Transgenic hepatocytes from a mouse with liver-specific expression of activated (S33Y) β-catenin displayed high levels of GSTm. Again, GSTm and GS expressions (F) were highly correlated (see arrows). (G) Cells from transgenic Tglox(pA)H-ras* mice expressing mutant Ha-ras were strongly stained for phosphorylated ERK1/2. Phospho-ERK1/2–positive hepatocytes expressed neither GSTm (H) nor GS (I). Expression of both GSTm (J) and GS (K) proteins was absent in mice with a liver-specific knock out of Ctnnb1, which is in strong contrast to the zonal expression pattern of these proteins in wild-type control mice (see A and B). Original magnification of images: ×2.5 (A–D), ×40 (E–I), and ×10 (J and K). p, portal vein; c, central vein.
GSTm Expression in Transgenic Mice Expressing a Point-Mutated β-CateninS33Y Allele
To analyze whether the observed induction of GSTm isoenzymes in response to β-catenin activation was a feature unique to hepatocellular tumors, GSTm expression in transgenic mice with liver-specific expression of an activated (S33Y) version of the β-catenin protein was analyzed. Livers of these mice display a scattered pattern of transgenic hepatocytes expressing the model β-catenin activation marker GS and other β-catenin target genes (Braeuning and Schwarz, 2009; Schreiber et al., unpublished data). Figures 2E–F shows immunostainings for GSTm and GS in serial slices from the liver of a transgenic β-cateninS33Y mouse. It becomes apparent that GSTm is ectopically expressed in a fraction of periportal hepatocytes, which normally should not express this enzyme (Fig. 2E). Comparison to GS staining in a parallel liver section revealed coexpression of GSTm and the β-catenin activation marker GS in the transgenic hepatocytes (Fig. 2F), indicating that ectopic activation of β-catenin signaling was able to override physiogical restriction of GSTm expression to perivenous hepatocytes.
GSTm Expression Is Abolished by Activation of Ha-ras in Transgenic Tglox(pA)H-ras* Mice
In contrast to what was seen in mice with liver-specific expression of activated β-catenin, hepatocytes from transgenic mice expressing mutant Ha-ras after Cre-mediated recombination have been described to completely lack expression of GS, indicating a repression of β-catenin signaling upon activation of the Ras/ERK signaling pathway (Braeuning et al., 2007b). Expression of mutant Ha-ras can be easily demonstrated by immunohistochemical staining for phosphorylated extracellular signal-regulated kinase (ERK1/2), which becomes activated by phosphorylation during Ras-mediated signal transduction through the mitogen-activated protein kinase pathway. As shown in Figure 2G, large dysplastic phospho-ERK1/2–positive hepatocytes appeared in the livers of Tglox(pA)H-ras* mice after activation of the transgene. Exactly, these phospho-ERK1/2–positive hepatocytes showed complete absence of GSTm and GS, irrespective of their position within the liver lobule, as can be taken from the immunostains of parallel serial sections (Figs. 2H–I).
GSTm Expression in Transgenic Mice with Liver-Specific Knock out of Ctnnb1
The role of β-catenin in the expression of GSTs was further examined by the analysis of livers from transgenic mice with liver-specific knock out of Ctnnb1. Knock out of Ctnnb1 in mouse liver results in a loss of GS expression, as recently published by Sekine et al. (2006). We now analyzed the effect of conditional β-catenin knock out on the expression of GSTm isoforms. The concomitant loss of GSTm and GS expression was demonstrated by immunostaining (Figs. 2J–K) and Western blotting (Fig. 3A). This effect was clearly related to alterations at the mRNA level, where expression of all GSTm isoenzymes analyzed was markedly reduced when compared to wild-type controls (Fig. 3B). Lack of GSTm expression in Ctnnb1 knockout livers was associated with significantly diminished levels of CAR mRNA, whereas levels of the CAR heterodimerization partner RXR remained more or less constant (data not shown).
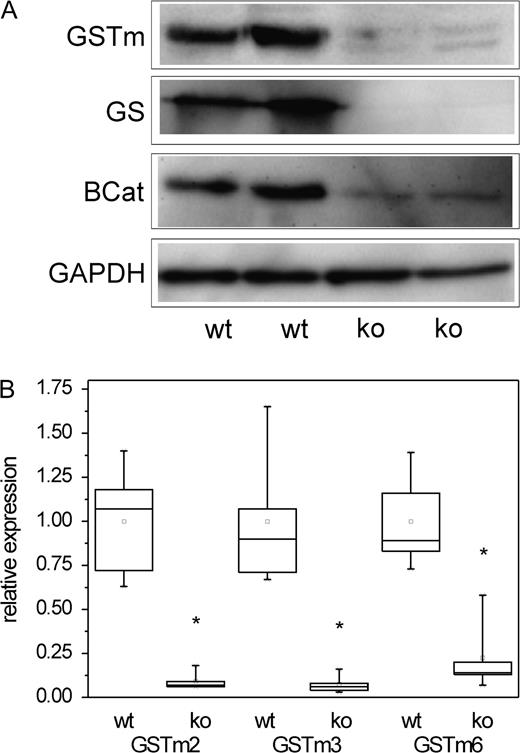
Analysis of GSTm expression in livers of mice with hepatocyte-specific conditional knock out of β-catenin compared to wild-type controls. (A) GSTm protein is almost indiscernible in lysates from livers of Ctnnb1 knockout mice. GAPDH was used as a loading control. (B) Significant reduction of GSTm2, GSTm3, and GSTm6 mRNA levels in Ctnnb1 knockout mice was observed for all three GSTm isoenzymes. Number of samples analyzed: n = 5 (wt) and n = 5 (ko). Significant differences (p < 0.05, Student’s t-test) are indicated by asterisk. wt, wild type; ko, Ctnnb1 knock out; BCat, β-catenin. Please note that remnant β-catenin protein detected in the knockout mice stems from nonparenchymal liver cells, which are not affected by the hepatocyte-targeted knock out.
Zonal-Specific Expression of GSTm Isoenzymes
Several GSTs are known to show zonal differences in expression within the liver lobule (Braeuning et al., 2006). We therefore determined mRNA levels of GSTm2, GSTm3, and GSTm6 in periportal and perivenous hepatocyte isolates by real-time RT-PCR. As shown in Table 3, mRNA expression levels of all three isoforms were threefold to fourfold higher in perivenous than in periportal hepatocyte populations, which is in accordance with previous results from a microarray experiment conducted in our laboratory (Braeuning et al., 2006). To further confirm these results, GSTm protein contents in perivenous and periportal Ctnnb1 wild-type hepatocytes were studied by Western blotting and compared to the expression of the perivenous marker protein GS (Gebhardt and Mecke, 1983). As evident from Figure 4, GSTm protein was predominantly detected in perivenous hepatocytes, confirming the results of the mRNA expression analyses. It has been recently shown that physiologically activated β-catenin is present in perivenous but not in periportal hepatocytes (Benhamouche et al., 2006; Sekine et al., 2007). We thus investigated zonation of GSTm mRNAs in isolated periportal and perivenous hepatocyte subpopulations in mice with hepatocyte-specific knock out of β-catenin. In contrast to their wild-type littermates, the low levels of mRNAs for GSTm isoenzymes expressed in Ctnnb1 knockout mice did not exhibit remarkable zonal preferences (Table 3).
Differential Expression of GSTm Isoforms in Perivenous and Periportal Hepatocytes
| Transcript | Log2 ratio perivenous versus periportal | ||
| RT-PCR (Ctnnb1 wt) | Microarray (Ctnnb1 wt) | RT-PCR (Ctnnb1 ko) | |
| GSTm2 | 3.18* | 4.19* | 0.07 |
| GSTm3 | 3.98* | 4.36*/2.31* | −0.12 |
| GSTm6 | 3.07* | 3.13* | −0.22 |
| Transcript | Log2 ratio perivenous versus periportal | ||
| RT-PCR (Ctnnb1 wt) | Microarray (Ctnnb1 wt) | RT-PCR (Ctnnb1 ko) | |
| GSTm2 | 3.18* | 4.19* | 0.07 |
| GSTm3 | 3.98* | 4.36*/2.31* | −0.12 |
| GSTm6 | 3.07* | 3.13* | −0.22 |
Notes. Expression of the indicated GSTm isoforms in perivenous and periportal hepatocyte subpopulations from Ctnnb1 wild-type (wt) and knockout (ko) mice was analyzed by RT-PCR. Results from a previous microarray experiment (Braeuning et al., 2006) using the same hepatocyte preparations are shown for comparison. As GSTm3 was represented by two probe sets on the array, their individual log2 ratios are shown. Significant zonal differences (p < 0.05, Student’s t-test) are indicated by asterisk.
Differential Expression of GSTm Isoforms in Perivenous and Periportal Hepatocytes
| Transcript | Log2 ratio perivenous versus periportal | ||
| RT-PCR (Ctnnb1 wt) | Microarray (Ctnnb1 wt) | RT-PCR (Ctnnb1 ko) | |
| GSTm2 | 3.18* | 4.19* | 0.07 |
| GSTm3 | 3.98* | 4.36*/2.31* | −0.12 |
| GSTm6 | 3.07* | 3.13* | −0.22 |
| Transcript | Log2 ratio perivenous versus periportal | ||
| RT-PCR (Ctnnb1 wt) | Microarray (Ctnnb1 wt) | RT-PCR (Ctnnb1 ko) | |
| GSTm2 | 3.18* | 4.19* | 0.07 |
| GSTm3 | 3.98* | 4.36*/2.31* | −0.12 |
| GSTm6 | 3.07* | 3.13* | −0.22 |
Notes. Expression of the indicated GSTm isoforms in perivenous and periportal hepatocyte subpopulations from Ctnnb1 wild-type (wt) and knockout (ko) mice was analyzed by RT-PCR. Results from a previous microarray experiment (Braeuning et al., 2006) using the same hepatocyte preparations are shown for comparison. As GSTm3 was represented by two probe sets on the array, their individual log2 ratios are shown. Significant zonal differences (p < 0.05, Student’s t-test) are indicated by asterisk.
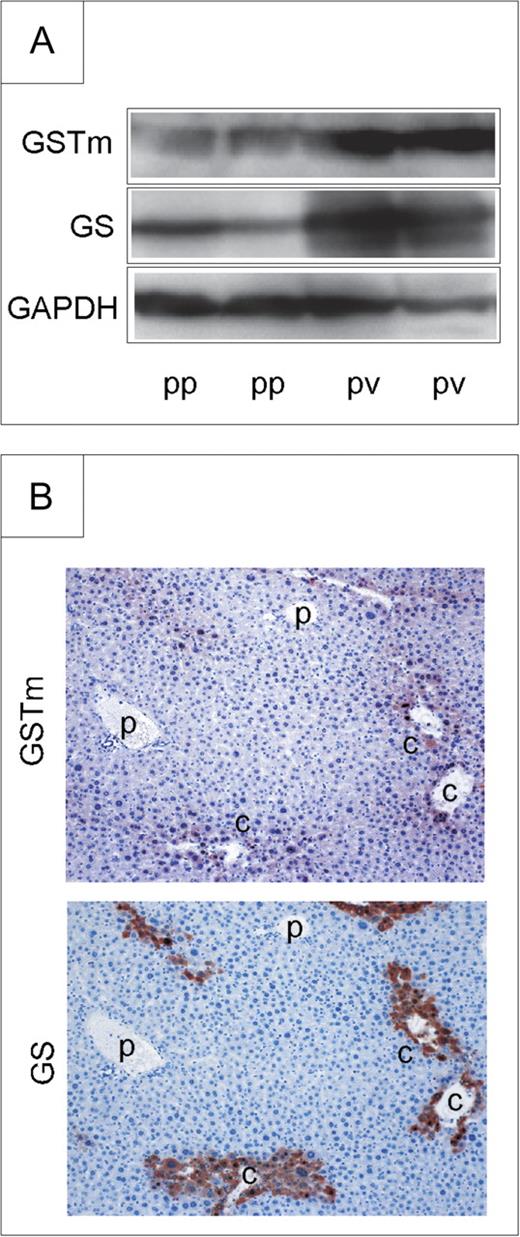
Zonation of GSTm protein expression. (A) GSTm protein is preferentially expressed in perivenous hepatocytes from Ctnnb1 wild-type mice, as determined by Western blotting. GAPDH was used as a loading control. (B) Preferential expression of GSTm protein is also detected by immunohistochemistry. Perivenous GS staining is shown for comparison. Original magnification of images: ×20. p, portal vein; c, central vein; pp, periportal; pv, perivenous.
Expression of GSTm Isoforms Is Induced by β-Catenin Signaling In Vitro
Recently, we have published data demonstrating that a variety of perivenous mRNAs overexpressed in Ctnnb1-mutated hepatomas including several cytochrome P450s can be induced in primary mouse hepatocytes by activation of β-catenin signaling (Hailfinger et al., 2006). To test whether the GSTm isoforms investigated in this study are also regulated by β-catenin in vitro, β-catenin signaling was activated in primary mouse hepatocytes by incubation with medium conditioned by a Wnt3A-producing 3T3 fibroblast cell line using the same cell culture conditions and incubation times as in our previous study (Hailfinger et al., 2006) to allow direct comparison of cytochrome P450 and GSTm induction. Expression of GSTm isoforms was analyzed by real-time RT-PCR, and activation of the β-catenin pathway was monitored by determination of mRNA levels of the β-catenin target genes Axin2 and Gpr49. Average induction of Axin2 was ∼90-fold, whereas Gpr49 levels were increased 35-fold (data not shown). As depicted in Figure 5A, Wnt3a-mediated activation of β-catenin signaling led to a significant increase in mRNA levels of all three GSTs analyzed. However, the observed upregulation was strikingly lower for the GSTs compared to the “classic” Wnt target genes: 2.70-fold for GSTm2, 2.25-fold for GSTm3, and 2.03-fold for GSTm6. This fact points toward the existence of different mechanisms by which activated β-catenin induces, on the one hand, direct TCF-dependent target genes like Axin2/Gpr49 and, on the other hand, the rather slightly regulated GSTs. At the protein level, upregulation of GSTm by Wnt3A was clearly demonstrated by Western blotting (Fig. 5B). Comparable effects were seen when enriched fractions of periportal hepatocytes, which do not express GSTm in vivo, were used (data not shown). Wnt3A was not able to induce transcription of CAR neither in hepatocyte preparations from whole liver nor in periportal-enriched fractions (data not shown).
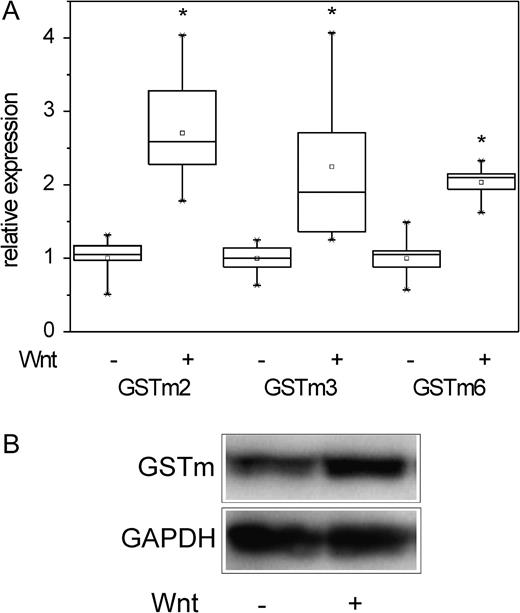
Activation of β-catenin signaling upregulates expression of GSTm mRNAs in vitro. (A) Incubation of primary hepatocytes with Wnt3A-conditioned medium significantly induced mRNA levels of GSTm2, GSTm3, and GSTm6. Number of samples analyzed: n = 9. Significant differences (p < 0.05, Student’s t-test) are indicated by asterisk. (B) Primary hepatocytes were incubated with Wnt3A, and GSTm protein levels were assessed by Western blotting. GAPDH was used as a loading control. Activation of the β-catenin signaling pathway by Wnt3A clearly induced GSTm protein levels.
For unknown reasons, GSTm expression rapidly declines during cultivation of primary hepatocytes, resulting in very low levels of the respective mRNAs. Potential inhibition of GSTm expression by activation of Ha-ras signaling could therefore not be studied properly in vitro.
β-Catenin Signaling Induces GSTm3 Promoter–Driven Luciferase Activity
To analyze whether the observed induction of GSTm isoenzymes by β-catenin signaling was a consequence of elevated transcription, two luciferase reporter vectors were created containing 1.0 and 4.9 kb of the 5′-regulatory sequence of the mouse GSTm3 gene, respectively. For a schematic representation of the 5′-regulatory sequence of the mouse GSTm3 gene showing the localization of putative transcription factor–binding sites and for an illustration of different deletion constructs used in the following experiments, see Figure 6A. The constructs were transiently transfected into mouse hepatoma cells from line 55.1c, which had been successfully used in a previous study to analyze the effects of β-catenin signaling on a luciferase reporter system driven by the rat cytochrome P450 2b1 promoter (Loeppen et al., 2005). Since 55.1c cells were insensitive to Wnt3A, cultures were incubated with 15mM of the widely used GSK3β inhibitor LiCl (Stambolic et al., 1996) for 24 h to activate β-catenin. A moderate but significant induction of GSTm3 promoter–driven luciferase activity was observed after stimulation of cells with the GSK3β inhibitor for both the 1 kb and the 5 kb wild-type constructs, with the 1 kb construct showing a more pronounced effect (approximately twofold, see Fig. 6B), indicating that transcriptional upregulation of GSTm3 upon activation of the β-catenin pathway was mediated by the proximal 1 kb promoter sequence. Background luciferase activity from the empty pT81luc backbone was not altered following LiCl treatment (data not shown).
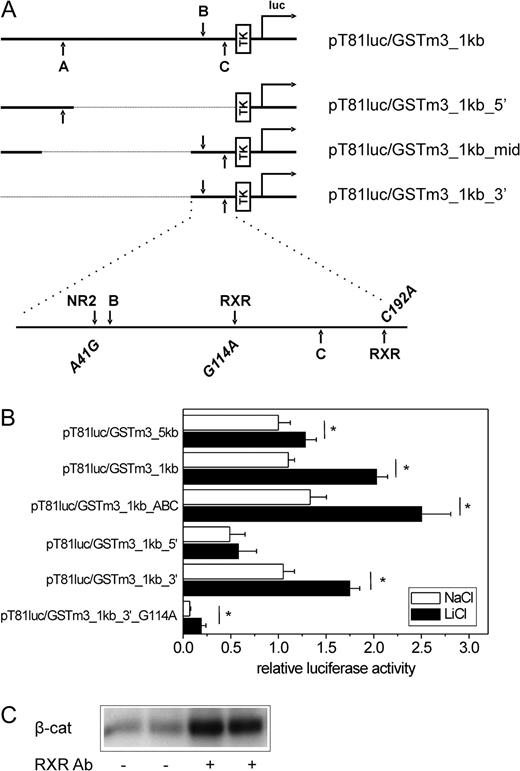
β-Catenin signaling activates the mouse GSTm3 promoter. (A) Schematic representation of the mouse GSTm3 gene 5′-regulatory region illustrating the promoter fragments contained in the different mGSTm3 promoter–driven pT81luc-derived luciferase reporter constructs and showing the positions of putative transcription factor–binding sites within the 1 kb promoter fragment. Upper characters indicate orientation of a motif on the sense strand; lower characters are used for sites located on the antisense strand. (B) Effect of β-catenin activation on GSTm3 promoter activity. Different luciferase reporter vectors containing fragments of the 5′-regulatory sequence of the mouse GSTm3 gene were transfected into mouse hepatoma cells from line 55.1c. Cells were treated with 15mM of the GSK3β inhibitor LiCl (or NaCl as a control) for 24 h prior to measurement of luciferase activity. Considerable induction in response to LiCl is monitored with the wild-type (pT81luc/GSTm3_1kb) and TCF-binding site–mutated (pT81luc/GSTm3_1kb_ABC) 1.0 kb construct and is only marginally diminished using the thereof-derived proximal 0.3 kb deletion mutant (pT81luc/GSTm3_3′). Deletion of the proximal 0.2 kb region (pT81luc/GSTm3_1kb_5′) and mutational inactivation of the therein-contained putative RXR homodimer/heterodimer–binding site (pT81luc/GSTm3_1kb_3′_G114A) dramatically decreases basal and LiCl-inducible luciferase activities. Mean ± SD from four independent experiments (each performed in quadruplicate) are given. Significant differences (p < 0.05, Student’s t-test) are indicated by asterisk. (C) Interaction of β-catenin and RXR proteins. Western analysis for β-catenin (β-cat.) after immunoprecipitation with anti-RXR antibody is shown. A–C, putative β-catenin/TCF-binding sites; Ab, antibody; luc, firefly luciferase gene; NR2, putative nuclear receptor subfamily 2–binding site; TK, thymidine kinase promoter.
We then analyzed whether the induction of GSTm3 promoter–driven luciferase activity was dependent on an activation of TCF/β-catenin sites. For this purpose, a mutant reporter plasmid was used lacking three putative β-catenin/TCF-binding sites (termed A, B, and C) that had been identified by in silico analysis using MatInspector software (Fig. 6A). After transfection of the triple-mutated plasmid pT81luc/GSTm3_1kb_ABC, basal and LiCl-inducible luciferase activities were comparable to or even slightly higher than those observed with the wild-type 1 kb construct, indicating that the putative TCF sites were not functional (Fig. 6B). The promoter was further analyzed by the use of several deletion mutants derived from the wild-type 1 kb plasmid pT81luc/GSTm3_1kb (Fig. 6A). Basal reporter signal and the inducing effect of LiCl were barely affected in the deletion mutant pT81luc/GSTm3_1kb_3′ containing only 0.2 kb of the 3′ terminus of the GSTm3 promoter (Fig. 6B). The same phenomenon was observed for pT81luc/GSTm3_1kb_mid lacking the middle 0.6 kb of the 1 kb construct (data not shown). In contrast, a mutant plasmid lacking the 0.2 kb 3′-terminal region (pT81luc_GSTm3_5′) showed significantly diminished basal activity and was barely inducible by activation of β-catenin (Fig. 6B). Taken together, these results indicated that basal and GSK inhibitor–inducible GSTm3 expression was, in its main part, mediated by the proximal 0.2 kb of the promoter but not by the putative TCF elements B and C located within that sequence.
Besides activation of TCF-binding sites, β-catenin is known to influence transcription via interaction with a variety of nuclear receptors; for review, see Mulholland et al. (2005). Since mutational inactivation of putative TCF sites had not altered GSTm3 promoter–driven luciferase activity, the proximal 0.2 kb promoter region was screened in silico for potential nuclear receptor–binding sites, which might be influenced by β-catenin. Mutations of putative binding sites for candidate transcription factors (i.e., nuclear receptor subfamily 2 members and retinoid X receptor/vitamin D receptor [RXR-VDR] heterodimers [RXR-VDR DR3 sites]; see Fig. 6A) were introduced into pT81luc/GSTm3_1kb_3′. The potential binding sites for RXR were of special interest since it is known that mice with liver-specific knock out of RXRα lack expression of GSTm3 (Dai et al., 2005; Wu et al., 2004). Neither of the mutations A41G or C192A was able to significantly abrogate basal or LiCl-induced reporter activities, suggesting that these sites do not play a major role (data not shown). However, G114A mutation of a putative RXR-VDR heterodimer–binding site led to a significant decrease in basal luciferase activity, and only very minor inducing effects of LiCl treatment were still detectable (Fig. 6B). RXRα mRNA levels were not altered after incubation with the GSK3β inhibitor (data not shown).
Association of β-catenin and RXR proteins has been reported previously; for review, see Mulholland et al. (2005). To confirm this interaction in our cell system, extracts from 55.1c cells were prepared and subjected to immunoprecipitation with an antibody directed against the RXR. Subsequent Western analysis revealed that β-catenin is coprecipitated from the lysates by the anti-RXR antibody (Fig. 6C).
DISCUSSION
Our present data show that GSTm2, GSTm3, and GSTm6 are abundantly expressed, at the mRNA and protein level, in cells with activated β-catenin, demonstrating that expression of these GSTm isoforms is positively controlled by β-catenin signaling. This is evidenced by the fact that mouse liver tumors harboring activating mutations in the Ctnnb1 proto-oncogene and transgenic β-cateninS33Y hepatocytes express highly elevated levels of the GSTm isoforms analyzed. Furthermore, conditional knock out of Ctnnb1 leads to a dramatic decrease in GSTm expression in mouse liver. In vitro analyses of mRNA levels and GSTm3 promoter activity after stimulation of the β-catenin signaling pathway indicate that the observed elevated GSTm levels are due to a regulation of the respective genes at the transcriptional level.
Expression of GSTs is regulated by a variety of cellular factors (for detailed description, please refer to the “Introduction” section), and our results suggest that β-catenin–dependent signaling also plays a role. However, mutational inactivation of the putative β-catenin/TCF-binding sites within the luciferase reporter plasmid did not affect GSK3β inhibitor–mediated luciferase expression, demonstrating that β-catenin does not directly activate GSTm3 transcription but influences GSTm3 expression in an indirect manner via activation of transcription factors different from TCF. The fact that fold induction of GSTm expression by β-catenin differs from that of classic TCF-dependent Wnt/β-catenin target genes also argues for that scenario. Our results obtained with mutant GSTm3 promoter constructs indicate that GSTm3 expression is, to its major part, mediated through a RXR-VDR heterodimer–binding site within the proximal 0.2 kb of the upstream promoter region. This is consistent with results from a previous report demonstrating a crucial role of RXRα in the regulation GSTm3: Using transgenic mice, Wu et al., (2004) convincingly demonstrated a massive loss of GSTm3 mRNA and protein expression after liver-specific knock out of RXRα. Moreover, vitamin A deficiency reduces GST activity in hepatocytes (Gad, 1994). Since several other drug-metabolizing enzymes are also downregulated in RXRα knockout mice, it is tempting to speculate that β-catenin signaling may indirectly impact via cross talk with retinoid receptors on the expression of a larger battery of genes encoding enzymes involved in drug metabolism. Signaling via the VDR has also been shown to positively influence GST activity in rat and mouse cells (Banakar et al., 2004; Karmakar et al., 2002; Sardar et al., 1996).
As our in vivo data demonstrate a concomitant regulation of CAR (but not of RXRα) and GSTm mRNA levels in response to β-catenin signaling in Ctnnb1-mutated tumors, in Ctnnb1 knockout mice, and in perivenous hepatocytes (for CAR zonation, see Braeuning et al., 2006), it is reasonable to assume that β-catenin induces higher CAR levels. This could lead to enhanced levels of CAR-RXR heterodimers, which may then mediate β-catenin–dependent regulation of GSTm expression. These data are, however, contrary to the situation observed in vitro: (1) CAR mRNA levels were not significantly altered upon Wnt3A treatment in hepatocytes, whereas GSTm expression was included. (2) This induction occurred in the absence of simultaneous treatment of cells with an activator of CAR signaling. (3) Gstm3 promoter–driven luciferase activity was inducible by activation of β-catenin signaling in 55.1c mouse hepatomas cells, where CAR mRNA is barely detectable and no induction of CAR target genes is observable after treatment of cell with CAR agonists (own unpublished data). (4) In silico analysis of the cloned GSTm3 promoter sequence provided good evidence for a binding of RXR-VDR but not of different RXR heterodimers, such as RXR-CAR.
This suggests that CAR is not crucially involved in induction of GSTm expression by β-catenin in cell culture. Furthermore, RXR mRNA levels were not induced by β-catenin signaling in mouse hepatocytes and hepatoma cells, and there are also no reports available from the literature describing elevated RXR levels after activation of the Wnt pathway in any context. Thus, it seems as if simple transcriptional upregulation of nuclear receptors by β-catenin is not responsible for elevated GSTm3 expression. The in vivo situation points toward complex regulatory mechanisms of expression of CAR target genes by β-catenin: In comparison to their wild-type littermates, Ctnnb1 knockout mice express lower levels of GSTm isoenzymes in their liver but higher levels of the CAR target gene cytochrome P450 2b10 (Tan et al., 2006) notwithstanding their lower levels of CAR. Nonetheless, elevated levels of CAR produced in response to β-catenin activation may be involved in the regulation of drug-inducible expression of GSTm3. Figure 7 shows a schematic delineation of possible mechanisms by which β-catenin influences GSTm3 expression in mouse liver.
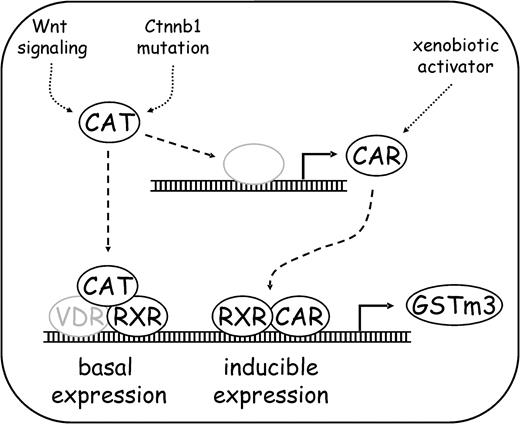
Schematic delineation of GSTm3 regulation by β-catenin. Basal (i.e., in the absence of xenobiotic enzyme inducers) expression of GSTm is augmented by activated β-catenin via interaction with RXR-dependent (and presumably VDR-dependent) transcription. On the other hand, β-catenin induces expression of CAR, which is involved in xenobiotic-inducible expression of GSTm3. CAT, β-catenin. Gray color: putative.
One example where an indirect regulatory mechanism by β-catenin seems to play a role is cytochrome P450 (Cyp) 2e1. Its gene is transcribed more or less constitutively, without involvement of nuclear receptors, such as AhR or CAR, under the control of the transcription factor hepatocyte nuclear factor 1α (HNF1α), which binds to a well-defined upstream regulatory sequence of the gene thus controlling ∼90% of Cyp2e1 expression (Ueno and Gonzalez, 1990). Cyp2e1 mRNA expression is almost completely abrogated in Ctnnb1 knockout mice (Sekine et al., 2006; Tan et al., 2006) but is induced to high levels in mouse liver tumors with activated β-catenin (Loeppen et al., 2005; Stahl et al., 2005) and in vitro in response to agonists of the Wnt/β-catenin pathway (Hailfinger et al., 2006). However, up to now, there is no indication of generally altered function of HNF1α in response to β-catenin. Thus, a Cyp2e1 promoter–specific interplay between HNF1α and β-catenin may regulate the expression of the gene, perhaps by β-catenin–mediated recruitment of essential cofactors to DNA-associated HNF1α.
β-Catenin physically interacts with different nuclear receptors, including different retinoid receptors and the VDR, in a highly cell type– and context-specific manner; as reviewed by Mulholland et al., (2005): For example, active β-catenin associates with the retinoic acid receptor RAR, thus potentiating the activity of RAR-dependent promoters, whereas RAR inversely inhibits β-catenin/TCF-dependent transcription. VDR-dependent transcription is augmented by activated β-catenin, which physically interacts with the receptor.
In summary, published data regarding interaction of β-catenin and nuclear receptors, and especially the above-mentioned observation that RXRα knock out abolishes GSTm3 transcription, strongly support our conclusion that not direct β-catenin/TCF-dependent mechanisms but rather indirect stimulation of nuclear receptor–mediated signal transduction is responsible for the induction of GSTm3 expression by β-catenin.
According to a hypothesis recently established in our laboratory, two opposing signaling gradients are responsible for the differential gene expression along the portocentral axis in mouse liver. Growth factors activating the Ha-ras cascade were proposed to dictate the periportal gene expression profile, whereas a second signal-inducing β-catenin–dependent transcription was assumed to prevail in the perivenous area of the liver lobule (Hailfinger et al., 2006). Since then, several publications from different groups, including ours, have confirmed the essential role of β-catenin signaling for the induction of several perivenous genes in liver (Benhamouche et al., 2006, 2007a; Hailfinger et al., 2006; Sekine et al., 2006; Tan et al., 2006) and demonstrated physiological activation of β-catenin in the perivenous zone of the liver lobule (Benhamouche et al., 2006; Sekine et al., 2007).
Data from our present study show that three perivenous-zonated GSTm isoforms are also regulated by β-catenin–dependent signals. Moreover, our data demonstrate that stimulation of the β-catenin pathway is sufficient to induce GSTm expression in all hepatocytes since transgenic hepatocytes with activated β-catenin signaling overexpress GSTm irrespective of their position within liver lobule and since GSTm expression was inducible in cell cultures with periportal hepatocyte subpopulations. Completely different effects were observed in transgenic hepatocytes expressing a constitutively activated version of Ha-ras and in tumor cells with Ha-ras mutations, both showing strongly downregulated GSTm expression levels. These changes resemble a periportal expression phenotype and support our recent evidence for an involvement of Ha-ras–dependent signaling in the regulation of periportal gene expression (Braeuning et al., 2007a, 2007b). Taken together, these findings are in favor of our recent hypothesis of a physiological antagonism of the β-catenin and Ha-ras signaling pathways in the regulation of gene expression in liver (Hailfinger et al., 2006).
GSTs are a group of enzymes of the so-called phase II of drug metabolism and play an essential role in hepatic detoxification of xenobiotic compounds and biotransformation of endogenous substrates. Our present results on GSTm expression clearly demonstrate the involvement of β-catenin in the regulation of this group of enzymes in mouse liver. Recent publications from our and other groups suggest that the expression of phase I enzymes of xenobiotic metabolism, such as several Cyp isoforms, is also dependent on the β-catenin pathway since certain Cyps are overexpressed in Ctnnb1-mutated hepatoma cells (Loeppen et al., 2005; Stahl et al., 2005), in transgenic hepatocytes expressing activated β-cateninS33Y (Braeuning and Schwarz, 2009), and in Wnt3A-treated hepatocyte cultures (Hailfinger et al., 2006), while their expression is abolished in a transgenic mouse model with liver-specific knock out of β-catenin (Sekine et al., 2006). There is also evidence for a contribution of Wnt/β-catenin signaling to the expression of different sulfotransferases (own unpublished data). Finally, β-catenin seems to be involved in the regulation of expression of several nuclear receptors responsible for the inducible expression of an entire battery of xenobiotic-metabolizing enzymes (own unpublished data). It thus seems that the Wnt/β-catenin pathway functions as a master regulator of the expression of both phase I and phase II drug-metabolizing enzymes in mouse liver.
FUNDING
Deutsche Krebshilfe (106356); Deutsche Forschungsgemeinschaft (SFB773).
We greatly acknowledge the excellent technical assistance by Johanna Mahr, Silvia Vetter, and Elke Zabinsky. We also thank Dr R. Wolf (Dundee, UK) for gift of GSTm antiserum, Dr R. Kemler (Freiburg, Germany) for gift of Wnt3A-producing cells, Dr J. Huelsken (Epalinges, Switzerland) for the gift of Ctnnb1loxP mice, and Dr N. Harada and Dr Y. Tamai (Tokyo, Japan) for the gift of Tglox(pA)H-ras* mice.
References
Author notes
These authors contributed equally to this work and should both be considered first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments