





ABSTRACT
RNA aptamers that bind the opium alkaloid codeine were generated using an iterative in vitro selection process. The binding properties of these aptamers, including equilibrium and kinetic rate constants, were determined through a rapid, high-throughput approach using surface plasmon resonance (SPR) analysis to measure real-time binding. The approach involves direct coupling of the target small molecule onto a sensor chip without utilization of a carrier protein. Two highest binding aptamer sequences, FC5 and FC45 with Kd values of 2.50 and 4.00 μM, respectively, were extensively studied. Corresponding mini-aptamers for FC5 and FC45 were subsequently identified through the described direct coupling Biacore assays. These assays were also employed to confirm the proposed secondary structures of the mini-aptamers. Both aptamers exhibit high specificity to codeine over morphine, which differs from codeine by a methyl group. Finally, the direct coupling method was demonstrated to eliminate potential non-specific interactions that may be associated with indirect coupling methods in which protein linkers are commonly employed. Therefore, in addition to presenting the first RNA aptamers to a subclass of benzylisoquinoline alkaloid molecules, this work highlights a method for characterizing small molecule aptamers that is more robust, precise, rapid and high-throughput than other commonly employed techniques.
INTRODUCTION
Codeine is a naturally-occurring opium alkaloid, part of the larger class of benzylisoquinoline alkaloids (BIAs), found in the opium poppy, Papaver somniferum, and constitutes ∼0.5% of opium (1). It is one of the most widely used narcotic drugs for the treatment of mild to moderate pain, diarrhea and cough with relatively low side effects (2). Despite its extensive medical applications, codeine is often abused for its euphoric and depressant effects as well as to prevent opiate withdrawal (3). Due to increasing misuse, codeine has been incorporated into workplace and military drug testing programs, and a screening and confirmation cut-off concentration of 40 μg/l has been suggested for federally-mandated testing in oral fluid by the Substance Abuse and Mental Health Services Administration (3). Therefore, a sensor system that can precisely measure the concentration of codeine and effectively discriminate against its structural analogues is highly desired.
Aptamers are nucleic acid molecules that bind ligands with high specificity and affinity (4). There is increasing interest in utilizing aptamers as the target recognition elements in various sensing applications (5–8). In addition to the drug detection applications of a codeine-binding aptamer, there are other potential biotechnology applications for this aptamer. Codeine is a member of the BIA family and is a key product metabolite in the opium alkaloid biosynthesis pathway (9). The BIAs comprise a structurally diverse group of pharmacologically important compounds (10) and efforts are ongoing to engineer microbial and plant hosts for the production of some of the important BIA intermediates in the codeine synthesis pathway, such as (S)-reticuline and thebaine (9–11).
Aptamers to BIA molecules may prove to be useful tools for such engineering efforts. Recent research has highlighted the application of aptamers as components of synthetic and naturally-occurring cellular sensors and switches (12–17), which can regulate enzyme levels in response to small molecule ligand concentrations. Therefore, aptamer-based cellular sensors may be generated to act as ‘intelligent’ regulatory tools for metabolic engineering efforts to provide dynamic regulation of gene expression at specific enzymatic steps so that pathway fluxes are rewired to enable the accumulation of desired intermediate metabolites, which has proven to be difficult to achieve in natural plant hosts (9). A codeine-binding aptamer may be used to construct tools, such as synthetic riboswitches that can be employed to redirect flux through an engineered BIA metabolic pathway or in setting up rapid functional screens of pathway variants. In addition, while aptamers have been developed to several of the far upstream metabolites in this pathway, such as dopamine (18) and tyrosine (19), they have not yet been developed against any BIA compounds, which harbor bulky, nitrogen-containing ring structures. Prior work has demonstrated that aptamers to specific molecules within a family of compounds may be used to design doped libraries for the selection of aptamers to similar compounds within that family from smaller library sequence spaces (19), and thus codeine aptamers would be potentially useful for selecting aptamers to diverse BIA molecules.
This work describes the generation of novel RNA aptamers to the small molecule codeine and highlights a robust, high-throughput assay method for measuring small molecule-aptamer binding properties. RNA aptamers that bind codeine with high affinities were selected from a combinatorial library containing a 30 nt randomized region using an iterative in vitro selection procedure or SELEX (Systematic Evolution of Ligands by EXponential enrichment) (20,21). The binding properties of the generated codeine aptamers were measured by surface plasmon resonance (SPR) through a real-time binding assay (Biacore), similar to previously reported methods (22,23) where the small molecule ligand is directly coupled to a sensor chip through chemical modification of the ligand, eliminating the need to use protein linkers between the target small molecule and the sensor surface as described in other methods (24–26). This direct coupling method limits potential non-specific interactions or binding artifacts arising from the presence of the linker protein observed in previous studies (24,25), which may alter the determined binding affinities. Therefore, this method may provide a more accurate assessment of small molecule-aptamer binding affinities since the measured interaction more closely mimics the binding environment of the in vitro selection process.
MATERIALS AND METHODS
DNA template library preparation
A random DNA library was generated through PCR using the following oligonucleotide sequences: a 59 nt DNA template 5′-GGGACAGGGCTAGC(N30)GAGGCAAAGCTTCCG-3′, primer1 5′-TTCTAATACGACTCACTATAGGGACAGGGCTAGC-3′ and primer2 5′-CGGAAGCTTTGCCTC-3′. All DNA synthesis was performed by Integrated DNA Technologies, Inc. The template contains a 30 nt randomized region flanked by two fixed primer-binding regions (Figure 1A). Primer1 contains a 17 nt T7 promoter sequence (italic). NheI and HindIII restriction endonuclease sites (underlined) were included in primer1 and primer2, respectively, for cloning of aptamer sequences.
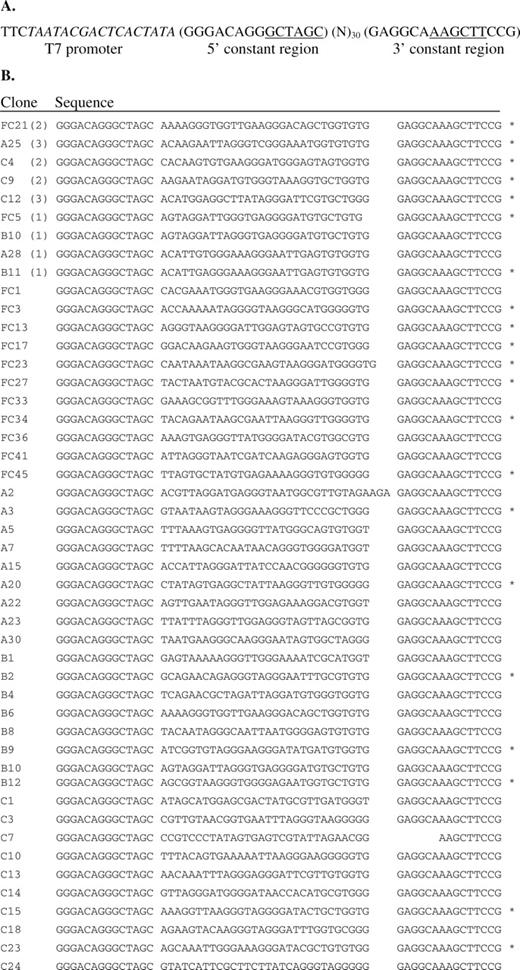
Aptamer clone sequences. (A) DNA template from which the initial RNA pool was generated. (B) Sequences of clones from the final aptamer pool. The codeine-binding properties of the sequences marked with an asterisk were characterized by the described direct coupling SPR assay. The number in parenthesis represents the frequency of a particular clone in the sequenced pool.
Codeine coupling and affinity chromatography matrix preparation
Approximately 300 mg of epoxy-activated Sepharose 6B (GE Healthcare) was hydrated and incubated with 2.5 mM codeine in coupling buffer [0.05 M Na2PO4 (pH 13)] overnight at 37°C according to the manufacturer's instructions. The coupled medium was washed three times with 2 ml of coupling buffer to remove uncoupled codeine. The medium was then incubated overnight with 1 M Tris–HCl (pH 8) at 40°C to block any remaining active groups. Finally, the medium was washed with a solution containing 0.1 M NaOAc (pH 4) and 0.5 M NaCl followed by a second solution containing 0.1 M Tris–HCl (pH 8) and 0.5 M NaCl. The wash was repeated twice and the matrix was resuspended in 10 mM Tris–HCl (pH 8) and stored at 4°C. The codeine affinity chromatography matrix was prepared by packing the coupled medium (500 μl) into a column following the manufacturer's instructions (Pierce). The packed column was washed with 10 column volumes of binding buffer [250 mM NaCl, 20 mM Tris–HCl (pH 7.4) and 5 mM MgCl2] and equilibrated prior to the selection process.
Initial RNA library pool preparation
The initial DNA library pool was generated by PCR conducted for 12 cycles on a mixture (100 μl) containing 20 pmol DNA template, 300 pmol each primer1 and primer2, 200 μM each dNTPs, 1.6 mM MgCl2, and 10 U Taq DNA polymerase (Roche). This DNA library pool (∼1.2 × 1014 molecules) was transcribed into an initial RNA library pool by incubating overnight at 37°C in the presence of 40 mM Tris–HCl (pH 7.9), 16 mM MgCl2, 10 mM DTT, 2 mM spermidine, 3 mM each rNTPs, 50 μCi [α-32P]UTP (GE Healthcare), 500 U RNase inhibitor and 50 U T7 RNA polymerase (New England Biolabs). The DNA template was subsequently degraded by incubating the reaction mixture with 10 U of DNase I (Invitrogen) at 37°C for 15 min. The unincorporated nucleotides were removed with a NucAway spin column (Ambion) following the manufacturer's instructions and binding buffer was added to the flow-through RNA to bring the total volume up to 500 μl.
In vitro selection of codeine-binding aptamers
Prior to incubation with the codeine-modified affinity column, the RNA pool was denatured at 70°C for 3 min and allowed to renature at room temperature for 30 min. To eliminate RNA molecules that non-specifically bind to the column matrix, the initial pool was first incubated with an unmodified column. The flow-through fraction from this incubation was subsequently transferred to a codeine-modified affinity column and incubated for 45 min. Following the incubation period, the affinity column was washed with 10 column volumes of binding buffer for cycles 1 to 5 to remove unbound RNAs. This wash volume was increased 10 column volumes for each of the subsequent cycles. Bound RNA was eluted with 7 column volumes of 5 mM codeine in binding buffer. The eluted RNA was recovered by ethanol precipitation in the presence of 20 μg/ml glycogen. Reverse transcription and cDNA amplification (15 PCR cycles) were performed in a single step using 200 U of SuperScript III reverse transcriptase (Invitrogen) and 5 U of Taq DNA polymerase in a 50 μl reaction volume. One-fifth of this DNA library was transcribed into an RNA library pool for the subsequent selection cycle. A total of 15 selection cycles were carried out during the in vitro selection process.
At the tenth cycle, a counter-selection against morphine was performed by eluting the bound RNA with 3 column volumes of 5 mM morphine in binding buffer prior to elution with codeine. Only RNA eluted with codeine was used to make the input DNA library pool for the subsequent selection cycle. Following the reverse transcription step of cycles 11, 12 and 13, an error-prone PCR was performed in a mutagenic buffer containing 40 pmol each primer1 and primer2, 7 mM MgCl2, 50 mM KCl, 10 mM Tris–HCl (pH 8.3), 0.2 mM dGTP, 0.2 mM dATP, 1 mM dCTP, 1 mM dTTP and 0.5 mM MnCl2. One-fifth of the error-prone PCR product from each of these cycles was used as the input DNA library pool for the subsequent selection cycle.
Aptamer library sequence analysis
The DNA pool from cycle 15 was amplified by PCR and cloned into a plasmid using the NheI and HindIII restriction sites present in the fixed regions of the aptamer sequence and the plasmid construct. This plasmid library was transformed into an electrocompetent Escherichia coli strain, DH10B (Invitrogen; F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 deoR recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ- rpsL nupG). Subcloning was confirmed by colony PCR, and a total of 58 positive colonies were sequenced by Laragen, Inc. The resulting sequences were aligned using the ClustalX sequence alignment program.
Qualitative binding affinity assay
Radiolabeled RNA was prepared from ∼1 μg of the final DNA pool (cycle 15) in the presence of 40 mM Tris–HCl (pH 7.9), 14 mM MgCl2, 10 mM DTT, 2 mM spermidine, 3 mM each rAGC mix, 150 μM rUTP, 50 μCi [α-32P]UTP, 40 U RNase inhibitor and 50 U T7 RNA polymerase. After allowing the transcription reaction to proceed for 3 h at 37°C, 5 U of DNase I were added to the mixture and the reaction was incubated for 15 min. The unincorporated nucleotides were removed with a NucAway spin column and the flow-through RNA was divided equally into two volumes. One of the radiolabeled RNA pools was incubated with a codeine-modified column, whereas the other pool was incubated with an unmodified column. After a 15 min incubation, each column was washed with 3 column volumes of binding buffer followed by elution with 7 column volumes of 5 mM codeine in binding buffer. The eluted RNA from each column was separated by electrophoresis on an 8% polyacryamide/7 M urea gel in 1× Tris–borate buffer. The gel was dried and the recovered radiolabeled RNA was imaged on a FX phosphorimager (BioRad).
Quantitative direct coupling small molecule-aptamer binding assay
A CM5 sensor chip was primed with RNase-free water followed by preconditioning with a 50 mM sodium hydroxide, 0.1% hydrochloric acid, 0.1% (w/v) SDS, 0.085% phosphoric acid solution prior to immobilization of codeine onto the chip surface. The chip was subsequently activated with a 0.2 M N-ethyl-N′-(dimethylaminopropyl)carbodiimide (EDC), 0.05 M N-hydroxysuccinimide (NHS) solution. An amine surface was created by injecting a solution of 0.1 M 1,8-diaminooctane dissolved in 50 mM sodium borate (pH 8.5) over the activated sensor chip at 5 μl/min for 10 min. In order to couple codeine to the amine surface, codeine was modified at its hydroxyl group with a succinimidyl group by placing 10 mM codeine in a pyridine solution containing 40 mM disuccinimidyl carbonate and 40 mM 4-dimethylamino pyridine. This modification reaction was allowed to take place for 30 min and the reaction mixture was subsequently diluted with 100 mM sodium borate (pH 7.0) in a 1:1 v/v ratio. Trenbolone (Figure 2B), a small molecule structurally distinct from codeine, was modified in the same manner for use as a background response. The modified trenbolone and codeine molecules were separately coupled onto flow cells 1 and 2 of the sensor chip, respectively, by alternating injections for 7 min at 5 μl/min for a total of 28 min for each molecule. After ligand coupling, the chip was deactivated with 1 M ethanolamine (pH 8.5) and primed twice with binding buffer.
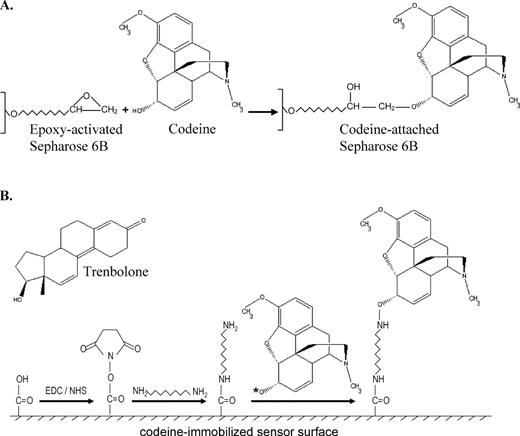
Schematics of the codeine-immobilized surfaces used in the in vitro selection process and SPR binding property assay. Illustration of the chemistries used for codeine coupling to the (A) Sepharose matrix and (B) Biacore CM5 sensor chip surface. Note that the codeine-immobilized sensor surface more closely mimics that of the affinity matrix used during the aptamer selection process versus coupling methods that employ a protein linker. The asterisk next to the oxygen group of codeine in (B) represents a succinimidyl group (the same group that is covalently attached to the carboxyl group of the sensor surface after EDC/NHS activation), which reacts with the amine group of the 1,8-diaminooctane linker. Codeine is thereby immobilized onto the chip surface through the same functional group used to attach it to the affinity matrix during the selection process. Trenbolone, the negative control molecule, is immobilized to the chip surface through the same chemistry and its structure is shown in (B).
RNA samples (initial pool, final pool and randomly-selected individual sequences from the final pool) were prepared for Biacore analysis using the Ampliscribe T7 High Yield Transcription Kit (Epicentre) following the manufacturer's instructions. Samples were sequentially injected over the sensor surface for 1.5 min at 5 μl/min with a 2 min dissociation time. For each sample, various RNA concentrations were injected by serially diluting samples from 48 to 0.375 μM along with two blank samples containing just binding buffer for use as double referencing. After each run, the surface was regenerated with 10 mM EGTA for 2 min at 5 μl/min. The raw data were processed and analyzed to determine the binding constant for each aptamer using Scrubber (Biologic Software, Pty, Australia, http://www.cores.utah.edu/interaction/).
Isocratic affinity elution and specificity assays
Radiolabeled FC5 and FC45 RNA were prepared using the Ampliscribe T7 High Yield Transcription Kit with minor modifications to the manufacturer's instructions (3 mM each rATP, rCTP, rUTP, 150 μM rGTP and 50 μCi [α-32P]GTP). After 3 h of incubation, DNase I was added to the transcription mixture and the reaction was incubated at 37°C for 15 min. Unincorporated nucleotides were removed with a NucAway spin column.
Isocratic affinity elution assays were performed on radiolabeled FC5 and FC45 as described previously (27,28). The binding affinities to codeine and morphine in solution were determined using the following equation: Kd = [Lel] × (Vel − Vn)/(Ve − Vel), where Lel is the free ligand concentration used to elute bound RNA, Vel and Ve are the elution volumes for RNA in the presence and absence of free ligand in binding buffer, respectively, and Vn is the column void volume.
Specificity assays were performed by equally dividing the flow-through radiolabeled FC5 and FC45 into three Sepharose columns (300 μl) modified with codeine. After a 30 min incubation, each column was washed with 7 column volumes of binding buffer. Columns were then eluted with a 5 mM solution of the different targets (codeine, morphine or thebaine) in binding buffer, and 5 column volumes of the elution were collected. Collected samples were added to 10 ml of Safety-Solve scintillation liquid (Reseach Products International Corp.) and radioactivity levels were measured on a liquid scintillation counter (Beckman Coulter).
Truncation experiments
Two full-length aptamers with the lowest determined Kd values were truncated primarily into four different sequences containing distinct regions of their parent sequences: (i) the random region (Ran), (ii) the cloning region (Cln), (iii) the random region and the 5′ constant terminus (L) and (iv) the random region and the 3′ constant terminus (R). Predicted secondary structures formed by these truncated sequences were examined using mfold (29) and RNAstructure (http://rna.chem.rochester.edu/RNAstructure.html). Sequences that adopt well-defined secondary structures were selected for subsequent Kd determination through the described small molecule-aptamer binding affinity SPR assay.
Structural probing assay
Structural probing of the FC5 and FC45 full-length aptamers was performed using a lead ion cleavage assay as described by Berens et al. (30) with the following slight modifications. 5′ end labeled RNA was incubated in binding buffer containing 0–250 μM codeine and 0.5 mM lead (II) acetate. After a 15 min incubation, the cleavage reactions were stopped by adding 0.5 mM EDTA and 1 μg/μl glycogen and the cleaved RNA was recovered by ethanol precipitation. Radiolabeled RNA was also subject to RNase T1 cleavage (Ambion) and alkaline hydrolysis (Ambion) following the manufacturer's instructions to be used as ladders. The recovered RNA samples were separated by electrophoresis on a 10% polyacryamide/8 M urea gel in 1× Tris–borate buffer. The gel was dried and the RNA cleavage patterns were imaged with an FX phosphorimager (BioRad).
Dopamine aptamer binding assay
For direct coupling of dopamine to the sensor surface, a CM5 sensor chip was activated with EDC/NHS as described above. Following the EDC/NHS activation step, a 10 mM dopamine, 50 mM sodium borate (pH 8.5) solution was injected over the activated sensor surface for 30 min at 5 μl/min to couple dopamine to the surface through its amino group. This is the same chemistry used in the selection of dopamine-binding aptamers described by Mannironi et al. (18). After dopamine immobilization, the sensor surface was deactivated with 1 M ethanolamine for 10 min at 5 μl/min to block the remaining unreacted succinimidyl groups. The previously selected dopamine-binding dopa2 RNA aptamer (18) was synthesized using a similar transcription procedure as described above. Various concentrations of this RNA sample were injected over the dopamine-coupled sensor surface and concentration-dependent binding responses were recorded and subsequently analyzed for binding properties as described previously.
For indirect coupling of dopamine to the sensor surface through a BSA protein linker, a CM5 sensor chip was activated with EDC/NHS as described above. Following the EDC/NHS activation step, BSA was injected over the activated surface at 5 μl/min until a signal of 12 500 response units (RU) was reached. A 0.2 M EDC and 0.1 M dopamine solution was injected over the BSA-immobilized surface for 30 min at 5 μl/min to couple dopamine to BSA. This chemistry couples dopamine to BSA through the same functional group as in the direct coupling chemistry. The remaining steps in the indirect coupling method are identical to those used in the direct coupling method.
RESULTS
Selection of codeine-binding RNA aptamers
A slightly modified in vitro selection procedure was used to isolate codeine-binding RNA aptamers from a library of RNA molecules containing a 30 nt random region flanked by constant primer-binding sequences (Figure 1A). Aptamers were selected on a codeine affinity column, which was made by immobilizing codeine to the epoxy-activated agarose through its hydroxyl group (Figure 2A). To enhance the stringency of the selection process, the wash volume was increased incrementally from cycles 6 to 15. To increase the specificity of the selected pool, a counter-selection with a 5 mM morphine solution was performed at cycle 10 prior to elution with codeine. In addition, a total of three error-prone PCR steps were carried out for the DNA template pools of cycles 11, 12 and 13, respectively, to potentially introduce sequences that are of slightly diverse nucleotide composition and search a larger sequence space for higher affinity binders. After cycle 15, the enriched pool was cloned and approximately 60 colonies were sequenced.
Sequence analysis of codeine aptamers reveals fairly conserved motifs
Sequence analysis of the selected clones revealed five sets of completely identical sequences, 2 with 1 nt difference (FC5/B10 and A28/B11), and the rest unique sequences (Figure 1B). Nucleotide deletions or insertions were observed in a few clones. No single consensus sequence that is conserved among the entire population of the selected clones was discovered. However, several fairly conserved but short sequences were found to exist in many clones. For instance, the motif AAGGG is present in over 50% of the sequenced aptamer population. In addition, most clones contain stretches of G's and/or UG's, which suggests that these stretches may be critical to codeine-binding. Similarly, none of the selected clones exhibit obvious predicted structural similarities. It was also observed that most of the sequences do not possess a predominant predicted structure based on analysis with folding programs, such as mfold and RNAstructure, and may adopt different motifs depending on the inclusion or exclusion of part or all of the constant regions in the structural analysis. As a result, further analysis through truncation experiments was conducted to determine the crucial sequences involved in codeine-binding for two aptamers (FC5 and FC45) that exhibited the highest binding affinities of those analyzed.
Qualitative assessment of codeine-binding affinity of the enriched final pool
The codeine-binding affinity of the final pool was qualitatively assessed by monitoring eluted levels of the radiolabeled aptamer pool using codeine affinity chromatography. Radiolabeled RNA from the enriched pool was incubated with codeine-modified and unmodified columns. The eluted RNA from each column was run on a polyacrylamide gel and visualized with a phosphorimager (Supplementary Figure 1). Significantly stronger radioactive signals were detected in the sample eluted from the codeine affinity column than that eluted from the unmodified column, indicating that the RNA aptamers in the final pool are highly enriched in codeine-binding affinity.
Determination of small molecule-aptamer binding constants using a direct coupling SPR assay
Quantitative assessment of the codeine-binding properties of the final pool, the initial pool and several aptamers from the final pool was performed using a modified SPR assay developed on a Biacore 2000. Previous studies where SPR was used to determine binding affinities between aptamers and non-protein targets involved the use of BSA or biotin/streptavidin as intermediate linkers between the sensor surface and the target molecules (24,26). Here we employ a direct coupling approach, similar to a previously described method (22,23), in which the small molecule target is directly coupled to the sensor surface without a supporting intermediate, such as BSA or biotin/strepavidin. Previous direct coupling strategies have used target molecules that contain an amine group (22,23), which is a commonly used functional group in Biacore sensor chip immobilization strategies. However, since codeine does not contain an amine group, a chemical modification strategy was developed to directly couple codeine to the chip surface through its hydroxyl group. In this coupling strategy codeine is first modified at its hydroxyl group with an amine-reactive succinimidyl group. This chemical modification enables codeine molecules to readily react with the amine groups attached to the activated chip surface (Figure 2B). Trenbolone was also immobilized onto the sensor surface in the same manner and used as a negative control molecule. Following the immobilization of codeine and trenbolone in their respective flow cells of the sensor chip, serial dilutions of RNA samples were injected into these flow cells. The response detected from the trenbolone-immobilized flow cell was used as the background subtraction in evaluating the binding constants. An equilibrium binding curve was generated from concentration-dependent binding response data for each sample to determine the corresponding Kd value.
The binding data from the SPR assay supports the qualitative binding data obtained from the chromatography-based assay. The data indicate that there was little to no detectable binding (Figure 3A) between the initial pool and codeine, whereas the final pool bound codeine with significant binding responses (Figure 3B). The overall Kd value of the final pool was evaluated to be ∼15 μM, whereas that of the initial pool was estimated to be in the high millimolar range. This latter value is only an estimate as no binding curve could be established for the initial pool due to its insufficient binding response. Therefore, codeine-binding affinity of the final pool was enhanced over 1000-fold from that of the initial pool. The Kd values of the analyzed aptamer clones are listed in Table 1. Several of the aptamer sequences have Kd values that are much lower than that of the enriched final pool. Two of the highest binding aptamers FC45 and FC5, with Kd values of 2.50 ± 0.06 μM and 4.00 ± 0.13 μM, respectively, were subject to further characterization studies (Figure 4). Despite their similar affinities for codeine, FC5 and FC45 may form different binding pockets since their corresponding mini-aptamers adopt different predicted secondary structures supported by structural studies described in a later section. In addition, FC5 and FC45 exhibit fairly different binding kinetics (Table 2), where the latter has faster kinetics (both binding and dissociation) than that of the former. Some clones, such as FC3, FC13, FC34 and C9 have observed dissociation constants on the same order as that of FC5, while other clones, such as FC23, A3, A20, B11, C15 and C23 exhibit similar dissociation kinetics to FC45 (data not shown). The kinetic data of the modified FC5 and FC45 sequences discussed in later sections are also reported in Table 2.
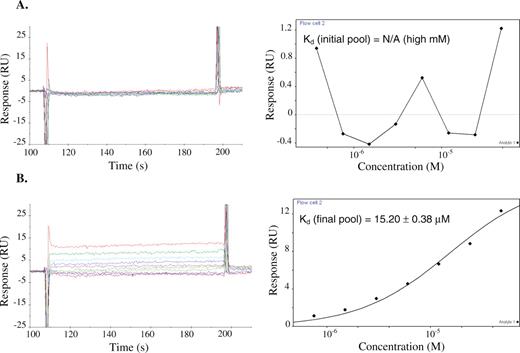
Concentration-dependent codeine-binding responses (left) and the corresponding equilibrium binding curve (right) of (A) the initial pool and (B) the enriched final pool. Codeine was coupled to the sensor chip as described. Serial dilutions of the appropriate RNA sample were injected across the sensor surface and binding responses were recorded over time. Kinetic rate constants were determined by examining the rate of change of binding response when the RNA samples were initially injected over the surface until equilibrium responses were reached (kon) and when a solution lacking the RNA sample was injected over the surface once equilibrium levels were bound to the chip surface (koff). Equilibrium binding constants (Kd) were determined by plotting the equilibrium binding response versus the RNA sample concentration and calculating the corresponding RNA concentration at which half of the maximal response was achieved. Binding responses were adjusted for background binding by subtracting responses of the corresponding RNA samples determined from a trenbolone-coupled sensor surface.
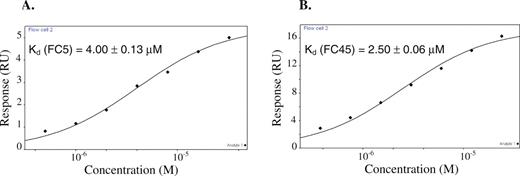
Equilibrium codeine-binding curves of (A) FC5 and (B) FC45.
Codeine-binding affinities of the full-length aptamer sequences as determined from the direct coupling SPR assay
| RNA sample | Kd (μM) | RNA sample | Kd (μM) | RNA sample | Kd (μM) |
| Final pool | 15.20 ± 0.38 | FC27 | 10.90 ± 0.95 | B11 | 5.80 ± 0.29 |
| Initial pool | N/A (high mM) | FC34 | 28.00 ± 1.42 | B12 | 8.80 ± 0.44 |
| FC3 | 28.60 ± 0.96 | FC45 | 2.50 ± 0.06 | C4 | 78.00 ± 4.15 |
| FC5 | 4.00 ± 0.13 | A3 | 11.50 ± 0.27 | C9 | 9.17 ± 0.33 |
| FC13 | 14.50 ± 0.58 | A20 | 13.00 ± 0.65 | C12 | 8.88 ± 0.39 |
| FC17 | 43.70 ± 1.23 | A25 | 7.23 ± 0.34 | C15 | 7.67 ± 0.26 |
| FC21 | 23.60 ± 1.22 | B2 | 4.75 ± 0.32 | C23 | 8.18 ± 0.23 |
| FC23 | 19.10 ± 1.49 | B9 | 5.77 ± 0.36 |
| RNA sample | Kd (μM) | RNA sample | Kd (μM) | RNA sample | Kd (μM) |
| Final pool | 15.20 ± 0.38 | FC27 | 10.90 ± 0.95 | B11 | 5.80 ± 0.29 |
| Initial pool | N/A (high mM) | FC34 | 28.00 ± 1.42 | B12 | 8.80 ± 0.44 |
| FC3 | 28.60 ± 0.96 | FC45 | 2.50 ± 0.06 | C4 | 78.00 ± 4.15 |
| FC5 | 4.00 ± 0.13 | A3 | 11.50 ± 0.27 | C9 | 9.17 ± 0.33 |
| FC13 | 14.50 ± 0.58 | A20 | 13.00 ± 0.65 | C12 | 8.88 ± 0.39 |
| FC17 | 43.70 ± 1.23 | A25 | 7.23 ± 0.34 | C15 | 7.67 ± 0.26 |
| FC21 | 23.60 ± 1.22 | B2 | 4.75 ± 0.32 | C23 | 8.18 ± 0.23 |
| FC23 | 19.10 ± 1.49 | B9 | 5.77 ± 0.36 |
Codeine-binding affinities of the full-length aptamer sequences as determined from the direct coupling SPR assay
| RNA sample | Kd (μM) | RNA sample | Kd (μM) | RNA sample | Kd (μM) |
| Final pool | 15.20 ± 0.38 | FC27 | 10.90 ± 0.95 | B11 | 5.80 ± 0.29 |
| Initial pool | N/A (high mM) | FC34 | 28.00 ± 1.42 | B12 | 8.80 ± 0.44 |
| FC3 | 28.60 ± 0.96 | FC45 | 2.50 ± 0.06 | C4 | 78.00 ± 4.15 |
| FC5 | 4.00 ± 0.13 | A3 | 11.50 ± 0.27 | C9 | 9.17 ± 0.33 |
| FC13 | 14.50 ± 0.58 | A20 | 13.00 ± 0.65 | C12 | 8.88 ± 0.39 |
| FC17 | 43.70 ± 1.23 | A25 | 7.23 ± 0.34 | C15 | 7.67 ± 0.26 |
| FC21 | 23.60 ± 1.22 | B2 | 4.75 ± 0.32 | C23 | 8.18 ± 0.23 |
| FC23 | 19.10 ± 1.49 | B9 | 5.77 ± 0.36 |
| RNA sample | Kd (μM) | RNA sample | Kd (μM) | RNA sample | Kd (μM) |
| Final pool | 15.20 ± 0.38 | FC27 | 10.90 ± 0.95 | B11 | 5.80 ± 0.29 |
| Initial pool | N/A (high mM) | FC34 | 28.00 ± 1.42 | B12 | 8.80 ± 0.44 |
| FC3 | 28.60 ± 0.96 | FC45 | 2.50 ± 0.06 | C4 | 78.00 ± 4.15 |
| FC5 | 4.00 ± 0.13 | A3 | 11.50 ± 0.27 | C9 | 9.17 ± 0.33 |
| FC13 | 14.50 ± 0.58 | A20 | 13.00 ± 0.65 | C12 | 8.88 ± 0.39 |
| FC17 | 43.70 ± 1.23 | A25 | 7.23 ± 0.34 | C15 | 7.67 ± 0.26 |
| FC21 | 23.60 ± 1.22 | B2 | 4.75 ± 0.32 | C23 | 8.18 ± 0.23 |
| FC23 | 19.10 ± 1.49 | B9 | 5.77 ± 0.36 |
Dissociation rate constants (koff) for codeine binding of the final pool, FC5, FC45 and their corresponding truncated sequences
| RNA sample | koff (1/s) % | RNA sample | koff (1/s) % |
| Final pool | 7.62×10−3 ± 5.81 | Initial pool | N/A |
| FC5 | 6.50×10−3 ± 3.78 | FC45 | 1.14×10−2 ± 3.35 |
| FC5L | 6.70×10−3 ± 2.80 | FC45L | 1.03×10−2 ± 3.61 |
| FC5L-S1 | 6.65×10−3 ± 2.79 | FC45L-S1 | 6.84×10−3 ± 3.98 |
| FC5L-S2 | 6.54×10−3 ± 2.44 | FC45L-S2 | 2.43×10−3 ± 3.29 |
| FC5L-S3 | 4.79×10−3 ± 5.00 | FC45L-S3 | 2.69×10−3 ± 2.23 |
| RNA sample | koff (1/s) % | RNA sample | koff (1/s) % |
| Final pool | 7.62×10−3 ± 5.81 | Initial pool | N/A |
| FC5 | 6.50×10−3 ± 3.78 | FC45 | 1.14×10−2 ± 3.35 |
| FC5L | 6.70×10−3 ± 2.80 | FC45L | 1.03×10−2 ± 3.61 |
| FC5L-S1 | 6.65×10−3 ± 2.79 | FC45L-S1 | 6.84×10−3 ± 3.98 |
| FC5L-S2 | 6.54×10−3 ± 2.44 | FC45L-S2 | 2.43×10−3 ± 3.29 |
| FC5L-S3 | 4.79×10−3 ± 5.00 | FC45L-S3 | 2.69×10−3 ± 2.23 |
The corresponding association rate constant (kon) is equivalent to koff/Kd.
Dissociation rate constants (koff) for codeine binding of the final pool, FC5, FC45 and their corresponding truncated sequences
| RNA sample | koff (1/s) % | RNA sample | koff (1/s) % |
| Final pool | 7.62×10−3 ± 5.81 | Initial pool | N/A |
| FC5 | 6.50×10−3 ± 3.78 | FC45 | 1.14×10−2 ± 3.35 |
| FC5L | 6.70×10−3 ± 2.80 | FC45L | 1.03×10−2 ± 3.61 |
| FC5L-S1 | 6.65×10−3 ± 2.79 | FC45L-S1 | 6.84×10−3 ± 3.98 |
| FC5L-S2 | 6.54×10−3 ± 2.44 | FC45L-S2 | 2.43×10−3 ± 3.29 |
| FC5L-S3 | 4.79×10−3 ± 5.00 | FC45L-S3 | 2.69×10−3 ± 2.23 |
| RNA sample | koff (1/s) % | RNA sample | koff (1/s) % |
| Final pool | 7.62×10−3 ± 5.81 | Initial pool | N/A |
| FC5 | 6.50×10−3 ± 3.78 | FC45 | 1.14×10−2 ± 3.35 |
| FC5L | 6.70×10−3 ± 2.80 | FC45L | 1.03×10−2 ± 3.61 |
| FC5L-S1 | 6.65×10−3 ± 2.79 | FC45L-S1 | 6.84×10−3 ± 3.98 |
| FC5L-S2 | 6.54×10−3 ± 2.44 | FC45L-S2 | 2.43×10−3 ± 3.29 |
| FC5L-S3 | 4.79×10−3 ± 5.00 | FC45L-S3 | 2.69×10−3 ± 2.23 |
The corresponding association rate constant (kon) is equivalent to koff/Kd.
The affinities of the two highest binding aptamers, FC5 and FC45, to codeine in solution were also determined through a standard isocratic affinity elution method (27,28). This control enables the comparison of the surface-based binding affinities determined with the described SPR assays to the solution-based affinities. The determined solution binding affinity of FC45 (Kd = 4.5 μM) was very similar to its surface binding affinity (Kd = 2.5 μM), whereas FC5 was determined to bind free codeine with an ∼10-fold lower affinity (Kd = 47 μM) than that to surface-immobilized codeine (Kd = 4.0 μM). For a given aptamer-ligand pair, the binding affinities for free target in solution and a target immobilized onto a solid support may differ, as has been observed in previous studies (19,31,32). For the aptamers studied here, FC5 shows differing affinities for free and immobilized codeine, whereas FC45 exhibits similar binding affinities.
Assays reveal distinct specificities of the codeine-binding aptamers to other benzylisoquinoline alkaloid targets
The ability of FC5 and FC45 to distinguish between three similar BIA molecules, codeine, thebaine and morphine, was determined using a chromatography-based assay. Radiolabeled RNA aptamers were eluted with codeine, morphine and thebaine, which are all closely related structural analogues (Figure 5A). Eluted FC5 and FC45 demonstrated ∼4- and 6-fold increases in radioactivity counts, respectively (Figure 5B), when eluted with codeine versus morphine. The semi-quantitative molecular specificities of these aptamers were supported by isocratic affinity elution experiments in which the solution affinities of these aptamers were determined and observed to differ by similar magnitudes. The solution affinity for FC45 was determined to be ∼4.5 μM to codeine and 25 μM to morphine, whereas the solution affinity for FC5 was determined to be ∼47 μM to codeine and 212 μM to morphine. These results demonstrate that the single morphine counter-selection performed during the in vitro selection process was effective at enhancing the specificity of the aptamers in the final pool to codeine over morphine. While aptamers that discriminate between molecules that differ by a single methyl group have been described previously for purine alkaloid targets (33,34), these results indicate that aptamers can exhibit this level of molecular discrimination in spite of the presence of the bulky 4 six-membered rings in the BIA targets examined here.
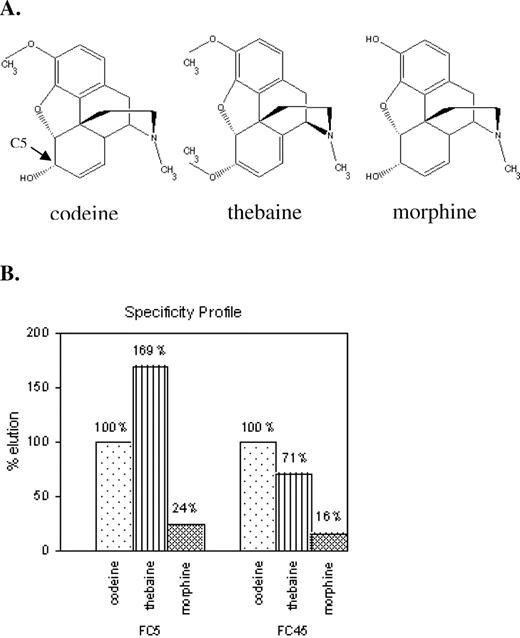
The FC5 and FC45 aptamers exhibit differing specificities to BIA structural analogues. (A) Structures of the three BIA molecules, codeine, thebaine and morphine, used in examining aptamer specificity. (B) Specificity elution profiles of the FC5 and FC45 aptamers. Radiolabeled aptamers were incubated with a codeine-modified Sepharose matrix. The bound aptamers were subsequently eluted with the different BIA targets and radioactivity levels in the eluted fractions were measured. Radioactivity levels were normalized with respect to values obtained from the codeine elutions for each aptamer.
These assays also demonstrate that these two aptamers exhibit differing specificities to thebaine. The eluted FC5 exhibited nearly a 2-fold increase in radioactivity counts when eluted with thebaine versus codeine, whereas FC45 exhibited an ∼30% decrease in signal. These results indicate that FC5 exhibits higher specificity for thebaine over codeine, whereas FC45 exhibits higher specificity for codeine over thebaine. It should be noted that during the selection process codeine was coupled to the Sepharose column in such a way that there was no differentiable functional group between codeine and thebaine. With the attachment chemistry used in these studies through the functional group at C5, these two molecules exhibit conformational differences in that the former has one double bond in the C5-six-membered ring, whereas the latter contains two (Figure 5A). These results suggest that aptamers can potentially perform molecular discrimination at the level of conformation, as the difference between these two targets is at the level of torsional structure of the ring backbone.
Characterization of mini-aptamers that demonstrate binding affinities similar to the full-length aptamers
Truncation experiments were systematically performed on the full-length FC5 and FC45 aptamers to identify minimal aptamer domains, or mini-aptamers. Various truncated aptamer sequences were characterized for their codeine-binding properties. Truncated sequences that form well-defined secondary structures as predicted by mfold or RNAstructure were selected for further analysis. The described SPR small molecule-aptamer binding assays were employed to determine the codeine-binding affinities of these truncated sequences.
An FC5 mini-aptamer was identified by characterizing three truncated sequences of the FC5 full-length aptamer. The codeine-binding properties of the random region (FC5Ran), which is the N30 region of the aptamer library; the cloning region (FC5Cln), which includes the random region, most of the 3′ constant terminus, and part of the 5′ constant terminus; and FC5L, which includes the random region and the 5′ constant terminus, were analyzed using the described SPR binding assay. No binding was observed between FC5Ran and the codeine-immobilized sensor surface, indicating that the FC5 random region is not sufficient for the codeine-binding properties of this aptamer. FC5Cln demonstrated a significantly reduced affinity to codeine (Kd = 39.50 ± 2.27 μM), suggesting that the remainder of the 5′ constant terminus of FC5 may play an important role in the formation of the correct binding pocket for codeine. FC5L binds codeine with an affinity similar to that of its full-length (59 nt) parent sequence (Kd = 4.55 ± 0.14 μM) despite its significantly reduced length (41 nt). These results indicate that FC5L, referred to as FC5 mini-aptamer, contains the necessary and sufficient sequence within FC5 for binding codeine (Figure 6A and C).
An FC45 mini-aptamer was identified by characterizing two truncated sequences of the FC45 full-length aptamer. The codeine-binding properties of the cloning region (FC45Cln), which includes the random region, most of the 3′ constant terminus, and part of the 5′ constant terminus; and FC45L, which includes the random region and the 5′ constant terminus, were analyzed using the described SPR binding assay. FC45Ran, harboring the N30 region of the library, was not analyzed in this set of truncation experiments, as there was no well-defined secondary structure predicted for this sequence by mfold or RNAstructure. FC45Cln did not exhibit binding to codeine, suggesting that the codeine-binding pocket was not correctly formed within the secondary structure adopted by this sequence. However, FC45L (44 nt) binds codeine with an affinity (2.59 ± 0.09 μM) that is almost identical to that of the full-length FC45 sequence (Figure 6B and D). Therefore, this FC45 mini-aptamer includes the sequence within FC45 required to form the correct binding pocket for codeine in contrast to that of FC45Cln.
These truncation experiments support the importance of the formation of the correct binding pocket for aptamer molecular recognition capabilities. In addition, both FC5 and FC45 mini-aptamers lack the 3′ constant terminal sequence, indicating that the 3′ terminus is not involved in binding codeine. Secondary structure predictions from mfold and RNAstructure indicate that the 3′ terminus forms a small hairpin (Supplementary Figure 3C), isolating itself from the remaining sequences of FC5 and FC45. The proposed secondary structures of the FC5 and FC45 mini-aptamers (Figure 6) are supported by the structural modification and structural probing experiments described in the next section.
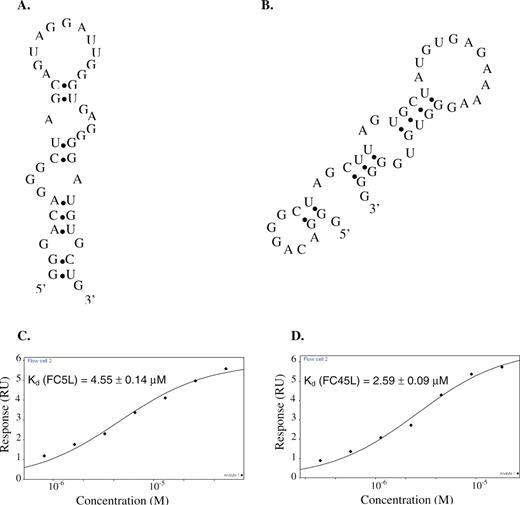
Codeine-binding mini-aptamer characterization. Proposed secondary structures from mfold of (A) the FC5 mini-aptamer (FC5L) and (B) the FC45 mini-aptamer (FC45L), and the corresponding equilibrium codeine-binding curves of (C) FC5L and (D) FC45L.
Characterization of modified mini-aptamer sequences supports the proposed secondary structures
The proposed secondary structures of the FC5 and FC45 mini-aptamers do not possess a strong base stem (Figure 6) in comparison to other reported aptamer structures. For instance, the tetracycline minimer (30) has a base stem that is comprised of five base-pairs, which contribute to the stability of the overall secondary structure of the minimer. Sequences lacking strong or stabilized base stems may adopt a number of possible secondary structures, whereas a stabilized base stem can significantly reduce presumed structural variability and therefore restrict a given aptamer sequence to adopt a very few, and in some cases just one, distinct structures. Therefore, the proposed secondary structures of the FC5 and FC45 mini-aptamers may be evaluated by examining the binding properties of these aptamers modified with stabilized base stems.
The base stems of the mini-aptamers were modified with an extension of GC base pairs to stabilize the proposed structures of these mini-aptamers. The FC5 mini-aptamer (FC5L) was stabilized by extending the existing 3 bp stem with two GC base pairs (Figure 7B), based on the assumption that a few nucleotides present on each end of the original mini-aptamer are unessential for codeine-binding. Similarly, the FC45 mini-aptamer (FC45L) was stabilized by extending the base stem formed by the 5′-CUU and 3′-GGG pairing with two GC base pairs (Figure 8B), excluding several nucleotides from the 5′ end. Following the modification, the structures of these stabilized mini-aptamers were further analyzed in mfold using the DotPlot Partition Function, which confirms these structures to be the most favorable ones to adopt among others. The codeine-binding properties of the resulting mini-aptamers, referred to as FC5L-S1 and FC45L-S1, respectively, were determined using the described SPR assay. FC5L-S1 and FC45L-S1 were determined to bind codeine with Kd values of 5.51 ± 0.23 μM and 4.18 ± 0.48 μM, respectively (Supplementary Figure 2A and B). These results indicate that the modified mini-aptamers bind the target molecule codeine with affinities similar to the corresponding unmodified mini-aptamers. Therefore, these results support the proposed secondary structures of the FC5 and FC45 mini-aptamers (Figure 6) and that their codeine-binding affinities were minimally affected by extending the original base stems. Structural probing studies were performed on FC5 and FC45 full-length aptamers using a standard lead-based cleavage assay to confirm the structures predicted through the SPR analysis. Lead-induced and RNase T1 cleavage patterns were observed to be in agreement with the corresponding proposed structures (Supplementary Figure 4).
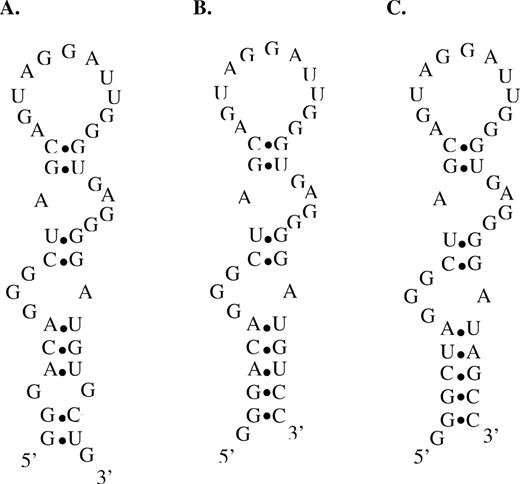
Structural stabilization and sequence requirements of the FC5 mini-aptamer stems. Proposed secondary structures from mfold of (A) the original FC5 mini-aptamer (FC5L), (B) the FC5 mini-aptamer with a stabilized base stem (FC5L-S1), (C) the FC5 mini-aptamer with a stabilized base stem composed of randomly-selected nucleotides (FC5L-S2).
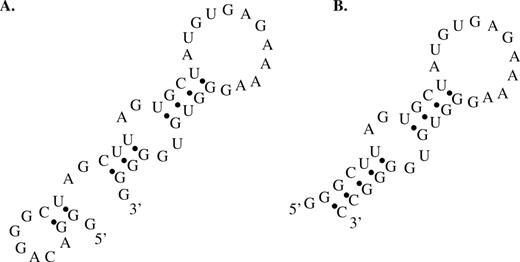
Structural stabilization of the FC45 mini-aptamer. Proposed secondary structures from mfold of (A) the original FC45 mini-aptamer (FC45L) and (B) the FC45 mini-aptamer with a stabilized base stem (FC45L-S1) in which several nucleotides at the termini of the original mini-aptamer are truncated.
The sequence requirements and flexibility of the mini-aptamer base stems were examined with directed mutational analysis coupled with characterization of the effects of these sequence changes on the codeine-binding properties of these aptamers by the described SPR assays. Two of the three original base pairs in the base stem of the FC5 mini-aptamer were replaced with randomly-selected base-pairs (Figure 7C). This new sequence (FC5L-S2) was determined to bind codeine with an affinity (Kd = 5.39 ± 0.28 μM) (Supplementary Figure 2C) comparable to that of the original aptamer sequence, indicating that while the presence of the base stem is essential for codeine-binding, its sequence is not. The sequence space flexibility demonstrated for the aptamer base stem of FC5L has been reported in other aptamers, such as the theophylline aptamer (33).
Studies were also conducted to demonstrate that the formation of the correct binding pocket within a given aptamer sequence is highly dictated by the formation of the correct base stem. The base stems of two alternative secondary structures for the FC45 mini-aptamer were extended with two GC-pairs to stabilize these proposed secondary structures (Supplementary Figures 3A and B), in the same way as previously described for FC45L-S1. Binding assays revealed that these structures did not bind codeine with as high affinity as the initially proposed structure. The Kd values of these alternative FC45 mini-aptamer structures were increased ∼10-fold (∼25 μM), indicating that the codeine-binding pocket may be somewhat disrupted in these structures. These results indicate that the formation of the correct base stem can have significant influence on the formation of the correct binding pocket for aptamer recognition events.
Validation of the direct coupling SPR assay for characterization of small molecule-aptamer binding properties
Biacore assays are widely used to study a variety of molecular interactions, such as RNA–protein and protein–protein interactions. While these assays are applicable to a broad range of target molecules, proteins have most often been used as the primary targets. Although, Biacore assays have been used to measure the interaction between aptamers and non-protein targets, these assays often include a carrier or linker protein between the target and the sensor surface (24,26). However, significant discrepancies have been observed in Kd values determined from these assays and other commonly used methods potentially due to the use of a linker protein between the dextran surface and the small molecule. While SPR assays in which the small molecule target is directly coupled to the sensor surface without inclusion of a linker protein have been reported previously (22,23), the observed binding properties have not been validated or proven to potentially eliminate non-specific interactions or artifacts that may arise from the presence of a linker protein used in the assay. Therefore, experiments were conducted to examine the reproducibility, accuracy and versatility of these direct coupling assays.
The reproducibility of the assay method was confirmed through several means. Binding assays were repeated for several samples: two randomly-selected sequences (FC34 and A25), the initial pool, FC5 and FC45. The codeine-binding affinities determined from these replicate experiments were nearly identical, thereby confirming the reproducibility of the assay method (Supplementary Table 1). In addition, the assay was repeated for the initial pool, FC5 and FC45 such that the concentration series sets of these samples were injected into the flow cells in a random order. Consistent Kd values (data not shown) were obtained from the random-injection experiments for all three of the tested RNA samples when compared to the values obtained from injecting them sequentially from lowest to highest concentrations. These results demonstrate the reproducibility and the robustness of this direct coupling assay method. FC45 was used as a positive control when performing the described assays on the remainder of the RNA aptamer sequences.
The potential elimination of non-specific interactions between an aptamer and the linker protein by the direct coupling small molecule-aptamer binding assay was demonstrated on a previously characterized RNA aptamer to a different small molecule target. The described SPR binding assay was performed on a previously characterized dopamine aptamer (dopa2) (18), whose reported Kd value was determined through commonly used solution-based affinity methods. Dopamine was immobilized onto the sensor chip through the same coupling chemistry that was used in the original selection of this dopamine-binding aptamer. The binding affinity determined through the direct coupling SPR assay of the dopa2 RNA aptamer to dopamine (Kd = 2.71 ± 0.06 μM) was nearly identical to the reported value of 2.8 μM (18) (Supplementary Figure 5A).
An indirect coupling assay was performed on the dopamine aptamer using BSA as a protein linker to demonstrate that the presence of a linker protein in a SPR small molecule-aptamer binding assay may generate non-specific interactions or artifacts. It was observed that aptamer samples at the same concentrations take considerably longer to reach an equilibrium binding response in the BSA linker assay versus the direct coupling assay, which is indicative of non-specific interactions. The two highest concentration samples reached a near equilibrium response after 50 min of injection, approximately 33 times longer than that employed in the direct coupling assay. Data analysis revealed that the aptamer binding affinity was significantly affected and resulted in a false assessment as the observed Kd value was substantially higher than the reported value of 2.8 μM (Supplementary Figure 5B). To better analyze the data, Clamp (35) was used to fit the kinetic binding responses, as the two highest concentration samples did not completely reach equilibrium. Kinetic data analysis suggested that multiple binding events are present in the BSA linker assay since the data were well-fit with a multiple binding site model and did not satisfy a one-to-one binding model (Supplementary Figures 5C and D). This finding was further supported by Scatchard plot analysis, which also suggested this indirect coupling assay as a multiple binding site system represented by a curvature in this plot, a hallmark of a multiple binding site model (Supplementary Figure 5F). In contrast, the direct coupling data fit a single binding site system represented by a linear fit to this data (Supplementary Figure 5E). These results indicate that the presence of a protein linker can cause an aptamer to bind to its surface-immobilized target molecule in a non-specific manner, leading to an inaccurate assessment of the binding affinity of the aptamer to its small molecule target.
DISCUSSION
In this study, we employed in vitro selection strategies to isolate RNA aptamers with high affinity and specificity to a subclass of BIA molecules, including codeine, within 15 selection cycles. A counter-selection with morphine and three error-prone PCR steps were incorporated into the selection process to enhance the specificity and affinity of the selected aptamers for their target molecule. The qualitative binding assays revealed that the final aptamer pool was highly enriched with codeine-binding affinity. The binding affinity of the enriched aptamer pool was determined to be 15 μM when characterized through the described Biacore assay; however, several of its member sequences, including FC5 and FC45, were determined to have higher affinities to codeine. In addition both of these aptamers were shown to be highly specific to codeine over morphine, indicating that the morphine counter-selection performed during the selection process was effective at enhancing the desired target specificity of the aptamers. Interestingly, while FC45 maintains codeine-binding specificity over another structural analogue, thebaine, FC5 demonstrates higher specificity to the latter. Therefore, these aptamers exhibit differing specificities to BIA alkaloid molecules, displaying molecular discrimination between targets differing by a single methyl group or structural conformation.
This work also highlights a direct coupling SPR binding assay for accurately and robustly determining the binding properties of aptamers to small molecule ligands. The described method is based on the direct immobilization of the target small molecule onto the sensor chip surface without inclusion of a linker protein as is commonly used. This direct coupling may provide a more accurate assessment of the binding affinity between the small molecule target and the aptamer by eliminating potential non-specific binding between the nucleic acid aptamer and the protein linker and more accurately reproducing conditions used in the selection process. Significant discrepancies have been observed between reported Kd values obtained from Biacore assays that employ a protein linker connecting the target molecule to the sensor surface and other methods that involve direct target coupling. In one example, BSA was used as a linker between a target carbohydrate and the sensor surface (24). The binding affinity of a selected aptamer was reported as 85 pM using this assay method. However, when the aptamer was immobilized onto the sensor surface and target molecules were injected over the surface, the binding affinity to the BSA-linked target was similar to that observed with the earlier experimental setup (Kd = 57 pM), whereas the binding affinity to the target molecule alone was determined to be ∼60-fold lower (Kd = 3.3 nM). In another example, an existing tobramycin aptamer, characterized with a Biacore binding assay using a streptavidin linker, showed a lower degree of selectivity and significantly reduced affinity (25) from previously reported binding properties for this aptamer determined using a number of different assay methods (36–39). These results indicate that the presence of a protein linker may introduce artifacts or non-specificity in the small molecule-aptamer interaction, preventing an accurate assessment of the intact affinity of the aptamer to its target molecule. Direct coupling of the small molecule target onto the sensor surface may provide a more accurate assessment of small molecule-aptamer binding properties by eliminating potential non-specific interactions or artifacts introduced when using a linker protein.
The direct coupling SPR small molecule-aptamer binding assay has the additional benefit of providing a rapid characterization assay. In comparison to other commonly used binding assays, such as isocratic elution or equilibrium filtration, Biacore assays offer a rapid, high-throughput platform, which provides information about both equilibrium and kinetic binding properties. Using the Biacore 2000 and the serial dilution method described in this work, the binding properties of as many as eight aptamer sequences may be accurately and precisely determined in one day on a single chip. It should be noted, that the binding properties determined through this assay correspond to ligand-immobilized binding properties, which may differ from free ligand binding properties depending on the particular aptamer-ligand pair as demonstrated in this and previous work. However, this high-throughput assay strategy may be particularly useful when applied to the screening of libraries for aptamers that exhibit particular binding properties. From this initial screen, those aptamers exhibiting desired binding affinities for surface-immobilized target may be further analyzed with standard solution affinity assays to determine and verify the corresponding binding affinities of those selected aptamers to free target in solution. Furthermore, the high-throughput nature of this platform may be used to rapidly determine the mini-aptamers for selected aptamers through truncation experiments, eliminating the need to perform time-consuming and labor-intensive chemical probing experiments (18,30). In addition, while traditional binding assays involve the use of radiolabeled aptamers or often rare and expensive radiolabeled target molecules, Biacore assays eliminate this requirement. The same assay methodology may be employed to perform structural stabilization studies, which were used to develop mini-aptamers with stabilized base stems. Aptamers with stabilized, modifiable and extendable base stems are more functionally attractive for applications in downstream molecular design strategies that involve exploiting structural rearrangements associated with the base stem formation (17). Therefore, the FC5 and FC45 mini-aptamers may be readily employed in molecular engineering applications as their stems are extendable and modifiable. Finally, the versatility of this direct coupling SPR assay to the study of small molecule-aptamer interactions was demonstrated through several means and validated on a previously characterized dopamine RNA aptamer. Elimination of non-specific interactions was demonstrated in the direct coupling assay compared to the indirect coupling assay for the same aptamer, where non-specific interactions or binding artifacts arose in the presence of the linker protein. Therefore, the SPR assay discussed here is proven to be a rapid, versatile, accurate and robust method for quantitative measurement of small molecule–RNA interactions.
ACKNOWLEDGEMENTS
The authors thank Professor Pamela J. Bjorkman for generously allowing us the use of the Biacore 2000 instrument, which was used to develop the direct small molecule-aptamer characterization assays. The authors also gratefully acknowledge Dr Laure Jason-Moller and William Hunter (Biacore Life Sciences) and Katie Saliba (Caltech Chemistry Department) for their technical support and advice regarding codeine coupling chemistry to the sensor chip. The authors also thank Dr Kevin Hoff (Caltech Chemical Engineering Department) for his critical reading of the manuscript. This work was funded by Caltech startup funds, the Center for Biological Circuit Design at Caltech (fellowship support for M.N.W.), and the National Institutes of Health (training grant for J.S.K.).
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments