





Abstract
Awadalla, Eyre-Walker, and Maynard Smith (1999) recently argued that there might be recombination in human mitochondrial DNA (mtDNA). Their claim was based on their observation of decaying linkage disequilibrium (LD) as a function of physical distance. Their study was much criticized, and follow-up studies have failed to find any evidence for recombination. We argue that the criticisms levied, even if correct, could not possibly explain the findings of Awadalla, Eyre-Walker, and Maynard Smith (1999) . Nonetheless, the test proposed by Awadalla, Eyre-Walker, and Maynard Smith (1999 ) is not robust because recombination is not the only explanation for decay of LD. We show that such a pattern can be caused by mutational hot spots as well. However, a closer look at the data suggests that the pattern observed was not caused by mutational hot spots but rather by chance. Thus, there appears to be no evidence for recombination in the mtDNA polymorphism data. In conclusion, we discuss the possibility of detecting recombination in mtDNA and the implications of its existence.
Introduction
The claim that there may be recombination in human mitochondrial DNA (mtDNA) (Awadalla, Eyre-Walker, and Maynard Smith 1999 ; Eyre-Walker, Smith, and Maynard Smith 1999 ) has caused a great deal of controversy. The argument for recombination is based on the observation that the pattern of polymorphism in mtDNA is incompatible with a single genealogical tree and unique mutations. The simplest example is the presence of all four possible haplotypes for a pair of diallelic loci. It is easy to show that such a pattern cannot exist in the absence of recombination unless at least one of the loci experienced multiple mutations. In the language of phylogenetics, recurrent mutation to the same allele is an example of “homoplasy” or convergent evolution.
Because incompatibilities can be created by recurrent mutation as well as by recombination, it is necessary to rule out the former explanation in order to conclude that the latter is correct. Eyre-Walker, Smith, and Maynard Smith (1999) argued that, given what is known about mutation rates, there were simply too many homoplasies in the data for multiple mutations to be the explanation. Clearly, such an argument is always dependent on what is assumed about mutation rates. Recognizing this weakness, Awadalla, Eyre-Walker, and Maynard Smith (1999) proposed a much simpler test that seemingly involves fewer assumptions (we will discuss the extent to which this is true later). Their test looks at the spatial behavior of a statistic of association between the alleles at pairs of polymorphic sites. Such associations, also known as “linkage disequilibrium” (LD), will decay as a result of recombination or recurrent mutation. The rationale behind the proposed test is that because the frequency of recombination increases with physical distance, the strength of association should go down with distance. In contrast, the probability of recurrent mutation on either site should be independent of the distance between the sites. Thus, if the strength of association is negatively correlated with distance, we may conclude that recombination is responsible. Awadalla, Eyre-Walker, and Maynard Smith (1999 , 2000) found such a correlation and established its significance by means of a permutation test.
The study of Awadalla, Eyre-Walker, and Maynard Smith (1999) was immediately challenged on two major grounds: In their response, Awadalla, Eyre-Walker, and Maynard Smith (1999) argued that both these objections were, in essence, irrelevant. We agree. Consider first the problem with errors in data. Awadalla, Eyre-Walker, and Maynard Smith (2000) admitted that there were indeed errors in their data but pointed out that errors cannot explain their finding—on the contrary, random sequencing errors would behave like multiple mutations and would therefore tend to obscure any correlation of LD with distance, not establish one.
Errors in data. Kivisild and Villems (2000) showed that several of the polymorphisms analyzed by Awadalla, Eyre-Walker, and Maynard Smith (1999) were likely to be the result of errors in genotyping or data handling (or both). The same point had previously been made by Macaulay, Richards, and Sykes (1999) in response to Eyre-Walker, Smith, and Maynard Smith (1999) .
The choice of LD statistic. Awadalla, Eyre-Walker, and Maynard Smith (2000) used the squared correlation coefficient, r2 (Hill and Robertson 1968 ); several researchers argued that they should have used |D′| (Lewontin 1964 ) instead, because the latter is less dependent on allele frequencies than the former (Jorde and Bamshad 2000 ; Kumar et al. 2000 ).
The argument about the choice of LD statistic is equally unconvincing. Leaving aside the issue of what it means for one LD statistic to be less frequency dependent than another (Lewontin 1988 ; Nordborg and Tavaré 2002 ), it is, as Awadalla, Eyre-Walker, and Maynard Smith (2000) noted, hard to see why frequency dependence should matter. Under the null hypothesis that there is no recombination in mtDNA, there is a single underlying genealogical tree relating all mtDNA copies, and the distribution of allele frequencies must be the same for all sites (modulo differences in the mutation rate). In any case, the argument of Awadalla, Eyre-Walker, and Maynard Smith (1999) was that r2 is not expected to decay with distance unless there is recombination; therefore, the observed decay implies recombination. This argument is not contradicted by the finding that some other statistic which might also be expected to decay with distance does not appear to do so.
In this article, we address two questions. First, in general, is the permutation test proposed by Awadalla, Eyre-Walker, and Maynard Smith (1999) really a robust test of recombination or are there alternative explanations for patterns such as those observed? Second, what explains the pattern they observed; in particular, what explains the discrepancy between their results and those of subsequent studies that have found no evidence for recombination (Ingman et al. 2000 ; Elson et al. 2001 )?
Does a Decay of LD Imply Recombination?
The test proposed by Awadalla, Eyre-Walker, and Maynard Smith (1999) is attractive because it initially appears to be nonparametric (Hey 2000 ). Unfortunately, as is often the case, there are hidden assumptions behind the apparent simplicity. The permutation test randomizes the position of the loci and recalculates the correlation between LD and distance. The rationale for this is that under the null hypothesis of no recombination, position should not matter because every site has the same genealogical history. However, this procedure is not valid in general unless we also assume that the distribution of mutations is the same for all positions (Sawyer 1989 ). Specifically, the test of Awadalla, Eyre-Walker, and Maynard Smith (1999 ) requires that the distribution of r2 (or whatever LD statistic we choose to use) does not depend on the distance between the loci under the null hypothesis that there is no recombination. This may seem like a reasonable assumption, but it is in fact violated if the mutation rate varies regionally.
An example should make this clearer. Consider a chromosome that is divided into two regions, one of which contains multiple mutational hot spots. Because multiple mutations erode LD, LD between pairs of loci in this “hot” region will be much lower than LD between pairs of loci in the “cold” (non–hot spot) region. Significantly, LD between pairs of loci in different regions (one hot, one cold) will also be low. Because the distance between loci in different regions is on an average greater than the distance between loci in the same region, the result is a pattern where high LD is associated with short distance. Thus, as illustrated in figure 1 , mutational hot spots can give rise to a pattern where LD decays with distance, just like the one observed by Awadalla, Eyre-Walker, and Maynard Smith (1999) . If their test were used in such a case, we would falsely conclude that recombination was responsible for the pattern.
Should We Expect LD to Decay in mtDNA?
We have seen that mutational hot spots can, in principle, give rise to a negative correlation between LD and distance. Should we expect such a pattern in human mtDNA? The existence of mutational hot spots in mtDNA is not in doubt. Figure 2 shows the spatial pattern of pairs of sites that show evidence of either recombination or recurrent mutation. Such plots can be used to look for traces of recombination as well as mutational hot spots (Jakobsen and Easteal 1996 , although it should be noted that the significance of patterns is difficult to evaluate for reasons analogous to those described in the previous section, see Does a Decay of LD Imply Recombination?). The pattern in figure 2 strongly suggests the existence of clusters of mutational hot spots. One of these corresponds to the D-loop, which is known to be hypermutable (e.g., Meyer, Weiss, and von Haeseler 1999 ; Ingman et al. 2000 ; Markovtsova, Marjoram, and Tavaré 2000 ). It should also be noted that nonrandom sequencing errors (perhaps caused by particular regions being difficult to sequence) would give rise to hypermutable regions.
However, even though hot spots exist, they do not appear to be as extreme as those assumed in the example of the previous section (see Does a Decay of LD Imply Recombination?). In that example, r2 was low for all pairs of loci that included at least one hot site (see fig. 1 ) and very high only for pairs of cold sites. The most complete mtDNA data set is that of Ingman et al. (2000) . We calculated r2 for all pairs of polymorphic sites in their data. Dividing the comparisons into those involving sites in the D-loop, those involving sites in the coding region, and those involving one site in each region, we found that average r2 was 0.0525, 0.0996, and 0.0705, respectively. These differences do not appear to be large enough to generate a negative correlation between LD and distance, especially when it is taken into account that there will be more polymorphic sites in hot regions, generating a large number of pairs of loci that are close together and have low LD. In fact, for simple models involving two regions with different mutation rates on a circular chromosome, we found that the expected correlation between LD and distance is weakly positive rather than negative, regardless of the length of the hot region (fig. 3 ). This conclusion is in agreement with the data of Ingman et al. (2000) : the plot of r2 against distance for all 287 informative sites shows a slight positive correlation (0.015), which disappears (−0.002) if the 89 sites in the D-loop are excluded (fig. 4 ).
What Explains the Discrepancy Between Different Studies?
Although mutational hot spots can, in principle, generate a negative correlation between LD and distance, they do not appear to do so in human mtDNA, at least not on a chromosome-wide scale. We are left with the question of why Awadalla, Eyre-Walker, and Maynard Smith (1999) observed a decay of r2 with distance, whereas follow-up studies (Ingman et al. 2000 ; Elson et al. 2001 ) did not. Note that this is not a matter of simply failing to replicate a finding, because there is no true replication here; there is a single history of mtDNA, and all samples should reflect it.
To understand the reason for the discrepancy, we compared the data of Awadalla, Eyre-Walker, and Maynard Smith (1999) with those of Ingman et al. (2000) . Awadalla, Eyre-Walker, and Maynard Smith (1999 ) found 49 synonymous informative sites in their data set of 45 nearly complete mtDNA sequences (table 1 ). More than one-third of these sites (18/49) are not polymorphic in the data of Ingman et al. (2000) . Given that both are samples from the same genealogy, we would expect a larger overlap (the probability of the observed difference is less than 3% under a standard coalescent model, but this is not really an appropriate comparison, given the star-like genealogy of mtDNA). This supports the notion that there are errors in the data, and it seems likely that most of the errors are in the data used by Awadalla, Eyre-Walker, and Maynard Smith (1999 ) (Macaulay, Richards, and Sykes 1999 ; Kivisild and Villems 2000 ).
However, these errors cannot explain the different conclusions reached by these studies. As noted previously, random sequencing errors should erase, rather than create patterns in the data. Furthermore, Awadalla, Eyre-Walker, and Maynard Smith (1999 ) used only 14 highly polymorphic sites for which the minor allele frequency is more than 10%. For these sites, the patterns of polymorphisms in the two data sets are not as different from each other, with the exception of two sites (4985 and 6455) that are monomorphic in the data of Ingman et al. (2000) (table 1 ). The possibility of sequencing error for these two sites was pointed out by Kivisild and Villems (2000) . Because three of the remaining 12 sites are singleton polymorphisms in the data of Ingman et al. (2000) , we investigated the decay of LD using the remaining nine informative sites (fig. 5 ). In the data of Ingman et al. (2000) the correlation coefficient is ρ = −0.295, which is very close to that obtained using the data of Awadalla, Eyre-Walker, and Maynard Smith (1999 ) (ρ = −0.248). The negative correlation is almost significant using the permutation test of Awadalla, Eyre-Walker, and Maynard Smith (1999 ) (P = 0.079). It should be noted that there are no incompatible pairs among the sites investigated (in the sense of fig. 2 ), so that |D′| = 1 for all pairs.
Thus, the data of Awadalla, Eyre-Walker, and Maynard Smith (1999 ) and Ingman et al. (2000) do not, in fact, disagree. For the small subset of sites used by the former, the data of the latter also reveal a negative correlation. If we increase the number of sites, the negative correlation quickly disappears. For example, if we add the remaining 12 informative sites listed in table 1 (for a total of 21 sites), the correlation coefficient is nearly zero (ρ = −0.003), and in the entire data set, the correlation is positive (fig. 4 ). It would appear that Awadalla, Eyre-Walker, and Maynard Smith (1999 ) were simply unlucky and happened to pick sites that gave rise to a negative correlation.
How unlucky were they? Figure 6 shows the distribution of ρ when nine sites are randomly sampled from the 49 highly polymorphic sites in the data of Ingman et al. (2000) . The distribution has a positive mode but is skewed toward negative values. Simulations (1,000 randomly chosen samples of nine sites, each followed by 1,000 permutations to assess significance) show that the probability of obtaining a significant negative correlation (at the 5% level) is approximately 4%.
Discussion
We have shown that although the test used by Awadalla, Eyre-Walker, and Maynard Smith (1999) is not robust to the presence of mutational hot spots, the simplest explanation for their finding is chance: they happened to pick sites that gave a negative correlation between r2 and distance. When more sites are used, there is no relationship between r2 and distance (Ingman et al. 2000 ; Elson et al. 2001 ). Awadalla, Eyre-Walker, and Maynard Smith (1999) also found a negative correlation between r2 and distance in three RFLP data sets. However, these data sets include a very small number of sites and are furthermore not independent (because they share some sites). We tried to investigate whether the sites analyzed in these studies also show a negative correlation between r2 and distance in the data of Ingman et al. (2000) , but we were unfortunately unable to identify the sites. We conclude that there is no evidence for recombination in the pattern of LD. However, there is no evidence against recombination either. The test used by Awadalla, Eyre-Walker, and Maynard Smith (1999 ) is based on the assumption that LD should decay with distance in the presence of recombination. This is correct when recombination occurs as a part of crossing-over in a linear chromosome, but it is by no means obvious how LD would be affected by whatever recombinational mechanism might be envisioned to take place in mitochondria (Wiuf 2001 ). Recombination in the mitochondria, if it indeed occurs, may well be almost impossible to detect using polymorphism data. It should also be noted that there appears to be some evidence for recombination in non-human mtDNA. Using data from the mitochondrial control region and the ND2 locus, Awadalla, Eyre-Walker, and Maynard Smith (1999) found a negative correlation between r2 and distance in chimpanzees. If the control region in chimps were hypermutable, such a correlation could easily be caused by the phenomenon illustrated in figure 1 . However, in contrast to the human data, r2 within the chimp control region is actually higher than average, which suggests that the chimp control region is not particularly hypermutable. The negative correlation is attributed to the fact that values of r2 between sites in the control region and sites in the ND2 locus are, on an average, considerably lower than they are within either region. This suggests recombination (unless it is caused by something like sample mix-up or contamination during sequencing). At considerably greater phylogenetic distances, there appears to be evidence for mtDNA recombination in several organisms (Ladoukakis and Zouros 2001 ).
Finally, we think that the implications of the existence of recombination in mtDNA have been misunderstood. Recombination would clearly have important implications for the evolution of mitochondria, particularly in the context of the long argument about the evolutionary advantages of sex. Without recombination, mitochondria might be expected to decay because of the pressure of deleterious mutations (Moran 1996 ). However, the main reason for the attention given to this question has been the perceived implications for our understanding of human evolution. This seems misguided. From the point of view of analyzing polymorphism data from populations, recombination matters because it allows different sites to have different genealogical histories or trees. The extent to which the tree for one site differs from the tree for another site depends on the rate of recombination between the sites (e.g., Nordborg and Tavaré 2002 ). If recombination does indeed occur in mitochondria, it is surely not very common, and the trees for different parts of the mtDNA would thus be strongly correlated. Certainly, they would be much more strongly correlated to each other than to trees for nuclear sites, with which they are genetically unlinked. It follows logically that any conclusion about human evolution that is not robust to a small amount of recombination in mtDNA cannot be robust to recombination in the rest of the genome. Putting it in another way, if recombination causes different parts of the mitochondrial genome to tell different stories, then these stories are certainly independent of the stories told by the rest of the genome. Awadalla, Eyre-Walker, and Maynard Smith (1999) argued that if recombination in mtDNA existed, then many inferences about human evolution would have to be reconsidered. A more correct statement is that any inference about human evolution that would have had to be reconsidered had recombination in mtDNA existed should in fact be reconsidered in any case.
Brandon Gaut, Reviewing Editor
Keywords: coalescent linkage disequilibrium gene conversion
Address for correspondence and reprints: Magnus Nordborg, Molecular & Computational Biology, University of Southern California, 835 W 37th Street, SHS 172, Los Angeles, California 90089-1340. magnus@usc.edu
Table 1 Comparison of the Data Sets of Awadalla, Eyre-Walker, and Maynard Smith (1999) and Ingman et al. (2000)
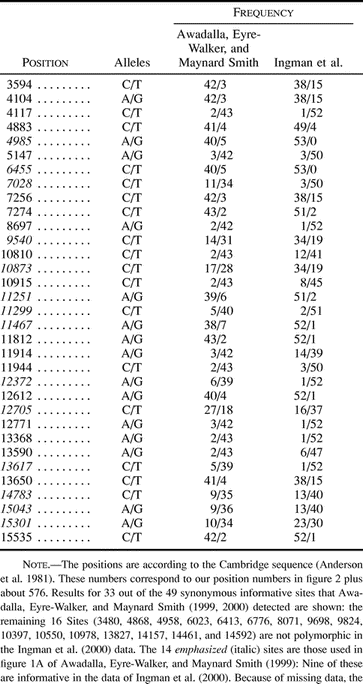
Table 1 Comparison of the Data Sets of Awadalla, Eyre-Walker, and Maynard Smith (1999) and Ingman et al. (2000)
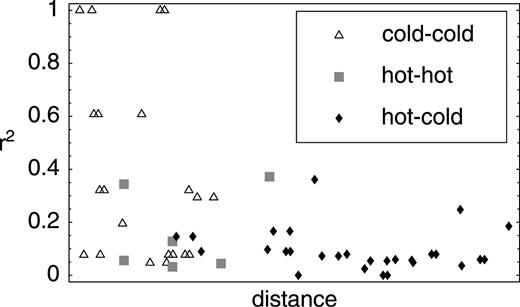
Fig. 1.—LD can decay with distance even in the absence of recombination. The figure shows r2 as a function of distance between sites in a data set simulated without recombination but with highly hypermutable (hot) sites in one half of the chromosome only. As explained in the text, this can give rise to a negative correlation between LD and distance. The simulation parameters were chosen to illustrate the point, not to be realistic: each of the hypermutable sites experienced multiple mutations, whereas none of the others did
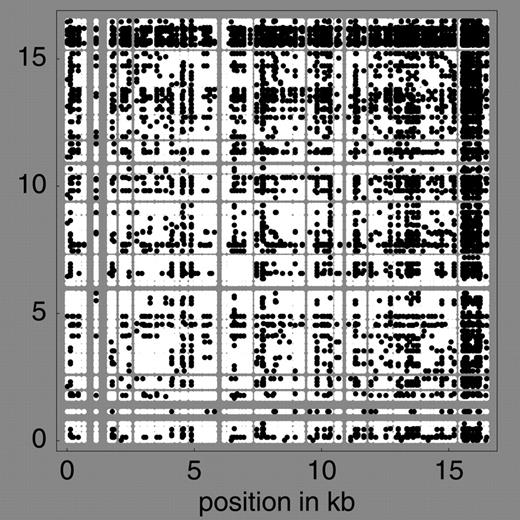
Fig. 2.—Evidence for recurrent mutation or recombination (or both) in human mtDNA (data of Ingman et al. 2000 ). Each point represents the comparison between a pair of polymorphic sites. The point is black if the pattern of polymorphism for the pair of loci is such that either recombination must have occurred between the loci or recurrent mutation affected at least one of the loci. The point is white otherwise. If recombination has occurred, and (importantly) the probability of recombination increases with distance between sites, white points are expected to be clustered along the diagonal (because recombination is less likely to have effected closely linked sites). Recurrent mutations, on the other hand, might be expected to give rise to a pattern that does not depend on the distance from the diagonal, leading to black “crosses” against a white background. The D-loop is visible as a cluster of such crosses in the upper right corner (position 0 corresponds to the first position after the D-loop)
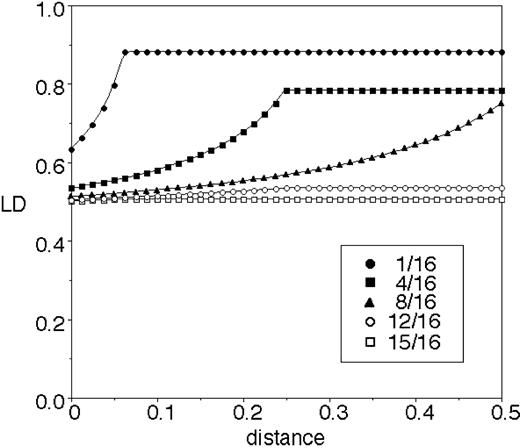
Fig. 3.—Expected LD (on relative scale) as a function of distance for a circular chromosome with a hot (high mutation) and cold (low mutation) region under different assumptions about the relative length of the hot region (1/16, 4/16, …). The per site mutation rate in the hot region is assumed to be six times greater than that in the cold region. The relative values of LD in the cold region, in the hot region, and between them are assumed to be 1, 0.5, and 0.75, respectively, reflecting the data of Ingman et al. (2000)
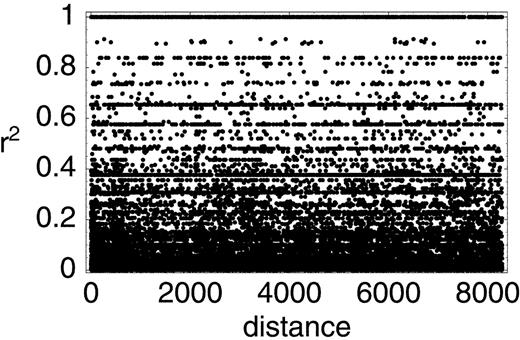
Fig. 4.—r2 as a function of distance in the data of Ingman et al. (2000) . All sites were included in the analysis; eliminating sites where the minority allele has frequency less than 5% or 10% yields very similar results. This is expected, given the relative insensitivity of the rate of decay of r2 to allele frequencies (Nordborg and Tavaré 2002 )
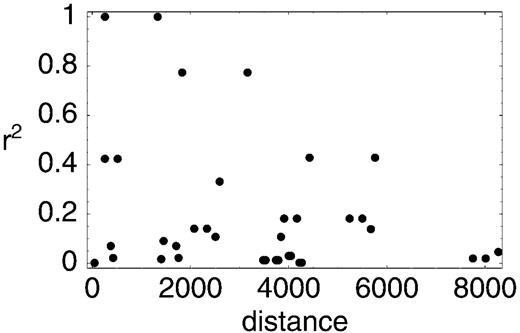
Fig. 5.—The relationship between r2 and the data of Ingman et al. (2000) for the sites used by Awadalla, Eyre-Walker, and Maynard Smith (1999) . See table 1 for details
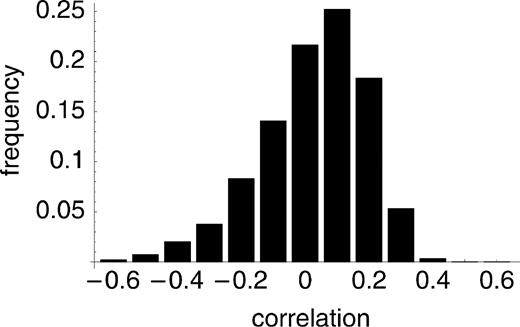
Fig. 6.—The distribution of the correlation between r2 and distance under repeated sampling of nine sites from the data of Ingman et al. (2000)
We thank Adam Eyre-Walker and Max Ingman for sending their respective data sets, Maarit Jaarola for discussions about mtDNA, Noah Rosenberg, Simon Tavaré, Carsten Wiuf, Brandon Gaut, and two anonymous reviewers for comments on the manuscript.
References
Anderson S., A. T. Bankier, B. G. Barrell, et al. (14 co-authors).
Awadalla P., A. Eyre-Walker, J. Maynard Smith,
Elson J. L., R. M. Andrews, P. F. Chinnery, R. N. Lightowlers, D. M. Turnbull, N. Howell,
Eyre-Walker A., N. H. Smith, J. Maynard Smith,
Hill W. C., A. Robertson,
Ingman M., H. Kaessmann, S. Pääbo, U. Gyllensten,
Jakobsen I. B., S. Easteal,
Jorde L. B., M. Bamshad,
Kivisild T., R. Villems,
Kumar S., P. Hedrick, T. Dowling, M. Stoneking,
Ladoukakis E. D., E. Zouros,
Lewontin R. C.,
Macaulay V., M. Richards, B. Sykes,
Markovtsova L., P. Marjoram, S. Tavaré,
Meyer S., G. Weiss, A. von Haeseler,
Moran N. A.,

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}