





Abstract
SJL mice are highly susceptible to proteolipid protein (PLP) 139–151-induced experimental allergic encephalomyelitis (EAE). The disease is characterized by a relapsing–remitting type of paralysis. However, the mechanism by which animals recover from EAE is poorly understood. Here, we investigated the role of regulatory T cells in the recovery from disease. We found that Forkhead box P3-expressing CD4+CD25+ T cells were increased in the blood, draining lymph node and spleen of EAE-recovered SJL mice. These cells were anergic and inhibited proliferation of CD4+CD25− T cells to PLP 139–151 or anti-CD3 antibody stimulation. Depletion of CD4+CD25+ T cells during the recovery phase exacerbated disease, resulted in the expansion of IAs/PLP 139–151-tetramer-positive cells and enhanced IFN-γ production. In addition, transforming growth factor-β (TGF-β) was shown to be involved in the recovery from EAE as the percentage of CD4+ cells expressing TGF-β latency-associated peptide (LAP) on the cell surface increased significantly in blood and spleen of EAE-recovered mice as compared with the naive mice and in vivo neutralization of TGF-β abolished recovery from disease. Taken together, our results demonstrate that both CD4+CD25+ and CD4+LAP+ regulatory T cells mediate recovery from PLP 139–151-induced EAE in SJL mice in which TGF-β plays an important role.
Introduction
Experimental allergic encephalomyelitis (EAE) is an animal model for human demyelinating disease, multiple sclerosis. EAE can be induced in several rodent species by either active immunization with myelin protein antigens such as proteolipid protein (PLP), MBP and myelin oligodendrocyte glycoprotein or adoptive transfer of myelin-specific Th1 cells into naive mice. The histologic hallmark of EAE is a perivascular infiltrate in the CNS comprising primarily macrophages and CD4+ T cells (1).
Immunization with PLP 139–151 induces EAE in SJL mice. The disease is characterized by a relapsing–remitting type of paralysis. It has been suggested that disease progression is associated with the process of epitope spreading, wherein T cells reacting to endogenous myelin epitopes are responsible for mediating clinical relapses (2). Although a number of theories including Fas-mediated apoptosis and immune deviation have been proposed for disease remission (3–8), the mechanisms associated with the recovery remain poorly understood.
Accumulating evidence suggests that CD4+CD25+ regulatory T cells play an important role in the maintenance of immunological self-tolerance and prevent the onset of autoimmune models including diabetes and inflammatory bowel disease (9, 10) by suppressing the potentially autoreactive T cell (11–14). CD4+CD25+ regulatory cells also play a role in affecting the onset of EAE as transfer of these cells ameliorates disease and depletion of these cells in vivo exacerbates disease (15–17). However, their regulation of activated pathogenic T cells during spontaneous remission of EAE is largely unknown.
Immunoregulatory cytokines such as IL-4, IL-10 and transforming growth factor-β (TGF-β) have been shown to contribute to the recovery from EAE (5–8). Oral administration of autoantigens suppresses EAE by inducing regulatory cells that mediate active suppression by producing these immunoregulatory cytokines (5, 7, 18). In addition, we have previously identified TGF-β-secreting Th3 regulatory cells (7) and CD8+ regulatory cells (19), both were associated with oral tolerance and dependent on TGF-β in vivo. Furthermore, Karpus and Swanborg (20) have identified CD4+ regulatory cells secreting TGF-β in Lewis rats recovered from EAE that suppress EAE via adoptive transfer. These results suggest that TGF-β-dependent regulatory cells participate during recovery from EAE.
Latency-associated peptide (LAP) is the amino-terminal domain of TGF-β precursor peptide. It remains non-covalently associated with TGF-β peptide after cleavage and forms the latent TGF-β complex. We and others have recently identified TGF-β-dependent regulatory cells characterized by surface expression of LAP that is active in the animal model of colitis and EAE (21–23). The presence of membrane-bound TGF-β or LAP on the surface of regulatory cells has linked TGF-β with regulatory T cell suppressive function. In addition, TGF-β-induced regulatory CD4+CD25+ T cells express increased TGF-β on the surface that mediates their suppressive function (24). We thus hypothesized that surface-bound TGF-β might play a role in regulatory T cell-mediated recovery from EAE and investigated the role of regulatory cells in natural recovery from EAE. We have found that both CD4+CD25+ and CD4+LAP+ regulatory cells play a role in recovery via a TGF-β-dependent mechanism.
Methods
Mice
Female SJL and C57BL/6 (8–10 weeks) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and housed in a pathogen-free animal facility at The Harvard Institutes of Medicine according to the animal protocol guidelines of Harvard University.
Antibodies and reagents
Anti-CD25 antibody (clone, PC 61) was prepared as previously described (16) and purified by BioExpress (West Lebanon, NH, USA). Anti-TGF-β (clone, 1D11) and mouse IgG were purified as described (21). The endotoxin activity of purified antibodies was measured with Limulus Amebocyte Lysate (0.3–0.6 endotoxin units mg−1, BioWhittaker, Walkersville, MD, USA). The following mAbs were purchased from PharMingen (San Diego, CA, USA): PE-conjugated anti-CD25 (PC 61), FITC-conjugated anti-CD25 (clone, 7D4), PE- or FITC-conjugated or purified anti-CD4 (clone, L3T4), streptavidin–allophycocyanin (APC), anti-TCR antibody (clone, H57-597), anti-CD28 (clone, 37.51) and anti-CD4 (clone, RM 4.5-APC). Cytokine antibodies and their corresponding isotype controls were obtained from PharMingen. Their respective clones were as follows: IL-2, JES6-5H4 and A-95-1; IL-4, 11B11 and R3-34; IL-6, MP5-20F3 and RS-34 and IFN-γ, XMG1.2 and R3-34. Biotinylated anti-LAP polyclonal antibody was purchased from R&D Systems (catalog no. BAF 246, Minneapolis, MN, USA). PLP peptide 139–151 (HSLGKWLGHPDKF) and Theiler's murine encephalomyelitis virus (TMEV) VP2 70–86 (WTTSQEAFSHIRIPLP) were synthesized (QCB, BioSource, Hopkinton, MA, USA).
Induction of EAE and depletion of CD4+CD25+ cells in vivo
SJL mice were immunized subcutaneously in the flank with 60 μg PLP peptide 139–151 in CFA containing 400 μg Mycobacterium tuberculosis H37 RA (Difco, Detroit, MI, USA). The mice also received two doses intra-peritoneally (i.p.) of 150 ng pertussis toxin (List Biological Laboratories, Campbell, CA, USA) on days 0 and 2 post-immunization. For CD4+CD25+ cell-depletion experiments, mice were injected i.p. on days 15, 17 and 19 with 400 μg of anti-CD25 antibody (PC 61) or 400 μg of rat IgG (ICN Biomedicals, Inc., Costa Mesa, CA, USA). Animals were monitored daily for symptoms of EAE and scored as follows: 1, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 4, hind limb plus forelimb paralysis, and 5, moribund or dead.
Flow cytometric analysis
Briefly, 0.5–1.0 × 106 cells were incubated in PBS with 2% normal FCS and 0.1% sodium azide for 5 min. Cells were incubated with a mixture of PE-, FITC- or APC-conjugated mAb on ice for 30 min and washed twice and then fixed in 1% of formaldehyde. The analysis was performed on a FACScan flow cytometer using CellQuest (Becton Dickinson, Mountain View, CA, USA) or FlowJo (Tree Star, Inc., Ashland, OR, USA) software.
Intracellular cytokine staining
Lymph node (LN) cells from SJL mice immunized with PLP 139–151 with or without anti-CD25/control antibody treatment were stimulated with PLP 139–151 (50 μg ml−1) for 4 days. Viable cells were harvested by Ficoll-Hypaque density-gradient centrifugation. After two washes in 1× PBS, cells were re-stimulated with anti-TCR antibody (5.0 μg ml−1), anti-CD28 (4.0 μg ml−1) and 2 mM monensin (GolgiStop, PharMingen) for 6 h at 37°C. Cells were washed and stained with anti-CD4 (clone, RM 4.5-APC) and 7-amino-actinomycin-D (7-AAD) (PharMingen) by incubating at room temperature for 20 min. After two washes in 1× PBS containing 2% FCS and 0.1% sodium azide, the cells were treated with 4% PFA and fixed with buffer containing saponin according to the manufacturer's recommendations (PharMingen). Following permeabilization, the PE-conjugated cytokine antibodies were added and 20 min after incubation at room temperature, cells were washed and analyzed by using FACSCalibur flow cytometer (Becton Dickinson).
IAs tetramer staining
To determine the precursor frequency of PLP 139–151-specific cells, we used IAs tetramers for PLP 139–151 and TMEV 70–86 as described previously (25). Briefly, CD3+ T cells were enriched from single-cell suspensions derived from LNs and spleens and treated with neuraminidase (0.7 U ml−1) in serum-free medium (DMEM, BioWhittaker) at a density of 1 × 107 cells ml−1 for 1 h at 37°C. After washing, cells were incubated with PE-conjugated, PLP 139–151 and TMEV 70–86 tetramers (30 μg ml−1) for 3–4 h at 37°C and stained with anti-CD4–APC (clone, RM 4.5) and 7-AAD (PharMingen). Cells were acquired by using a FACSCalibur flow cytometer and the data were analyzed by using FlowJo software. The tetramer-positive cells were then analyzed in the live CD4 population after eliminating the dead (7-AAD+) cells. To determine the frequency of PLP 139–151-reactive T cells in the cultures stimulated with PLP 139–151, viable cells were collected by Ficoll-Hypaque density-gradient centrifugation and used for tetramer staining without neuraminidase treatment.
Real-time (TaqMan) reverse transcription–PCR
Total RNA was extracted from CD4+CD25− and CD4+CD25+ cell fractions using RNeasy kit (Qiagen, Valencia, CA, USA) followed by DNase I treatment (Qiagen). cDNA was synthesized using TaqMan reverse transcription reagents and as primers, both random hexamers and oligo dT were used (Applied Biosystems, Blanchburg, NJ, USA). Expression of Forkhead box P3 (Foxp3) and glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA was measured by PCR in separate tubes in duplicates using probes labeled with 6-carboxyfluorescein and VIC®, respectively, and with TAMRA® (Applied Biosystems) as a quencher using TaqMan® universal PCR master mix and the ABI Prism 7700 sequence detection system (Applied Biosystems). Primers and probe for GAPDH were purchased from Applied Biosystems and for Foxp3, the sequences were as described previously (26). A comparative threshold cycle (CT) was used to determine mRNA expression of Foxp3 and GAPDH relative to no-template control (calibrator). CT value was normalized for each sample using the formula: ΔCT = CT(Foxp3) − CT(GAPDH) and the relative expression of Foxp3 was then calculated using the expression
Cell purification and adoptive transfer experiments
Single-cell suspensions were prepared from spleen and LN (axillary, inguinal and mesenteric). CD4+ T cells were then enriched by negative selection using T cell subset enrichment columns (R&D Systems, Inc.) according to the suggested protocol. To separate CD4+CD25+ T cells and CD4+CD25− T cells, the enriched CD4+ T cells were incubated with biotin-conjugated anti-CD25 (10 μg per 108 cells) in PBS with 4% BSA on ice for 30 min and washed twice. The cells were then incubated with streptavidin MicroBeads (Miltenyi Biotec, Inc., Auburn, CA, USA) for 15 min at 4°C. Magnetic separation was performed with LS separation columns (Miltenyi Biotec) according to the manufacturer's instruction. For adoptive transfer experiments, purified CD4+CD25+ or CD25− T cells were suspended in PBS and injected intravenously into mice.
Proliferation assay
To analyze the proliferation in response to anti-CD3 or PLP 139–151 stimulation, purified CD4+CD25− or CD4+CD25+ T cells (2.0 × 105 cells per well) were cultured in 10% FCS DMEM in triplicate wells in the presence of APCs (2.0 × 105 cells per well) with 0.5 μg ml−1 of soluble anti-CD3 antibody or 20 μg ml−1 of PLP 139–151 peptide. To test the suppressive function of CD4+CD25+ T cells to anti-CD3 or PLP 139–151 stimulation, freshly isolated CD4+CD25− T cells (2.0 × 105 cells per well) were co-cultured at a 1:1 ratio with CD4+CD25+ T cells (2.0 × 105 cells per well) in the presence of APCs (2.0 × 105 cells per well) with 0.5 μg ml−1 of soluble anti-CD3 antibody or 20 μg ml−1 of PLP 139–151 peptide. Cultures were pulsed with 1 μCi [3H]thymidine per well (NEN, Boston, MA, USA) for 48 h (for anti-CD3 antibody stimulation) or for 72 h (for PLP 139–151 stimulation) and the cells were harvested 12 h later using a Wallac liquid scintillation counter (Perkin Elmer, Boston, MA, USA). Purified splenic CD4+ T cells (5.0 × 105 cells per well) were cultured with equal numbers of APC in the presence of various concentrations of PLP 139–151 for 72 h and the proliferative response was measured as above.
Cytokine ELISA
For cytokine assays, splenocytes or LN cells were grown at 1.0 × 106 cells per well in 200 μl of serum-free medium X-VIVO 20 (BioWhittaker) with various antigen concentrations. Supernatants were collected after 24 h for IL-2 and 48 h for IL-4, IL-10 and IFN-γ. Purified CD25+CD4+ and CD25−CD4+ T cells were cultured in 48-well plates (0.5 ml per well) at 2.0 × 106 cells ml−1 in serum-free medium X-VIVO 20 and plate-bound anti-CD3 antibody (10 μg ml−1). Supernatants were collected 24 and 48 h after culture. Quantitative ELISA for IL-2, IL-4, IL-10 and IFN-γ was performed using paired antibodies and recombinant cytokines obtained from PharMingen according to their recommendations.
Statistical analysis
Differences were analyzed using Student's t-test. When there were more than two groups used for comparison, differences were analyzed by one-way analysis of variance test. P-values ≤0.05 were considered significant.
Results
Recovery from EAE is associated with an increase of CD4+CD25+ T cells
Immunization with PLP 139–151 induces EAE in SJL mice that is characterized by a relapsing–remitting type of paralysis. The first peak of clinical symptoms occurs on days 13–15 post-immunization followed by clinical recovery. Although the disease relapses, the disease severity is much lower than the first peak. Using our protocol for induction of EAE in SJL mice, we have always observed the lowest clinical score during recovery phase of the disease on days 28–30 post-immunization. Thus, we were able to ask whether recovery from EAE was associated with the induction of CD4+CD25+ regulatory T cells at a defined end-point in the disease. To address this question, SJL mice were immunized with 60 μg of PLP 139–151 in CFA and CD4+CD25+ T cells in blood, peripheral LN and spleen from naive, EAE-ongoing (day 10) and EAE-recovered mice (day 30) were analyzed by flow cytometry. As shown in Fig. 1(A) and Table 1, in recovered mice (day 30) percentages of CD4+CD25+ T cells were significantly increased in blood, draining LN and spleen. In contrast to CD4+CD25+ T cells, the percentage of CD4+CD25− T cells in blood and spleen was not significantly different in EAE-recovered mice compared with naive or EAE-ongoing mice. However, the percentage of CD4+CD25− T cells in draining LN was significantly reduced in EAE-recovered mice (36.2 ± 1.5%) as opposed to naive mice (48.5 ± 1.9%, P < 0.01) or EAE-ongoing mice (49.8 ± 1.7%, P < 0.01). There were no significant differences in CD4+CD25+ T cells in blood, LN and spleen from mice on day 10 compared with naive mice, although this population was slightly increased. We also found a significant increase in absolute numbers of CD4+CD25+ T cells in the spleens from EAE-recovered mice (8.5 ± 1.9 × 106) as compared with naive mice (3.1 ± 0.4 × 106, P = 0.002) or EAE-ongoing mice (3.7 ± 0.5 × 106, P < 0.01) (Fig. 1B). We did not detect activation markers CD44 or CD69 on CD4+CD25+ T cells from recovered mice.
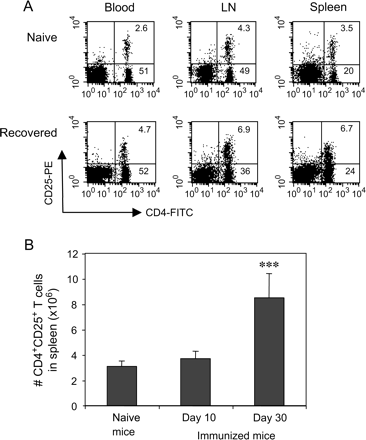
EAE-recovered mice have elevated CD4+CD25+ T cells. (A) Dot plots of CD4+CD25+ T cells in blood, draining LN and spleen of naive and EAE-recovered mice. Blood was lysed with ACK buffer and draining LN cells and splenocytes were prepared from naive or PLP 139–151-immunized mice (day 10 or day 30 post-immunization). These cells were stained with PE-conjugated anti-CD25 and FITC-conjugated CD4 and percentages of CD4+CD25+ T cells in blood, LN and spleen were analyzed with flow cytometry. (B) Absolute numbers of CD4+CD25+ T cells in spleens. CD4+CD25+ T cells in spleen of naive (n = 12), EAE-ongoing (day 10, n = 10) and EAE-recovered (day 30, n = 14) mice were calculated. The results shown were mean values ± SD (***P < 0.001).
Percentages of CD4+CD25+ T cells in blood, LN and spleen of naive and PLP 139–151-immunized SJL/J mice
Blood | LN | Spleen | |
|---|---|---|---|
| Naive | 2.8 ± 0.1 | 4.4 ± 0.6 | 2.7 ± 0.45 |
| Day 10 | 3.0 ± 0.4 | 5.4 ± 0.9 | 2.8 ± 0.27 |
| Day 30 | 4.8 ± 0.6** | 6.7 ± 0.6* | 6.3 ± 0.59** |
Blood | LN | Spleen | |
|---|---|---|---|
| Naive | 2.8 ± 0.1 | 4.4 ± 0.6 | 2.7 ± 0.45 |
| Day 10 | 3.0 ± 0.4 | 5.4 ± 0.9 | 2.8 ± 0.27 |
| Day 30 | 4.8 ± 0.6** | 6.7 ± 0.6* | 6.3 ± 0.59** |
SJL/L mice were immunized with PLP 139–151. Blood was collected and lysed with ACK buffer. Single cell suspensions were prepared from naive or immunized animals (on day 10 or day 30 after immunization). These cells were stained with PE-conjugated anti-CD25 and FITC-conjugated anti-CD4 and analyzed by flow cytometry. There were 15–18 mice in each group.
P < 0.05 versus naive;
P < 0.01 versus naive or day 10.
Percentages of CD4+CD25+ T cells in blood, LN and spleen of naive and PLP 139–151-immunized SJL/J mice
Blood | LN | Spleen | |
|---|---|---|---|
| Naive | 2.8 ± 0.1 | 4.4 ± 0.6 | 2.7 ± 0.45 |
| Day 10 | 3.0 ± 0.4 | 5.4 ± 0.9 | 2.8 ± 0.27 |
| Day 30 | 4.8 ± 0.6** | 6.7 ± 0.6* | 6.3 ± 0.59** |
Blood | LN | Spleen | |
|---|---|---|---|
| Naive | 2.8 ± 0.1 | 4.4 ± 0.6 | 2.7 ± 0.45 |
| Day 10 | 3.0 ± 0.4 | 5.4 ± 0.9 | 2.8 ± 0.27 |
| Day 30 | 4.8 ± 0.6** | 6.7 ± 0.6* | 6.3 ± 0.59** |
SJL/L mice were immunized with PLP 139–151. Blood was collected and lysed with ACK buffer. Single cell suspensions were prepared from naive or immunized animals (on day 10 or day 30 after immunization). These cells were stained with PE-conjugated anti-CD25 and FITC-conjugated anti-CD4 and analyzed by flow cytometry. There were 15–18 mice in each group.
P < 0.05 versus naive;
P < 0.01 versus naive or day 10.
CD4+CD25+ T cells from recovered mice express Foxp3 and have regulatory activity in vitro
To characterize CD4+CD25+ T cells from EAE-recovered mice, we examined the expression of transcription factor Foxp3 that controls the development of CD4+CD25+ regulatory T cells (27–29). CD4+CD25− T cells or CD4+CD25+ T cells from draining LN and spleen of naive or EAE-recovered mice were purified and the expression of Foxp3 mRNA was detected by using real-time PCR as described in Methods. As shown in Fig. 2(A), CD4+CD25− T cells from either naive or recovered mice expressed very low level of Foxp3 mRNA. Foxp3 expression in CD4+CD25+ T cells was 8- to 10-fold higher than CD4+CD25− T cells, however, it was not up-regulated in recovered mice compared with naive mice.
![CD4+CD25+ T cells from EAE-recovered mice express Foxp3 and have suppressive activity in vitro. (A) Foxp3 mRNA expression. Total RNA was extracted from CD4+CD25− and CD4+CD25+ cell fractions using RNeasy kit followed by DNase I treatment. Relative expression of Foxp3 was measured and calculated by using real-time PCR as described in Methods (**P < 0.01; ***P < 0.001). (B and C) Suppressive activity of CD4+CD25+ T cells with anti-CD3 or PLP 139–151 stimulation. CD4+CD25− or CD4+CD25+ T cells (2.0 × 105 cells per well) from EAE-recovered mice were purified as described in Methods and cultured either alone or mixed at a 1:1 ratio in the presence of APCs plus stimulation of 0.5 μg ml−1 of anti-CD3 (B) (*P < 0.05; **P < 0.01) or 20 μg ml−1 of PLP 139–151 (C) (*P < 0.05; **P < 0.01). Cells were pulsed with [3H]thymidine 48 h for anti-CD3 or 72 h for PLP 139–151 and these cells were harvested overnight. The results were shown as the mean ± SD of triplicate wells of a representative of three independent experiments. (D) Depletion of CD4+CD25+ cells during recovery exacerbates EAE. SJL mice were immunized with 60 μg of PLP 139–151 in CFA and received two injections of pertussis toxin. The mice were given i.p. either 0.4 mg of rat IgG (open triangles, n = 8) or 0.4 mg of anti-CD25 antibody (filled triangles, n = 8) on days 15, 17 and 19 post-immunization. Animals were scored daily for symptoms of EAE. Data were representative of three independent experiments.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/intimm/18/4/10.1093/intimm/dxh390/2/m_intimmdxh390f02_ht.gif?Expires=1716490067&Signature=QZi754qk3M-kOVBjPTy2uTWaGSQIHsi4DsrP9Q9t97JpOp4ByFCotWCWaqx2H7O0RSyg~ooQy3H9pxu~4jXq-VYw~tS-L-eDhgdpkHgKohU3TtI3eXIhnnnuUnQjxMLVdPCSVhU9NN~SbOMp801GyG1m3Gv~9ZXiT1Enxr5oJvopLyhXBJop3GcbQXb9sUSQduHdkmHGJzeA2zQmFyJWhJKuW063vKxl~ljbXgYRw6o8kjgfKZiNoKQGt8mOny0Oj4PjgJ3JbMT8NGhWU5S2~vUebxIPxtRkQAeICk~VbsXtSkquH6lzsyeGvqPM7WhgxTrg1k8Vt2x7BFCCRJ8gDw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CD4+CD25+ T cells from EAE-recovered mice express Foxp3 and have suppressive activity in vitro. (A) Foxp3 mRNA expression. Total RNA was extracted from CD4+CD25− and CD4+CD25+ cell fractions using RNeasy kit followed by DNase I treatment. Relative expression of Foxp3 was measured and calculated by using real-time PCR as described in Methods (**P < 0.01; ***P < 0.001). (B and C) Suppressive activity of CD4+CD25+ T cells with anti-CD3 or PLP 139–151 stimulation. CD4+CD25− or CD4+CD25+ T cells (2.0 × 105 cells per well) from EAE-recovered mice were purified as described in Methods and cultured either alone or mixed at a 1:1 ratio in the presence of APCs plus stimulation of 0.5 μg ml−1 of anti-CD3 (B) (*P < 0.05; **P < 0.01) or 20 μg ml−1 of PLP 139–151 (C) (*P < 0.05; **P < 0.01). Cells were pulsed with [3H]thymidine 48 h for anti-CD3 or 72 h for PLP 139–151 and these cells were harvested overnight. The results were shown as the mean ± SD of triplicate wells of a representative of three independent experiments. (D) Depletion of CD4+CD25+ cells during recovery exacerbates EAE. SJL mice were immunized with 60 μg of PLP 139–151 in CFA and received two injections of pertussis toxin. The mice were given i.p. either 0.4 mg of rat IgG (open triangles, n = 8) or 0.4 mg of anti-CD25 antibody (filled triangles, n = 8) on days 15, 17 and 19 post-immunization. Animals were scored daily for symptoms of EAE. Data were representative of three independent experiments.
To test the function of CD4+CD25+ T cells from EAE-recovered mice, we first measured the suppressive activity of this population in vitro. As shown in Fig. 2(B), CD4+CD25+ T cells did not proliferate to stimulation with anti-CD3 antibody in vitro, whereas CD4+CD25− T cells from EAE-recovered mice showed primary proliferative response. Furthermore, CD+CD25+ cells from EAE-recovered mice suppressed CD4+CD25− T cell proliferation when these cells were mixed with the culture of CD4+CD25− T cells at a 1:1 ratio. To determine whether CD4+CD25+ T cells from EAE-recovered mice mediated suppressive function in an antigen-specific manner, we used PLP 139–151 as a stimulus. While CD4+CD25+ T cells from EAE-recovered mice did not respond, the CD4+CD25− T cells proliferated well to PLP 139–151 stimulation whose response was suppressed when co-cultured with CD4+CD25+ T cells (Fig. 2C).
Depletion of CD4+CD25+ cells during recovery from EAE exacerbates disease
To investigate the role of CD4+CD25+ regulatory T cells in recovery from the disease, SJL mice were immunized with 60 μg PLP 139–151 in CFA. Mice were also treated with 0.4 mg normal rat IgG or 0.4 mg anti-CD25 antibody (PC 61) i.p. on days 15, 17 and 19 post-immunization. CD4+CD25+ T cells in blood (91%), LN (83%) and spleen (87%) of anti-CD25 antibody-treated mice were depleted as measured on day 25 after immunization. As shown in Fig. 2(D), anti-CD25 antibody treatment abolished recovery from the disease and the mean EAE clinical score on day 30 post-immunization was significantly higher than that of rat IgG-treated mice (2.3 ± 1.0 versus 1.1 ± 1.1, P < 0.05). The disease severity peaked on day 6 after the last dose of anti-CD25 antibody.
CD4+CD25+ regulatory T cells in recovery phase control PLP 139–151-specific T cell responses
To investigate the effect of in vivo depletion of CD4+CD25+ T cells on immune responses to PLP 139–151, we first measured CD4+ T cell proliferation in response to PLP 139–151. To avoid the effect of immunization on APCs, CD4+ T cells enriched from rat IgG- or anti-CD25 antibody-treated mice were cultured with APCs freshly isolated from naive mice in the presence of PLP 139–151. The proliferative response of CD4+ T cells from anti-CD25 antibody-treated mice was higher than that from rat IgG-treated or PBS-treated mice (Fig. 3A). We then used IAs/PLP 139–151 tetramers to determine the precursor frequency of PLP 139–151-reactive cells in the naive SJL mice and compared with those in PLP 139–151-immunized mice on days 10 and 30 ex vivo. As shown in Fig. 3(B), the frequency of PLP 139–151-reactive cells in the naive SJL mice was estimated to be 0.09 ± 0.02% of the CD4 population in their periphery. Ten days after immunization with PLP 139–151 in CFA, the frequency of PLP-reactive cells was increased significantly before showing a decline by day 30 (0.25 ± 0.05% on day 10 versus 0.09 ± 0.05% on day 30, P < 0.01). In all the above groups, reactivity to control tetramers (TMEV 70–86) was negligible, although it tended to be higher at day 10 but the differences were not significant. We then asked whether CD4+CD25+ T cells controlled the frequency of antigen-specific reactive T cells during the recovery phase. SJL mice were immunized with PLP 139–151 in CFA followed by intra-peritoneal injections of rat IgG or anti-CD25 antibody during the recovery phase. CD3+ T cells were then prepared from mice on day 30 post-immunization and stimulated with PLP 139–151. Cells were stained with PE-conjugated, PLP 139–151 and TMEV 70–86 tetramers and with anti-CD4–APC and 7-AAD and analyzed as mentioned in Methods. Upon expansion in vitro, we found that PLP 139–151-tetramer-reactive T cells were significantly higher in the anti-CD25 antibody-treated group than those from rat IgG control antibody-treated group (1.0 ± 0.06% versus 0.6 ± 0.09%, P < 0.05) (Fig. 3C). These results indicate that T cell proliferative response to PLP 139–151 and expansion of PLP 139–151-tetramer-reactive T cells are under control of CD4+CD25+ T cells during the recovery phase.
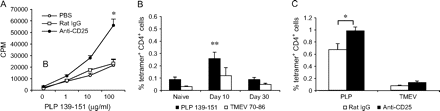
Depletion of CD4+CD25+ T cells during recovery phase of EAE enhances PLP 139–151-reactive T cell responses. (A) Proliferative responses of spleen cells to PLP 139–151 in vitro. Spleen cells (5.0 × 105 cells per well) from PBS- (open circles), rat IgG- (open squares) or anti-CD25 antibody- (filled circles) treated mice (on days 15, 17 and 19 after immunization) were cultured with indicated concentrations of PLP 139–151 for 72 h. Cells were harvested 12 h later. The result (mean values ± SD) was representative of three experiments (*P < 0.05). (B) Frequency of IAs/PLP 139–151 or TMEV 70–86 tetramers+CD4+ T cells in naive, EAE-ongoing (day 10) and EAE-recovered (day 30) mice. CD3+ T cells were enriched from single-cell suspensions derived from LNs and spleens and treated with neuraminidase. Cells were incubated with PE-conjugated, PLP 139–151 and TMEV 70–86 tetramers and stained with anti-CD4–APC and 7-AAD. The tetramer-positive cells were then analyzed in the live CD4 population by using FlowJo software. The results were mean values (±SD) of three experiments. There were 12 mice in each group. The P-values of the one-way analysis of variance test for PLP tetramers+ or TMEV 70–86 CD4+ T cells among three groups were equal to 0.0038 or 0.244, respectively. (C) Frequency of IAs/PLP 139–151 or TMEV 70–86 tetramers+CD4+ T cells from EAE-recovered mice (day 30) previously treated with rat IgG or anti-CD25 antibody on days 15, 17 and 19 after immunization. CD3+ cells were stimulated with PLP 139–151 peptides in vitro and viable cells were used for tetramer staining. The results were mean values (±SD) of three experiments. There were 10 mice in rat IgG-treated group and 12 mice in anti-CD25 antibody-treated group (*P < 0.05; **P < 0.01).
We then determined the frequency of cytokine-producing CD4 cells from rat IgG- or anti-CD25 antibody-treated mice by intracellular staining. As shown in Fig. 4(A), the IFN-γ-producing CD4+ T cells in the cultures derived from anti-CD25 antibody-treated group were significantly increased as compared with rat IgG-treated mice (2.4 ± 0.3% versus 1.4 ± 0.2%, P < 0.01). Similarly, IL-2-producing CD4+ T cells were also significantly increased as compared with rat IgG-treated group (1.1 ± 0.3% versus 0.4 ± 0.1%, P < 0.05). Interestingly, while IL-4-producing CD4+ T cells were also increased in anti-CD25 antibody-treated group (0.9 ± 0.2% versus 0.3 ± 0.1%, P ≤ 0.01), IL-10-producing CD4+ T cells although low were not significantly different from rat IgG-treated mice (data not shown). These results were verified by cytokine ELISA. As shown in Fig. 4(B), there was a significant increase in IFN-γ production with a concomitant decrease in IL-10 production. IL-2 production was slightly increased but not significant and IL-4 production in culture from anti-CD25 antibody-treated mice was significantly higher than those from rat IgG-treated mice (P < 0.05). These data suggest that CD4+CD25+ T cells in the recovery phase of EAE inhibit both Th1 and Th2 cells.
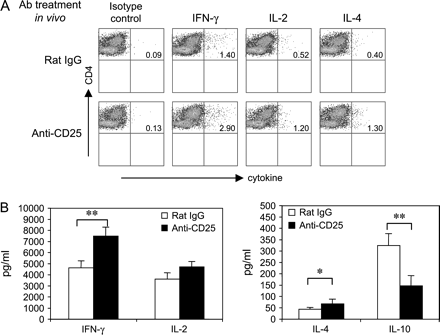
CD4+CD25+ T cells in recovery phase of EAE suppress Th1 and Th2 cytokine production. (A) Intracellular cytokine staining. LN cells from PLP 139–151-immunized SJL mice that were pre-treated with rat IgG or anti-CD25 antibody were stimulated with PLP 139–151 in vitro. After 4 days, viable lymphoblasts were re-stimulated with anti-TCR and anti-CD28 antibody for 6 h. After staining with anti-CD4 antibody and 7-AAD, cells were fixed and permeabilized to stain with antibodies specific for IFN-γ, IL-2 and IL-4. Cells were acquired by flow cytometry and the frequency of cytokine-secreting cells was determined in the live (7-AAD−) CD4 subset. (B) Cytokine ELISA. LN cells from PLP 139–151-immunized SJL mice that were pre-treated with rat IgG or anti-CD25 were cultured with PLP 139–151. The levels of IFN-γ, IL-2, IL-4 and IL-10 in the supernantant 2 days after activation were measured by using ELISA. Data were the mean values ± SD of three independent experiments (*P < 0.05; **P < 0.01).
TGF-β plays a role in recovery from EAE
A fraction of CD4+CD25+ T cells are positive for TGF-β LAP on the cell surface and the percentage of positive cells is increased after in vitro activation (22). We and others previously reported that the membrane-bound TGF-β, positive for LAP, was involved in suppression of CD4+CD25+ T cells (21–23). To investigate whether TGF-β plays a role in recovery from EAE, we first examined LAP expression on CD4+CD25+ T cells in blood, LN and spleen of naive and EAE-recovered mice. As shown in Fig. 5(A), there was a significant increase in the percentage of LAP+ cells within CD4+CD25+ population in blood and spleen of EAE-recovered mice as compared with the naive mice (12.6 ± 1.5% versus 5.9 ± 1.5% in blood, P < 0.001, and 23.8 ± 3.8% versus 9.3 ± 0.7% in spleen, P < 0.01). But, the percentage of CD4+CD25+LAP+ T cells in draining LN was not significantly different (11.5 ± 1.8% versus 8.3 ± 3.2%, P > 0.05). Furthermore, the absolute number of CD4+CD25+LAP+ cells in spleens of EAE-recovered mice was 6-fold higher than that of naive mice (1.9 ± 0.4 versus 0.3 ± 0.1 × 106, P < 0.01). Similarly, percentages of LAP+ cells within CD4+ subset in blood and spleen from EAE-recovered mice were significantly higher than those from naive mice (6.1 ± 1.6% versus 1.4 ± 0.3% in blood, P < 0.001, and 8.3 ± 2.1% versus 5.0 ± 0.5% in spleen, P < 0.01) (Fig. 5B and C). However, the percentage of CD4+LAP+ T cells in draining LN from EAE-recovered mice did not change significantly compared with those from naive mice (2.2 ± 0.5% versus 2.0 ± 0.7%, P = 0.6). Importantly, the absolute number of CD4+LAP+ T cells in spleen from EAE-recovered mice was 2-fold higher than those from naive mice (3.2 ± 0.5 versus 1.3 ± 0.1 × 106, P < 0.001). To neutralize TGF-β in vivo, EAE mice were injected i.p. 0.4 mg of anti-TGF-β1, β2, β3 antibody (1D11) or isotype control antibody (mouse IgG) on days 24, 26, 28 and 30 after induction of EAE and the disease was monitored for 36 days. We found that in vivo neutralization of TGF-β abolished the recovery from EAE and the disease severity was higher than control antibody-treated group (Fig. 5D). These results indicate that LAP+ T cells play a role in recovery from EAE by a TGF-β-dependent mechanism.
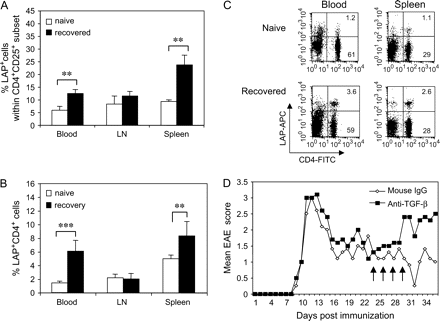
TGF-β is involved in recovery from EAE. Frequency of CD4+CD25+LAP+ T cells (A) and CD4+LAP+ T cells (B) in naive and EAE-recovered mice. Cells (1.0 × 106) of blood, LN and spleen from naive or EAE-recovered mice (on day 30) were first incubated with anti-CD16/CD32, then stained with 1.0 μg of biotinylated anti-LAP (TGF-β1) polyclonal antibody for 30 min. Cells were washed twice and incubated with PE-conjugated anti-CD25, FITC-conjugated anti-CD4, APC-conjugated streptavidin (1:200 dilution) and 7-AAD. The analysis was performed on a FACScan flow cytometer with CellQuest software. The frequencies of LAP+ cells were determined in the live CD4+CD25+ or CD4+ population (naive mice, n = 12; recovered mice, n = 14, **P < 0.01; ***P < 0.001). (C) Dot plots of CD4+LAP+ T cells in blood and spleen from naive or EAE-recovered mice. (D) Neutralization of TGF-β in vivo. EAE mice were given i.p. 0.4 mg of mouse IgG (open diamonds) or 0.4 mg of anti-TGF-β neutralizing antibody (filled squares) on days 24, 26, 28 and 30 after induction of EAE. Animals were scored daily for symptoms of EAE for 36 days. The result shown here was one of three independent experiments. There were 5–7 mice per group.
Discussion
In the present study, we have demonstrated that CD4+CD25+ regulatory T cells participate in recovery from EAE. First, EAE-recovered mice have an increased number of Foxp3-expressing CD4+CD25+ T cells that have a regulatory function in vitro. Second, depletion of CD4+CD25+ T cells during recovery phase of EAE exacerbates the disease and enhances PLP 139–151-reactive T cell responses in vitro. Third, TGF-β is involved in recovery from EAE as LAP+ cells increased and in vivo neutralization of TGF-β abolished recovery from the disease.
There are now reports on the role of CD4+CD25+ regulatory T cells in preventing the development of EAE (15–17), however, their role in spontaneous remission of EAE is still unknown. Here, we hypothesize that CD4+CD25+ regulatory T cells may be generated during the recovery phase of EAE and they may potentially contribute to the remission of disease. We found an increased number of Foxp3-expressing CD4+CD25+ T cells in EAE-recovered SJL mice. Importantly, these cells did not proliferate to PLP 139–151 stimulation and showed suppressive ability in an antigen-specific manner (Fig. 2C). We have recently reported that although CD4+CD25+ T cells from naive SJL mice were able to suppress responder proliferation to a polyclonal activator, they were unable to suppress PLP 139–151-specific proliferative response in vitro (30), and high frequency of PLP 139–151-reactive CD4+CD25+ T regulatory cells in B10.S mice mediate resistance to induction of EAE in B10.S mice. Taken together, antigen-specific CD4+CD25+ regulatory T cells may be generated in the periphery during the recovery phase of the disease and mediate natural remission. It is also consistent with previous observations that CD4+CD25+ regulatory T cells were induced and expanded in vivo in an antigen-specific manner by oral administration of antigen (31) or immunization with self-antigen (32). However, it is still unknown how CD4+CD25+ regulatory T cells are generated in the recovery phase of the disease. Given that the naive mice indeed possess PLP 139–151-tetramer-positive CD4+CD25+ T cells (25), we propose that this fraction may expand upon immunization and once they reach the threshold to limit the expansion of CD4+CD25− effector cells that react with PLP 139–151, recovery ensues. It may be possible that CD4+CD25+ regulatory T cells are generated in the thymus in order to control PLP 139–151-specific CD4+CD25− T cell responses, or they may be converted from CD4+CD25− T cell pool in the periphery. Reduced number of CD4+CD25− T cells in draining LN of EAE-recovered mice may be due to Fas-mediated apoptosis as previously reported (3).
Consistent with Bischof et al. (33), we found that the frequency of IAs/PLP 139–151-reactive cells in the periphery of mice during the effector phase of EAE (day 10 after immunization) was increased significantly compared with those in naive mice and that the frequency of these cells in EAE-recovered mice declined to a base level (Fig. 3B). The reduced IAs/PLP 139–151-reactive cells in the periphery of EAE-recovered mice may not be due to migration to the CNS because IAs/PLP 139–151-reactive cells in CNS were also decreased in EAE-recovered mice (33). Furthermore, depletion of CD4+CD25+ T cells resulted in an increase of IAs/PLP 139–151-reactive cells when CD4+ T cells were re-stimulated in vitro (Fig. 3C). These data suggest that upon immunization and stimulation with PLP 139–151 in vitro, expansion of IAs/PLP 139–151-reactive cells is under control of CD4+CD25+ regulatory T cells in the periphery. These are consistent with our observation that PLP 139–151-specific CD4+CD25+ regulatory T cells exist in naive SJL mice and prevent the expansion of PLP 139–151-reactive CD4+CD25− T cells in an antigen-specific manner (25). Depletion of CD4+CD25+ regulatory T cells in the recovery phase of the disease also resulted in an increase of IFN-γ-producing CD4+ T cells after in vitro stimulation with PLP 139–151. As it has been reported (33), CD4+CD25+ regulatory T cells suppress not only Th1 cells but also Th2 cells even though Th1 cells are more susceptible than Th2 cells. In this study, we found that frequency of IL-4-producing CD4 T cells and IL-4 production were elevated in CD4+CD25+ T cell-depleted mice, suggesting that CD4+CD25+ regulatory T cells in recovery phase of EAE suppress both Th1 and Th2 cells. An increase of IL-4 production may also explain that there is a potential to recover from the disease in CD4+CD25+ T cell-depleted mice 2 weeks after the last treatment with anti-CD25 antibody (data not shown). Of note, anti-CD25 treatment could theoretically also delete effector cells. If this were the case, we would have expected an opposite outcome with disease amelioration.
TGF-β is shown to play a role in mediating the suppressive activity of CD4+CD25+ regulatory T cells in some systems (10). In vivo, TGF-β regulates the expression of Foxp3 and expands Foxp3-expressing CD4+CD25+ regulatory T cells (34). In addition, TGF-β-induced Foxp3 expression in CD4+CD25− naive T cells mediates their transition toward a regulatory T cell phenotype (24). We and others have recently demonstrated that the presence of membrane-bound TGF-β or LAP on the surface of regulatory cells has linked TGF-β with regulatory T cell suppressive function (21–23). Furthermore, TGF-β-induced regulatory CD4+CD25+ T cells expressed increased TGF-β on the surface that mediates their suppressive function (24). We hypothesized that surface-bound TGF-β might play a role in regulatory T cell-mediated recovery from EAE. We found that percentage of LAP+ cells increased significantly with disease recovery. The exact relationship between CD4+CD25+ cells and CD4+LAP+ cells remains to be determined. LAP+ cells are positive for thrombospondin, thus they can convert latent TGF-β to the active form in a cell-free system (21). Thus, it is possible that LAP+ cells are involved in induction and expansion of CD4+CD25+ cells by producing active TGF-β. In addition, Ochi and Weiner have recently demonstrated that regardless of CD25 expression, CD4+LAP+ cells are anergic and are capable of inhibiting CD4+CD25− cell proliferation in vitro and in vivo (unpublished observation).
In summary, we show that Foxp3-expressing CD4+CD25+ regulatory T cells mediate recovery from PLP 139–151-induced EAE in SJL mice. Depletion of CD4+CD25+ T cells during recovery phase exacerbates disease and facilitates expansion of IAs/PLP 139–151-tetramer-positive cells and enhances IFN-γ production. CD4+LAP+ cells are significantly increased in EAE-recovered mice and in vivo neutralization of TGF-β abolishes recovery from the disease. Taken together, our data suggest that both CD4+CD25+ and CD4+LAP+ regulatory T cells mediate recovery from EAE in which TGF-β plays an important role.
Transmitting editor: L. Steinman
References
Zamvil, S. S. and Steinman, L.
McRae, B. L., Vanderlugt, C. L., Dal Canto, M. C. and Miller, S. D.
Suvannavejh, G. C., Dal Canto, M. C., Matis, L. A. and Miller, S. D.
Sabelko-Downes, K. A., Cross, A. H. and Russell, J. H.
Khoury, S. J., Hancock, W. W. and Weiner, H. L.
Kennedy, M. K., Torrance, D. S., Picha, K. S. and Mohler, K. M.
Chen, Y., Kuchroo, V. K., Inobe, J., Hafler, D. A. and Weiner, H. L.
Issazadeh, S., Ljungdahl, A., Hojeberg, B., Mustafa, M. and Olsson, T.
Salomon, B., Lenschow, D. J., Rhee, L. et al.
Read, S., Malmstrom, V. and Powrie, F.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. and Toda, M.
Asano, M., Toda, M., Sakaguchi, N. and Sakaguchi, S.
Takahashi, T., Kuniyasu, Y., Toda, M. et al.
Thornton, A. M. and Shevach, E. M.
Kohm, A. P., Carpentier, P. A., Anger, H. A. and Miller, S. D.
Zhang, X., Koldzic, D. N., Izikson, L. et al.
Montero, E., Nussbaum, G., Kaye, J. F. et al.
Nicholson, L. B., Greer, J. M., Sobel, R. A., Lees, M. B. and Kuchroo, V. K.
Miller, A., Lider, O., Roberts, A. B., Sporn, M. B. and Weiner, H. L.
Karpus, W. J. and Swanborg, R. H.
Oida, T., Zhang, X., Goto, M. et al.
Nakamura, K., Kitani, A. and Strober, W.
Nakamura, K., Kitani, A., Fuss, I. et al.
Chen, W., Jin, W., Hardegen, N. et al.
Reddy, J., Bettelli, E., Nicholson, L. et al.
Aluvihare, V. R., Kallikourdis, M. and Betz, A. G.
Hori, S., Nomura, T. and Sakaguchi, S.
Khattri, R., Cox, T., Yasayko, S. A. and Ramsdell, F.
Fontenot, J. D., Gavin, M. A. and Rudensky, A. Y.
Reddy, J., Illes, Z., Zhang, X. et al.
Zhang, X., Izikson, L., Liu, L. and Weiner, H. L.
Walker, L. S., Chodos, A., Eggena, M., Dooms, H. and Abbas, A. K.
Bischof, F., Hofmann, M., Schumacher, T. N. et al.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}