

Abstract
Recently, off-centering behavior has been discovered in a series of thermoelectric materials. This behavior indicates that the constituent atoms of the lattice displace from their coordination centers, leading to the locally distorted state and local symmetry breaking, while the material still retains its original crystallographic symmetry. This effect has been proved to be the root cause of ultralow thermal conductivity in off-centering materials, and is considered as an effective tool to regulate the thermal conductivity and improve the thermoelectric performance. Herein, we present a collection of recently discovered off-centering compounds, discuss their electronic origins and local coordination structures, and illuminate the underlying mechanism of the off-centering effect on phonon transport and thermal conductivity. This paper presents a comprehensive view of our current understanding to the off-centering effect, and provides a new idea for designing high performance thermoelectrics.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Thermoelectric materials offer a simple and environmentally friendly solution for directly converting heat into electrical power, which can be potentially employed in waste heat harvesting, thereby improving the energy conversion efficiency of fossil fuels utilization [1]. Furthermore, electricity can drive the thermoelectric materials to work as a solid-state heat pump, makes the thermoelectric cooling been a promising temperature control method [2]. Owing to these advanced and multifarious applications, thermoelectricity has attracted worldwide attention. The energy conversion efficiency of a thermoelectric device is primarily determined by the ZT value of thermoelectric material, which is given as ZT = (S2 σ)T/κ. It is obvious that good thermoelectric materials are generally required to have both high Seebeck coefficients (S), and high electrical conductivity (σ), along with low thermal conductivity (κ) [3–6]. However, the actual challenge is that these thermal and electronic transport properties are, in fact, strongly correlated with the carrier concentration and effective mass. Maximizing one of these transport parameters often leads to the degradation of the other two [7–10].
For most thermoelectric materials, the total thermal conductivity (κ) consists of two components: the electronic thermal conductivity (κe) and the lattice thermal conductivity (κL), which contributed by charge carriers' transport and the lattice vibrations, respectively [11]. Since lattice thermal conductivity is determined by the phonon characteristic and its transport mode in the material. It is relatively independent of the electronic transport properties, and can be minimized without altering the carrier concentration, but only through the crystal structure and microstructure design. This makes the reduction of lattice thermal conductivity an effective approach in improving thermoelectric performance [12], which has been widely used in the field of thermoelectrics. Therefore, understanding the mechanism of phonon transport in crystal solid and developing new strategies to regulate thermal conductivity is important for designing high performance thermoelectrics. This is also of fundamental interest in many other fields. In the past decades, numerous strategies have been proposed and successfully applied to suppress thermal conductivity, including the in-situ nanostructure [13–18], rattling atom filling [19–24], liquid-like lattice [25–28], high-entropy alloying [29–33] and recently, the off-centering effect [34–38]. Compared with other well-known mechanisms, the off-centering effect has been rarely discussed. However, recent research has shown that the off-centering effect could introduce low frequency optical mode and result in strong acoustic–optical (A–O) phonon scattering, making it an effective approach to reduce the thermal conductivity [39–42].
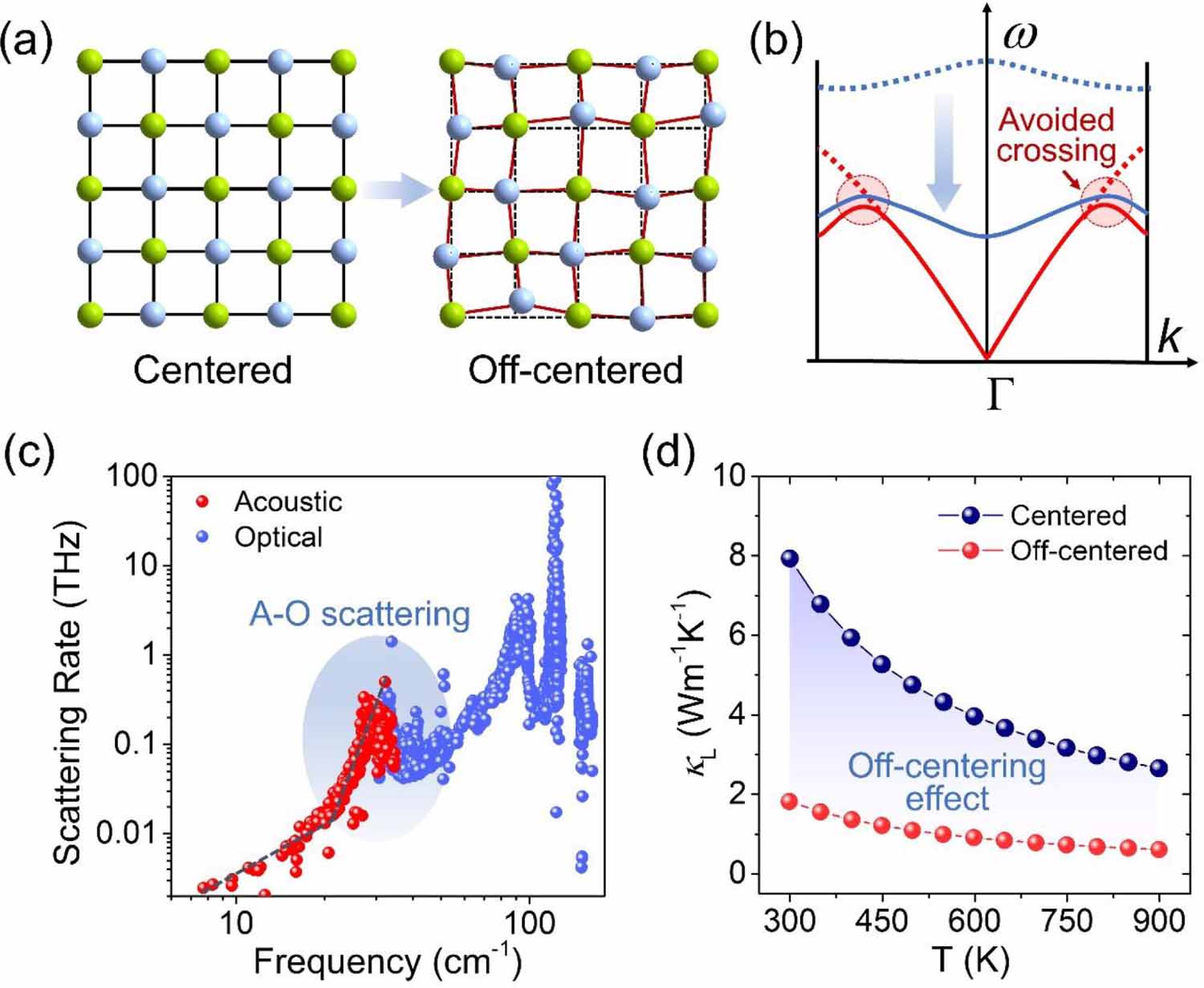
The schematic diagram in figure 1(a) illuminates the concept of off-centering behavior. As we know, in a solid crystal, the constituent atoms incessantly oscillate due to thermal energy, and the vibration amplitude increases with rising temperature. However, over a period of time, their equilibrium positions should always be at the lattice centers. While for the off-centering case, the discordant atom (depicted as the blue sphere in figure 1(a)), usually of electronic origin, needs its own space to stereochemically express itself [37, 39]. This distorts the chemical bonding and imposes a strong local geometric influence on the lattice. Thus, the equilibrium position of the discordant atom is off-centered from its ideal coordination site, leading to the distortion of the local structure and local symmetry breaking. Moreover, the discordant atoms dynamically fluctuate in different available distortion directions. As a result, the statistical average displacement of their equilibrium positions is net zero, meaning the global crystal structure of the material still retains its original crystallographic symmetry. Since the off-centering behavior and local symmetry breaking are hidden within the symmetrically global crystal structure, they are easily neglected in conventional x-ray diffraction inspections. An effective tool to detect off-centering behavior is x-ray atomic pair distribution function (PDF) analysis [37, 43], which is able to detect the local coordination structure and probe the local bonding state. When off-centering behavior appear, the local bonding distortion would be captured in the PDF results.
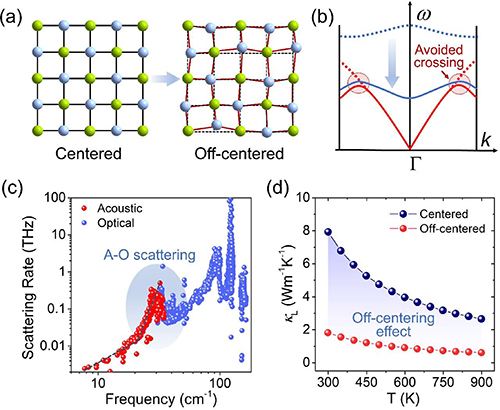
Figure 1. (a) The schematic diagrams show the concepts of ideal coordination structure and off-centering behavior. (b) The schematic diagram of phonon dispersion illuminates how off-centering behavior could introduce low-frequency optical mode and result in acoustic-optical phonon scattering; The blue and red curves indicate optical and acoustic modes, respectively. (c) The frequency dependent phonon scattering rate of off-centered diamondoid material, indicating the acoustic–optical (A–O) phonon scattering leads to the high scattering rate of heat-carrying phonons. (d) The calculated thermal conductivities of ideal diamondoid structure and off-centered diamondoid structure. [37] John Wiley & Sons.© 2022 Wiley-VCH GmbH.
Download figure:
Standard image High-resolution imageHowever, this hidden off-centering behavior could have a considerable influence on phonon transport and heat conductivity. Generally, acoustic phonons are the primary heat carriers in crystal solids, and the contribution of optical phonons to thermal conductivity is negligible due to their stationary wave character [44]. Besides, the large difference in frequency (energy) between acoustic and optical phonons makes their interaction to be difficult [45]. Thus, the A–O phonon scattering mechanism is usually very weak in most materials. Things change in the off-centering case. As we know, off-centering behavior can lead to local distortion in the structure, which in fact, implies the distortion in chemical bonding. This distorted and weak bonding can actually lower the frequency of optical phonons, as shown in figure 1(b). These low frequency optical modes reduce the energy gap between acoustic and optical phonons, greatly increasing the chance of their interaction. Consequently, the low frequency optical modes can involve in the acoustic phonon scattering processes and cause A–O phonon scattering [39, 46–48]. The avoided crossing phenomenon in the phonon dispersion is usually a sign of this additional phonon scattering. Figure 1(c) depicts the frequency dependent phonon scattering rate of an off-centered diamondoid material [37], which serve as a canonical example. Generally, the phonon scattering rate increases with the rising phonon frequency, because of the increasing energy and phonon density of states. However, in the off-centering case, the acoustic and optical phonons exhibit anomalously exponential growth in the scattering rate within the frequency overlap region (the blue area in figure 1(c)), this is the key feature of the A–O phonon scattering. Thus, although the low frequency optical phonons do not directly carry heat, they significantly enhance the scattering rate of heat-carrying phonons, then results in a low thermal conductivity. Figure 1(d) depicts the theoretical calculated thermal conductivities of CuGaTe2 with an ideal diamondoid structure and an off-centered diamondoid structure [37], which distinctly shows the off-centering behavior is effective in reducing thermal conductivity.
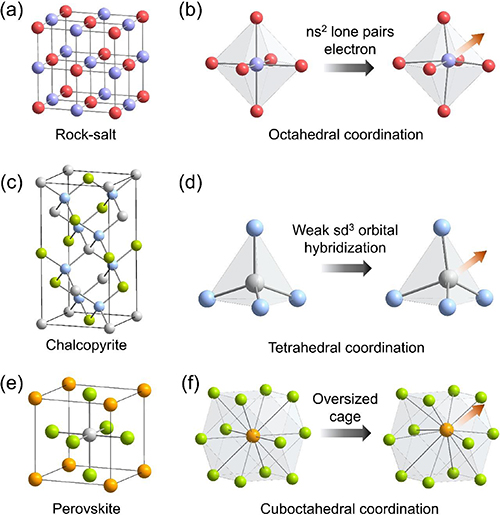
Figure 2 shows a series of off-centered lattices, including their local coordination structures and the corresponding driving forces. A representative case of off-centered thermoelectric material is the rock-salt PbTe (figure 2(a)). For a long time, PbTe was believed to adopt the ideal rock-salt structure and be perfect packing, until Božin, Kanatzidis and Billinge employed synchrotron x-ray atomic PDF analysis to investigate its local structure [34], and found the Pb2+ is actually off-centered from the Te octahedral coordination cage, as shown in figure 2(b). This is because the lone pair electron of Pb2+ needs its own space to stereochemically express itself, causing the Pb2+ ion shift along the crystallographic 〈100〉 direction, with a maximum distortion value of 0.24 Å [34, 49]. This results in the local structure distortion and the ferroelectric instability of PbTe. Additionally, the off-centering behavior of Pb2+ leads to a strong phonon coupling between low frequency transverse optical modes and longitudinal acoustic modes [46], resulting in the low lattice thermal conductivity of PbTe. In addition to PbTe, cation off-centering behavior had also been identified in many IV–VI compounds and I-V-VI2 rock-salt materials, such as PbSe, SnSe [50], SnTe [51], GeTe [52] and AgSbSe2 [53, 54]. The origin of cation off-centering in these compounds is attributed to the ns2 lone pair electron of the main-group metals, for example, the Pb2+, Sn2+, Ge2+ and Sb3+.
{kind=link}
Figure 2. (a) Rock-salt structure and (b) the octahedral coordination structure, illustrates the cation off-centering behavior caused by the ns2 lone pairs electron. (c) Chalcopyrite structure and (d) the tetrahedral coordination structure, illustrates the cation off-centering behavior caused by the weak sd3 orbital hybridization. (e) Perovskite structure and (f) the cuboctahedral coordination structure, illustrates the cation off-centering behavior caused by the oversized coordination environment and weak chemical bonding.
Download figure:
Standard image High-resolution image{kind=link}
Another driving focus behind the off-centering behavior is the weak electronic orbital hybridization, which has been first discovered in the I-III-VI2 chalcopyrite (diamondoid compound, figure 2(c)). One representative case is AgGaTe2 [37]. In a diamondoid structure, the transition metal should adopt the sd3 electronic orbital hybridization to construct a tetrahedral coordination geometry, see figure 2(d). This hybridization requires the atomic electron orbitals with similar energy. While for the Ag element, the energy difference between its 5 s and 4d electronic orbitals is large, which is almost four times higher compare to the difference of 4 s and 3d orbitals in Cu (which adopts the sd3 hybridization in diamondoid structure). This substantial energy difference between the interacting orbitals lead to a weak sd3 hybridization in Ag, makes it amenable to deviate from the tetrahedral coordination geometry and shifts toward one of the Te–Te edges within the tetrahedral unit. In response, the Te atoms would displace along the Te–Te edge direction, leading to weak chemical bonding and strong A–O phonon scattering. As a result, the AgGaTe2 compound exhibits an extremely low intrinsic lattice thermal conductivity, which is 0.26 Wm−1K−1 at 850 K [37]. Since the off-centering behavior of Ag is electronic in origin, which is believed to exist in other silver diamondoid compounds, and has been considered as the root cause of low thermal conductivity of these materials.
Another fascinating off-centered case is the cubic metal halide perovskite [38, 55], as shown in figure 2(e). The constitution of the metal halide perovskite is expressed as ABX3, (A = Cs+; B = Pb2+, Sn2+; X = Cl−, Br−, I−), which has been widely investigated in the photoelectricity, and more recently, in thermoelectricity. In the perovskite structure, the B-site cation sits on an octahedral coordination geometry, since the Pb2+ and Sn2+ possess the ns2 lone pairs [55], it is stereochemical activity and causes the cation to move away from the ideal octahedron center. Moreover, the Cs+ is also off-centering because of the oversize effect [38]. In the cubic perovskite structure, Cs atom resides in a 12-coordinated cuboctahedral site, bonding with 12 halide anions. Through careful investigation of the local structure, Xie and Kanatzidis found that Cs is actually located inside a relatively oversized cuboctahedral cage [38], see figure 2(f). For example, the bond distance of Cs-Br in CsSnBr3 is 4.0977 Å, while in cubic CsBr, Cs resides in an eight-coordinated hexahedral geometry, and the bond distance of Cs-Br is 3.6979 Å. The significantly longer bonding in cubic perovskite implies that the 12-coordinated cuboctahedral cage is oversized for the Cs atom, and the bonding strength of Cs-Br is relatively weak. This oversized cage and weak chemical bonding make Cs tend to move away from the cuboctahedral center and shift along the crystallographic 〈111〉 direction. Thus, the off-centering effect of A-site and B-site atoms leads to the intrinsic low thermal conductivity of halide perovskite.
In summary, the off-centering effect is an intriguing phenomenon in crystal lattice, which is able to suppress lattice thermal conductivity through A–O phonon scattering, and is the root cause of intrinsic low thermal conductivity in many materials. The driving force behind the off-centering effect could be the lone pairs electron, weak electronic orbital hybridization, oversized coordination and weak chemical bonding. Additionally, we want to note that, the origins of the off-centering effect are not necessary confined to one type of coordination structure. For example, the lone pairs electron could generate the off-centering effect in both octahedral and tetrahedral coordination geometries. Moreover, the off-centering effect is not limited exclusively to those intrinsic off-centered compounds it could also be introduced to other materials as an effective tool to reduce thermal conductivity and enhance thermoelectric performance. Some successful cases have been demonstrated, such as the Ag-alloyed CuGaTe2 and CuInTe2 (weak sd3 hybridization) [41, 47, 48]; In+ doped CuFeS2 [56] and Ge2+ doped PbSe [35] (lone pairs electron); Hg2+ doped [57] and Cd2+ doped [36] PbSe (discordant dopant). All these suggest that the off-centering effect provides a new path to regulate heat conductivity and could be an effective strategy for designing high performance thermoelectrics.
Acknowledgments
This work was supported by National Natural Science Foundation of China (52250090, 52371208, 51571007, 51772012), the Beijing Natural Science Foundation (JQ18004), 111 Project (B17002). L D Z appreciates the National Science Fund for Distinguished Young Scholars (51925101).
Conflict of interest
The authors declare no competing interests.