
Abstract
Photodynamic therapy (PDT) is a well-established treatment of cancer that uses the toxic reactive oxygen species, including singlet oxygen (1O2), generated by photosensitiser (PS) drugs following irradiation of a specific wavelength to destroy the cancerous cells and tumours. Visible light is commonly used as the excitation source in PDT, which is not ideal for cancer treatment due to its reduced tissue penetration, and thus inefficiency to treat deep-lying tumours. Additionally, these wavelengths exhibit elevated autofluorescence background from the biological tissues which hinders optical biomedical imaging. An alternative to UV–Vis irradiation is the use of near infrared (NIR) excitation for PDT. This can be achieved using upconverting nanoparticles (UCNPs) functionalised with photosensitiser drugs where UCNPs can be used as an indirect excitation source for the activation of PS drugs yielding to the production of singlet 1O2 following NIR excitation. The use of nanoparticles for PDT is also beneficial due to their tumour targeting capability, either passively via the enhanced permeability and retention (EPR) effect or actively via stimuli-responsive targeting and ligand-mediated targeting (i.e. using recognition units that can bind specific receptors only present or overexpressed on tumour cells). Here, we review recent advances in NIR upconverting nanomaterials for PDT of cancer with a clear distinction between those reported nanoparticles that could potentially target the tumour due to accumulation via the EPR effect (passive targeting) and nanoparticle-based systems that contain targeting agents with the aim of actively target the tumour via a molecular recognition process.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Photodynamic therapy (PDT) is a minimally invasive treatment that combines the use of light, oxygen and photosensitiser (PS) drugs to generate reactive oxygen species (ROS) [1, 2], mainly singlet oxygen (1O2) [3], that are able to destroy tumours by oxidising biological substrates [3]. PS drugs are photoactive molecules able to produce 1O2 or other ROS after being excited with light of a specific wavelength, most commonly visible or ultraviolet light [2]. Excited PS can lose their energy via fluorescence emission and/or heat [4]; or following a non-radiative pathway, intersystem crossing, forming a long-lived excited triplet state. From the PS excited triplet state, the energy may decay emitting light or can undergo two different types of reactions yielding radicals and ROS [5]. Type I reaction is based on an electron or hydrogen transfer and produces radical molecules such as hydroxyl radical (HO•). In contrast, Type II reaction involves an energy transfer from the PS excited triplet state to the ground triplet state of molecular oxygen (O2) leading to the formation of 1O2 [4, 5].
Apoptosis, necrosis and autophagy are the main cell death pathways that occur by immediate consequence of PDT [6, 7]. Factors such as cell type, light dosage, and photosensitiser intracellular localisation and concentration define the mechanism of cell death induced by PDT [8]. For example, if the PS accumulates in the mitochondria or the endoplasmic reticulum (ER), apoptosis is the main path for cell death [7, 8]. If, by contrast, the PS is targeted either in the plasma membrane or lysosomes, the cells will die via necrosis [5]. An elevated concentration of PS drug or light dose has also been related to a necrotic cell death [7]. Mitochondrial damage yielding to cytochrome c release and formation of the protein apoptosome has been reported due to the acute stress response produced by PDT [9].
PDT is currently clinically used to treat different types of early-stage cancers [10, 11] including skin [12], oesophageal [13], mouth [14] and lung [15] cancer. One of the major advantages of PDT compared to conventional cancer treatments, such as chemotherapy and radiotherapy, is that it is a non-invasive, localised treatment and thus, its side effects are considerably reduced. The combination of PDT with other anticancer therapies has shown synergistic effects [16–18]. For example, PDT has been combined with low doses of x-ray radiation for the treatment of breast cancer cells (MCF-7) in in vitro experiments resulting more effective than each treatment alone [18]. The combination of PDT with chemotherapy has been studied in, for example, metastatic melanoma resulting in a higher decrease of metastatic melanoma tumorigenic cells than the decrease observed for the individual treatments [19].
Phthalocyanines, porphyrins, naphthalocyanines, chlorins, bacteriochlorins, texaphyrins, among others, are examples of organic PS drugs commonly reported in the literature for PDT of cancer [3, 10]. Inorganic PS, based on nanoparticles of semiconductor metal oxides (TiO2 and ZnO) have also been successfully employed [20, 21]. The effectiveness of PDT is dependent on the chemical properties, photostability, physiological stability, absorption coefficient and efficiency of 1O2 production of the PS drugs [1]. Despite the large number of examples of PS drugs reported in the literature, only a few of these are currently approved by the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for clinical use [11, 22–26]. Examples of clinically approved PS include Phtofrin® that was approved in the US in 1995 and can be used for the treatment of obstructing oesophageal cancer, obstructing lung cancer, microinvasive endobronchial cancer, gastric and papillary bladder cancer, and cervical dysplasia and cancer; Foscan® that was clinically approved by the EU in 2001 and can be used to treat head and neck cancer; NPe6 that was clinically approved in Japan in 2004 for the PDT of lung cancer; and Levulan® that was approved in the US in 1999 for the treatment of actinic keratoses. Photofrin® administration is at 2 mg kg−1 intravenously and the laser light dose and time scale depend on the cancer to be treated, for example for the treatment of oesophageal cancer, laser light dose of 300 J·cm−1 40–50 h following injection with Photofrin® is administered and the treatment is repeated, if needed, 96–120 h after initial injection. Foscan® is administered at 0.15 mg·kg−1 and light must be administered between 90 and 110 h following injection at a dose of 20 J·cm−2. The reduced number of PS approved for clinical use is most likely due to their limitations. The majority of them are hydrophobic molecules, thus easily internalised by cells but with restricted transportation through the body [1, 27]. Furthermore, most PS exhibit non-specific biodistribution [28], leading to severe side effects; and their activation to produce 1O2 requires UV–Vis light [29], which are not ideal for cancer treatment due to their reduced tissue penetration (ca. < 1 cm), and thus inefficiency to treat deep-lying tumours [30]. It is important to mention that PDT is more effectively achieved when the PSs are close enough to the tumour sites due to the short half-time (0.04 μs) and the high reactivity of 1O2 after being formed, thus resulting in a ratio of action of ca. 0.02 μm [1, 8]. Additionally, in the UV–Vis range, there is an elevated autofluorescence background from the biological tissues which hinders optical biomedical imaging. To overcome the limitations of UV–Vis excitation, and as it will be detailed later in this review, the use of near infrared (NIR) excitation for PDT has attracted extensive interest in the past decades since it shows minimal light absorption and scattering by biological tissue, thus reducing the photodamage and enabling deeper tissue penetration (ca. < 10 cm) [30, 31]. The unique advantages of NIR on deep tissue penetration include the reduced light-induced cytotoxicity which could permit the non-invasive PDT treatment of tumours located deep into the tissue [32, 33]. In the last decades, several NIR-excitable photosensitisers have been developed [30] and an example is IR780. This PS exhibits excellent optical properties and great photoconversion efficiency; and has been extensively used for in vitro and in vivo PDT [34]. One of the main drawbacks of this photosensitiser is its low water solubility and potential toxicity, thus, IR780 has been combined with nanoparticles yielding improved water dispersibility, stability and biocompatibility [35].
The use of nanoparticles as delivery systems of PS drugs for PDT has been extensively investigated offering many advantages over molecular PS and overcoming some of the listed drawbacks [36]. Incorporation of PS onto nanoparticles can increase the water-solubility of the drugs, elongating their blood circulation time and thus improving their cellular/tumour uptake, pharmacokinetics and biodistribution; and can protect the PS against, for example, enzymatic degradation [37]. Additionally, nanoparticles can pass through the leaky blood vessels of tumour sites and accumulate in the malign tissues in a process known as the enhanced permeability and retention (EPR) effect; yielding passive tumour-targeting [38–40]. The small size, tailored surface and multi-functionality characteristics of nanoparticles lead furthermore to high drug-loading capacity improving the photostability of the PS and reducing the photobleaching effect normally observed in molecular PS [40]. Examples of the most commonly used carrier nanoparticles for PDT include polymeric nanoparticles [41], liposomes [42], silica nanoparticles [43] and metallic nanoparticles (such as gold [44–47], silver [48] and iron oxide [49, 50]). For example, Wang et al functionalised gold nanostars with Chlorin e6 (Ce6) and reported higher PDT efficiency both in in vitro and in vivo when using the nanostars compared to free Ce6 [51]. The incorporation of PS drugs onto a nanoplatform has been shown to improve the biocompatibility of hydrophobic drugs [45, 52]. For instance, Lee et al developed chitosan nanoparticles loaded with Ce6 for tumour treatment via PDT [52]. Although the 1O2 generation was slower for the Ce6 loaded nanoparticles than for the free drug, the nanoparticles were biocompatible showing prolonged blood circulation times and enhanced tumour targeting compared to the free drug [52]. Nanoparticles have also been reported to increase the PDT effect of some hydrophobic and hydrophilic photosensitiser drugs. For example, Penon et al reported two different functionalised iron oxide nanoparticles containing either hydrophobic or hydrophilic porphyrin derivative ligands that, upon irradiation (400–500 nm), showed higher levels of 1O2 production than the corresponding free porphyrin-based ligands [49]. Alea-Reyes et al studied different types of gold-based vehicles including nanoparticles, nanorods and microparticles, as carriers of porphyrin derivatives for PDT [53]. Higher 1O2 production was observed when using gold nanoparticles and, when used for PDT in HeLa cervical cancer cells, these probed to be more efficient than the corresponding free porphyrin [53]. An example of polymeric nanoparticles for encapsulation of PS drugs was published by Brandhonneur et al in which poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles were used for the loading of a molybdenum cluster as PS drug leading to encapsulation efficiency higher than 80% [54]. The loaded PLGA nanoparticles were used for the PDT treatment (365 nm irradiation) of ovarian cancer cells (A2780 cell line) showing a higher cellular viability decrease (ca. 20% higher) compared to the free cluster [54]. All of these examples prove that nanoparticles are promising carriers of PS drugs for PDT of cancer, overcoming some of the limitations of the free PS drugs and enhancing the efficiency of the treatment. However, one of the persistent problems when using nanoparticles as drug delivery systems is their clearance by the reticuloendothelial system (RES) [55] which can be overcome by covering them with biocompatible ligands such as proteins, liposomes or polyethylene glycol (PEG). The incorporation of PEG is the most common approach to enhance the water dispersibility of the nanosystems, elongating their circulation time in blood and reducing their RES uptake [55]. Camerin et al reported PEGylated gold nanoparticles containing a hydrophobic Zn(II)-phthalocyanine derivative as PS for in vivo PDT of amelanotic melanoma that resulted in 40% of the mice completely recovered following treatment and no tumour regrowth following PDT [44].
Another advantage of using nanoparticles for the delivery of PS drugs is that they can be further functionalised with specific targeting molecules to ensure the targeted delivery to the tumour side and thus, the targeted treatment of cancer via PDT. Targeting agents such as antibodies, folic acid, transferrin, peptides, aptamers, and carbohydrates, amongst others, are frequently used. For example, Penon et al reported a water-soluble gold nanosystem functionalised with a hydrophobic porphyrin derivative and the antibody anti-erbB2 conjugated to PEG ligands to specifically target the erbB2 receptors expressed in SK-BR-3 breast cancer cells [56]. High cellular uptake of the antibody-porphyrin-functionalised nanoparticles was observed confirming the antibody-antigen interactions; and PDT induced cell death was achieved following irradiation using a blue light source [56].
In all the aforementioned examples, the nanoparticles are not directly involved in the photosensitisation process but act as passive carriers of the PS drugs. By contrast, nanoparticles such as quantum dots (QDs) [57, 58] and upconverting nanoparticles (UCNPs) [59–61] have been used not only as carriers of PS drugs but as active participants in the PS excitation for PDT of cancer [31, 57, 62, 63]. Although QDs are excellent candidates in PDT of cancer, this review paper will focus its attention on the use of UCNPs for PDT due to the advantages that indirect NIR excitation of PS drugs through UCNPs present.
2. UCNPs and their role in photodynamic therapy
UCNPs are nanomaterials capable of converting low energy excitation, NIR light, into high energy emitting photons through an anti-Stokes process which can be separated from other scattered light [64, 65]. UCNPs consist of a crystalline host lattice (hexagonal NaYF4 being the most efficient to date for green and blue upconversion [64]) containing trivalent lanthanide dopants that act as sensitisers and emitters of energy yielding the upconversion energy transfer process to occur [64, 66]. The sensitiser lanthanide ions (ytterbium ions (Yb3+) typically used for 980 nm excitation) can absorb NIR light and transfer two or more photons to the emitting lanthanide ions (the most common are erbium (Er3+), holmium (Ho3+) and thulium (Tm3+)) [64, 67]. Cubic or hexagonal NaYF4 nanoparticles doped with Yb3+ and Er3+ (NaYF4 (20%Yb, 2%Er)) result in green or red emissions (522, 541 and 655 nm peaks) [68], whereas the emission peaks for NaYF4 (25%Yb3+, 0.3%Tm3+) nanocrystal are in the blue spectral region (450 nm and 475 nm) [68]. Lanthanide-doped upconverting NaYF4:Yb3+,Er3+ nanoparticles are commonly synthesised via thermal decomposition of trifluoroacetates in solvent mixtures of oleic acid and octadecene [64, 69], but can be also synthesised following other procedures including co-precipitation and hydrothermal methods [70]. Importantly, the size, shape, crystal phase and morphology of the nanoparticles can be controlled during the synthesis taking into account parameters such as temperature, dopant concentrations and reaction time resulting in narrow particle size distribution and high reproducibility [69, 70].
The interest in using UCNPs is increasing in fields such as material science, bioimaging and biomedicine due to their intrinsic upconversion luminescent properties [71–80]. They present advantages compared to classical (luminescent) systems (quantum dots and fluorescent organic dyes) such as high signal-to-noise ratio, large anti-stokes shifts, non-autofluorescent background, narrow absorption and emission peaks, long lifetimes, high quantum yields and superior photostability, and they have shown to have low toxicity [64, 81]. These lanthanide-doped nanoparticles are chemically stable and in general, less toxic than QDs [81]. Several surface modification strategies have been investigated to enhance the biocompatibility of the UCNPs in vitro and in vivo, showing negligible cytotoxicities of the NaYF4-based UCNPs [82, 83]. Most of the NaYF4-based UCNPs developed for biomedical applications have demonstrated to be non-toxic in a broad range of cell lines. Animal studies with NaYF4-based UCNPs have also been performed to study their potential toxicity in vivo showing good biocompatibility [84]. In PDT, UCNPs have attracted considerable attention since the first proof-of-concept studies were reported by Zhang et al in 2007 [85, 86]. UCNPs can overcome limitations of molecular PS including hydrophobicity and non-specificity but also, when used as an indirect excitation source for the activation of PS drugs, can overcome the drawback of visible excitation. The energy transfer between the UCNPs and the PS can take place by radiative (i.e. direct absorption of the emitted luminescence photons by the PS drugs) or non-radiative (i.e. Föster resonance energy transfer (FRET) mechanism) transitions [87].
When designing UCNPs functionalised with PS drugs for PDT of cancer, there are several features to be considered (figure 1): (1) the spectral overlap between the light emitted by the UCNPs and the maximum absorption wavelength of the PS; (2) the distance between the inner core of the UCNP and the PS—which is key for an efficient energy transfer; (3) the loading of the PS—which plays an important role in the efficiency of PDT (an excess of PS could inversely affect the PDT effect) [63]; (4) the thickness of the shell, when a core-shell strategy is chosen, since the distance between the PS drug and the luminescence core affects the energy transfer process that activates the PS and thus, the PDT efficiency; (5) the biocompatibility of the nanosystem—which may be increased by introducing ligands such as PEG derivatives; and (6) the targeted PDT strategies—which can be introduced by further functionalisation of PS-loaded UCNPs with targeting agents, thus increasing the specificity and the selectivity of the treatment.
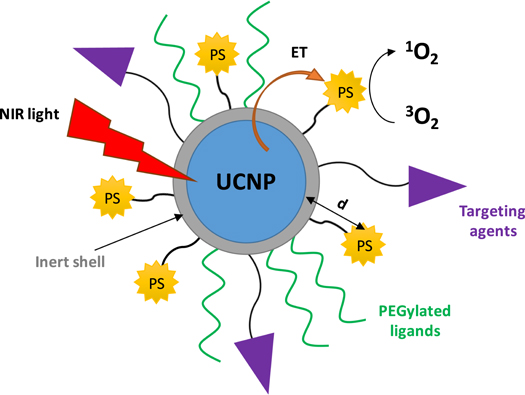
Figure 1. Schematic representation of a core-shell UCNPs functionalised with PS drugs, PEGylated ligands and targeting agents. NIR irradiation of UCNPs generates upconversion luminescence emission which activates the PS drug via energy transfer (ET) to produce singlet oxygen (1O2) from molecular oxygen (3O2).
Download figure:
Standard image High-resolution imageThe surface of UCNPs can be modified with ligands relevant for bioimaging, diagnostics or/and therapeutics via ligand exchange, ligand oxidation reaction, host-guest interactions, layer-by-layer self-assembly method, silica encapsulation or coating with amphiphilic polymers [88]. Loading of PS on the surface of the nanoparticles to obtain an effective electronic excitation energy transfer between the UCNPs and the drug can be achieved following: (1) non-covalent loading of the PS on the surface of the UCNPs using a silica shell coating (figure 2(a)) or physical adsorption which is achieved by using for example amphiphilic polymers (figure 2(b)) [63, 85, 89, 90]; or (2) covalent chemical linkage where PS drugs are anchored to the surface of the nanoparticle (figure 2(c)) [59, 91, 92].

Figure 2. Schematic representation of UCNPs functionalised with PS through (a) silica encapsulation, (b) physical adsorption and (c) covalent chemical bonding strategies.
Download figure:
Standard image High-resolution imageCe6 (Amax at ca. 400 nm), zinc phthalocyanine (ZnPc) (Amax at ca. 660 nm), merocyanine 540 (MC540) (Amax at ca. 540 nm), Rose Bengal (RB) (Amax at ca. 550 nm) and methylene blue (MB) (Amax at ca. 650 nm) are PS drugs commonly used for the functionalisation of UCNPs since their absorption spectrum overlaps the luminescence emission intensities of commonly used UCNPs. For instance, NaYF4:Yb,Er nanoparticles exhibit two main luminescence emission wavelengths in the green (522 and 541 nm) and the red (655 nm) regions. This can lead to an effective energy transfer between the NaYF4:Yb,Er UCNPs and the aforementioned PS drugs and to successful NIR-induced PDT of cancer. Although NaYF4:Yb,Er are the most commonly used UCNPs for PDT due to their high upconversion efficiency, UCNPs doped with Tm3+ have also been reported resulting in luminescence emissions in the blue region (450 and 475 nm) that overlap with PS such as riboflavin, fullerenes, ZnO or TiO2.
The first example of PS-UCNPs for PDT of cancer was reported by Zhang et al in 2007 and consisted of PS M-540 encapsulated onto silica coated NaYF4:Yb3+,Er3+ nanoparticles [85]. Upon irradiation at 974 nm, the UCNPs emitted light at two wavelengths, 537 and 635 nm, with the former effectively overlapping the absorption spectrum of M-540. The M-540-UCNPs were further functionalised with a mouse monoclonal antibody (anti-MUC1/episialin) to specifically target the MUC1 receptors overexpressed in MCF-7/AZ breast cancer cells. Phototoxicity studies performed for MCF-7/AZ cells incubated with the antibody-M-540-UCNPs and irradiated at 974 nm confirmed the effective PDT induced cell death [85]. Since then, a large number of articles have been published describing the use of PS-UCNPs for targeted PDT of cancer. This review paper provides an in-depth analysis of the most relevant articles reporting the use of UCNPs for targeted PDT. The review will differentiate between those reported nanoparticles that could potentially target the tumour due to accumulation via the EPR effect (passive targeting) and nanoparticle-based systems that contain targeting agents to actively target the tumour via a molecular recognition processes [93] (active targeting). A schematic representation of the engineering possibilities when designing UCNPs for PDT is given in figure 3 and examples of the different approaches will be given in the forthcoming sections of this review paper.

Figure 3. Schematic representation of an UCNP and its engineering possibilities: NIR excitable nanoparticle (λexc = 980 or 808 nm) capped with an inert shell, different functionalisation strategies for the incorporation of the PS on the surface of the UCNP, functionalisation with active targeting agents and further coating to enhance the biocompatibility of the nanosystem.
Download figure:
Standard image High-resolution image3. Targeted photodynamic therapy using UCNPs
3.1. Passive targeting
The passive targeting of tumour sites using nanoparticles has been extensively exploited for bioimaging and therapeutic applications, including PDT [37, 62, 94, 95]. The accumulation of the nanoparticles in the solid tumour enhances the specificity of the drug and reduces the side effects on the healthy tissue [45, 51, 52].
The first example of PS-UCNP incorporated M-540 PS drugs on the UCNPs via silica shell encapsulation [85]. Since then, a plethora of non-covalent strategies for the incorporation of PS on UCNPs have been described [76, 89, 90, 96–101]. PS drugs can be encapsulated onto silica shells surrounding the UCNPs using a mesoporous silica layer or a dense silica shell. The former provides a larger surface area than the dense silica coating allowing for higher PS loading, thus this has become the preferred approach on the preparation of silica layered-PS-UCNPs. For example, Zhang and co-workers reported core-shell NaYF4:Yb,Er@silica nanoparticles functionalised with a mesoporous silica layer loaded with zinc (II) phthalocyanine (ZnPc) (0.1 wt%) [89]. ZnPc was not released from the silica following soaking of the particles in deionised water, phosphate buffer saline or RPMI 1640 cell culture medium for one hour. Leaching was however observed when the particles were soaked in ethanol for one hour. The nanoparticles presented an average size of ca. 35 nm width and 60 nm length, equipped with a ca. 10 nm thick silica shell and a ca. 11 nm thick mesoporous silica layer. Their red emission band (ca. 660 nm) overlapped the absorption peak of the ZnPc (670 nm) allowing the activation of ZnPc to generate 1O2 upon irradiation at 980 nm, as confirmed measuring the photobleaching of the 1O2 probe 9,10-anthracenediyl-bis(methylene) dimalonic acid (ABMA) following the endoperoxide formation [89]. However, lifetime measurements to confirm the FRET process occurring between the UCNPs and the PS drug were not reported. The cellular uptake of the nanoparticles in MB49 bladder cancer cells following 24 h incubation was confirmed using confocal fluorescence microscopy with excitation at 980 nm (Δλem = 525 ± 50 nm and 640 ± 90 nm). Cell viability studies (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazoliumbromide, MTT assay) showed lower viability for cells incubated with the ZnPc-UCNPs and irradiated at 980 nm (output power of 500 mW, 5 min) than for cells treated with UCNPs without ZnPc and treated under the same conditions. The cellular uptake of the ZnPc-UCNPs was also investigated measuring the intracellular concentration of Y3+ over time using inductively coupled plasma-atomic emission spectroscopy (ICP-AES) (maximum Y3+ concentration after 6 h of incubation) [90]. In this work, the intracellular production of ROS was confirmed in MB49 cells incubated with ZnPc-UCNPs (100 μg·ml−1) and irradiated at 980 nm (500 mW) using a ROS fluorescent probe (5-(and-6)-carboxy-2'7'-dichlorodihydrofluorescein diacetate) [90]. The PDT efficiency of the ZnPc-UCNPs was confirmed by a drop in cell viability (MTT assay 48 h following irradiation) for MB49 cells treated with ZnPc-UCNPs and irradiated for 5 min at 980 nm compared to the corresponding controls (without irradiation and without ZnPc on the UCNPs)—although cell death was observed without irradiation at high dose nanoparticle concentration [90]. The photodynamic effect was further confirmed by changes in the nuclear morphology—by 4',6-diamidino-2-phenylindole (DAPI) staining; intranucleosomal DNA fragmentation—by DNA-ladder agarose gel electrophoresis; cytochrome c release—by fluorescence staining of cytochrome c; and inhibition of the expression of prostate-specific antigen (PSA)—by enzyme-linked immunosorbent assay (ELISA).
To increase the loading capacity of the PS drug on to the mesoporous silica layer and to avoid the PS leaching, a new strategy was proposed by Han et al based on an electrostatic-driven PS loading [96, 97]. NaYF4:Yb,Er,Nd nanoparticles were coated with a mesoporous silica shell and functionalised with NH2 groups resulting in positively charge silica coated UCNPs that could be further functionalised with the negatively charged Rose Bengal via electronic interactions. To prevent the leaching of the PS and to improve the biocompatibility of the nanoparticles, 1-adamantane carboxylic acid molecules were linked to the surface of the UCNPs as guest unit, and hydrophilic β-cyclodextrin was used to coat the surface of the RB-UCNPs acting as host molecules [96]. The resulting RB-UCNPs (64 ± 2 nm) were stable to RB leaching in PBS buffer for up to 70 h [96]. Upon irradiation at 808 nm, an effective energy transfer between the UCNPs and the RB was evidenced by the intensity decrease of the upconversion luminescence at 540 nm in the presence of PS. The optimal RB content for the 1O2 production upon 808 nm irradiation (power density of 4 W·cm−2) was determined to be 1 wt% since lower concentrations showed less 1O2 production and increasing RB concentration led to a reduced 1O2 generation possibly due to the self-quenching effect of the RB [96]. The RB-UCNPs were taken up by HeLa cervical cancer cells as confirmed using fluorescence microscopy and recording the intracellular emission of the RB on the UCNPs (whether the RB was excited directly or through the UCNPs was not indicated) [96]. Using a CCK-8 assay, a significant induced cell death was observed for HeLa cells treated with RB-UCNPs (250 μg·ml−1) following PDT treatment (808 nm, 1 W·cm−2, 10 min) compared to the non-irradiated cells treated with RB-UCNPs. A comparative study using 808 or 980 nm excitation indicated that the former was superior for penetration and for 1O2 generation, causing less photodamage [96]. This detailed work was preceded by an interesting delivery system reported by the same authors based also on UCNPs coated with a solid silica shell and a mesoporous silica shell containing pore channels [97]. The photosensitiser MB was embedded in the solid silica shell while RB (as model drug) was loaded in the pores. The novelty of this system lied on the further functionalisation of the particles with a 'gatekeeper' to avoid the release of RB from the mesoporous shell. The gatekeeper was a strategically placed 1O2-sensitive linker that would only open in the presence of 1O2, releasing the trapped RB. MB was chosen since its absorption spectrum overlaps the emission of the UCNPs at 660 nm which undergoes a decrease in intensity, due to the energy transferred to MB, following 980 nm irradiation. The 1O2 production by the MB-RB-UCNPs and the subsequent release of the RB was confirmed following 980 nm. Cytotoxicity of the nanosystem was observed in A549 lung cancer cells at concentrations higher than 64 μg·ml−1; however, PDT could be performed at lower concentrations (32 μg·ml−1 UCNPs, 980 nm irradiation for 50 s at 2.0 W·cm−2). The authors reported also the cell imaging capability of this nanosystem following irradiation at 980 nm [97].
Biocompatible PS drugs like Vitamin B12 (VB12), have also been used with UCNPs for PDT of cancer. Xu et al reported mesoporous-silica-coated NaYF4:Yb,Er nanoparticles functionalised with VB12 [98]. The NaYF4:Yb,Er UCNPs emit light at 545 nm (λexc = 980 nm) which overlaps the absorption of VB12. MDA-MB-231 breast cancer cells loaded with VB12-UCNPs exhibited a 40% cell viability decrease (MTT assay) following 10 min irradiation at 980 nm while no cell death was observed for non-irradiated cells incubated with VB12-UCNPs and irradiated cells incubated with non-functionalised UCNPs. This work would have benefited from stability studies confirming that the VB12 was still present on the UCNPS when stored over time [98].
Although mesoporous silica coated UCNPs functionalised with PS drugs have proven to be efficient for the energy transfer from the UCNPs to the loaded-PS drugs, other approaches to link PS drugs non-covalently to the UCNPs have been reported, including physical adsorption, which results in a more straightforward methodology. For example, the addition of biocompatible ligands or polymers, such as PEG, to the surface of the UCNPs could act as a hydrophobic environment for the incorporation of PS and increase the water dispersibility and stability of the nanosystem. These biocompatible PS-UCNPs are expected to have increased blood circulation times and consequently a higher chance to accumulate in the tumour through the EPR effect. These non-covalent physical adsorption strategies include the entrapping of the PS on amphiphilic polymeric [76, 99, 100, 102] or metal-organic framework (MOF) [103] coatings. For instance, the first publication reporting the potential use of UCNPs for in vivo PDT described the functionalisation of water-soluble PEGylated NaYF4:Yb,Er nanoparticles with Ce6 via hydrophobic interactions between the PEG chains and the PS, with a ca. 8% loading capacity reported [99]. The Ce6-UCNPs were stable under different physiological conditions (PBS, cell culture medium and foetal bovine serum) and only a 10% Ce6 release was observed in PBS (pH 7.4) after 50 h at room temperature. The energy transfer from the UCNPs to the Ce6 was confirmed by the quenching of the red emission band of the naked UCNPs after the loading of the PS. The efficiency of the Ce6-UCNPs for NIR light-activated PDT was investigated both in vitro and in vivo. Ce6-UCNPs were incubated in 4T1 murine breast cancer cells and a significant reduction in cell viability (MTT assay) was observed following 980 nm excitation (10 min, 0.5 W·cm−2), which was not observed when the cells were irradiated following treatment with naked UCNPs or with free Ce6. In vivo experiments were performed by intratumoral injection of Ce6-UCNPs in 4T1 murine breast tumour-bearing Balb/c mice and treatment in 980 nm NIR light exposure (0.5 W·cm−2, 30 min). Two weeks after treatment, the disappearance of the tumour was observed in 7 out of 10 mice and the mice remained alive for up to 60 d (when the mice were put down) [99]. Although the direct activation of the Ce6 showed higher 1O2 generation in solution, a greater tumour reduction was observed following 980 nm irradiation when an 8 mm pork tissue was placed between the laser source and the tumour—simulating deep lying tumours—thus, confirming the higher tissue penetration of NIR light over wavelengths at the visible range, and thus the superiority of using PS-UCNPs than the PS drug alone for PDT [99]. Quantitative biodistribution studies were performed showing that, although 1 d following treatment of the tumour with the Ce6-UCNPs the nanoparticles were found mostly on and close to the skin where they had been injected, their concentration was higher in the liver and spleen 15 d following injection and barely detectable in organs and tissues 60 d following injection. Ce6 was also used as the PS drug by the same authors to report an improved PS-UCNPs-based nanosystem in which the nanoparticles were dopped with Mn2+ to increase the luminescence intensity at 660 nm [102]. Ce6 was loaded following a layer-by-layer self-assembly approach yielding 2xCe6-UCNPs which exhibited higher 1O2 generation than Ce6-UCNPs. 2xCe6-UCNPs and 980 nm irradiation (0.5 W·cm−2, 30 min) were used to treat BALB/c mice bearing a 4T1 murine breast cancer tumour inducing, interestingly, a delay in tumour growth when the 2xCe6-UCNPs were injected intratumorally that was not observed when they were administered via intravenous injection [102].
Following a similar non-covalent strategy and using amphiphilic polymer coating, Cui et al reported NaYF4:Yb,Er nanoparticles, first coated with N-succinyl-N'-octyl chitosan via aqueous phase transfer, dopped with ZnPc for in vivo PDT [76]. The loading capacity achieved in this synthesis (10.8%) was higher than those obtained in the previously reported example of PS-UCNPs [99]. Using 1,3-diphenyllisobenzofuran (DPBF) as the 1O2 probe, a 65% decrease in the fluorescence emission of DPBF was observed following 60 min irradiation at 980 nm of the ZnPc-chitosan-UCNPs, whereas a 3% reduction was obtained for non-irradiated void nanoparticles. 1O2 production of non-irradiated ZnPc-chitosan-UCNPs was not reported in this paper. ZnPc-chitosan-UCNPs became toxic to human embryonic lung fibroblast (HELF) and human breast adenocarcinoma (MCF-7) cells when used at high doses (800 μg·ml−1). A decrease in cell viability was observed in MCF-7 cells loaded with ZnPc-chitosan-UCNPS after irradiation at 980 nm (600 mW, 10 min) compared to the control groups (cells incubated with ZnPc-chitosan-UCNPS and non-irradiated and untreated cells irradiated at 980 nm). Additionally, cell apoptosis in ZnPc-chitosan-UCNPs treated cells exposed to NIR light was confirmed by staining with Annexin V-FITC/PI [76]. In in vivo studies, a slower increase of tumour volume and higher survival rates were observed in S180 sarcoma tumour bearing mice treated with ZnPc-chitosan-UCNPs and irradiated with NIR light than for mice treated only with NIR light, only with ZnPc-chitosan-UCNPs or for non-treated mice. Fluorescence imaging of isolated organs and tumour tissues confirmed that, 14 d after the injection of the ZnPc-chitosan-UNPs, the UCNPs accumulated in the tumour tissue and not in the imaged organs (liver, lung, heart, spleen, intestine and kidney) of the mice [76]. Meng et al developed another amphiphilic conjugate for the surface modification of UCNPs which permits the adsorption of the PS and enhances the cellular permeability by inhibiting the P-glycoprotein (P-gp) which is overexpressed in cancer cells and obstructs the internalisation of therapeutic agents (figure 4) [100]. Oleate capped NaYF4:Yb,Er nanoparticles were modified with D-α-tocopherol (vitamin E) polyethylene glycol 1000 succinate-succinic acid-mercaptoethylamine (TPGS-SH) following sonication. The resulting TPGS-SH-UCNPs were water-dispersible containing an outer hydrophilic shell and a hydrophobic core due to the vitamin E segments and the oleate chains, respectively; and exhibited two emission bands at 540 and 654 nm. The functionalisation of the UCNPs with ZnPc was successfully achieved through hydrophobic interactions with a loading capacity of 3.05% [100]. The ZnPc-TPGS-SH-UCNPs exhibited relatively good size stability following 6 h incubation in simulated gastrointestinal media. The ability of the ZnPc-TPGS-SH-UCNPs to produce 1O2 was confirmed by the decrease in absorbance of DPBF following excitation at 980 nm, which was not observed when particles without ZnPc were irradiated. A control experiment measuring the consumption of DPBF in the presence of non-irradiated ZnPc-TPGS-SH-UCNPs was not reported. In vitro, a significant decrease in cell viability (MTT assay) was observed in Caco-2 human colon carcinoma cells incubated with ZnPc-TPGS-SH-UCNPs (100 μg·ml−1) and irradiated at 980 nm (800 mW, 10 min), which was not observed for control groups [100]. The mucosal penetration of the ZnPc-TPGS-SH-UCNPs, attributed to the thiol-based ligands, was investigated in Sprague-Dawley rats measuring the Y3+ concentration and showed the accumulation of the nanoparticles on the enterocytes of duodenum [100].

Figure 4. Schematic representation of (a) ZnPc-TPGS-SH-UCNPs and (b) intestinal distribution, mucosal penetration and subsequent PDT [100]. Reprinted from [100] with permission from American Chemical Society.
Download figure:
Standard image High-resolution imageAlthough non-covalent strategies for the loading of PS drugs onto the surface of UCNPs present a straightforward approach, the precipitate release of the PS can reduce the efficiency of the treatment and lead to side effects. For instance, Liu et al covalently attached RB hexanoic acid to amino-functionalised UCNPs via amide formation reaction and compared the RB desorption with that in UCNPs loaded with RB via electrostatic interactions [104]. The eluted RB was 1 order of magnitude higher for the non-covalent RB loading strategy confirming the higher stability of the covalently functionalised RB-UCNPs [104]. This demonstrates that the use of robust covalent bonds to bind PS—and other cargo—to UCNPs may improve the loading efficiency and stability of the nanosystems ensuring the higher control of the possible leaching and of the overall structure.
Additionally, a covalent approach may lead to a better control of the distance between the UCNPs core and the loaded PS, which is key for an efficient energy transfer and thus for the production of 1O2 in PDT applications. Longer distances have been related to a proportional decrease of the energy transfer resulting in a decrease in 1O2 generation [105, 106]. To control the distance between the core and the drug, Marín and co-workers covalently bonded RB to UCNPs through an L-lysine unit [59]. Oleate-capped core-shell NaYF4:Yb,Er,Gd@NaYF4 nanoparticles (17.2 ± 1.0 nm). were modified via a ligand exchange method yielding BF4- capped UCNPs and the L-lysine ligand was then incorporated replacing the BF4- ions. Finally, the lysine-capped UCNPs were functionalised with RB via classic N-ethyl-N'-(3-(dimethylamino)propyl)carbodiimide/N-hydroxysuccinimide (EDC/NHS) chemistry resulting RB-lysine-UCNPs [59]. ABMA was used to confirm the ability of the RB-lysine-UCNPs to generate 1O2 upon irradiation at 980 nm. Luminescence lifetime measurements were reported to confirm the efficiency of the FRET from the UCNPs to the RB. RB-lysine-UCNPs were internalised by SK-BR-3 breast cancer cells as evidenced by the emission of the RB and the UCNPs using confocal laser scanning and multi-photon microscopies. Using CellTiter Blue® assay, the cell viability of SK-BR-3 cells incubated with the RB-lysine-UCNPs and irradiated at 980 nm (200 mW, 6 min) showed a decrease of 67% whereas non-irradiated cells showed a 5% decrease due to the dark toxicity of the RB-lysine-UCNPs [59].
To obtain water-dispersible and highly biocompatible NIR excitable nanosystems for PDT, reports have shown the incorporation of biologically acceptable ligands to UCNPs covalently functionalised with PS. For example, Sun et al covalently functionalised PEG-UCNPs with protoporphyrin IX as PS drug resulting in a water-dispersible nanosystem [107]. The protoporphyrin IX structure was modified with jeffamine molecules, to obtain a hydrophilic porphyrin derivative, and a thiolated terminal group was incorporated. Then, PEGylated UCNPs were prepared from core-shell NaGdF4:Yb,Er@NaGdF4 UCNPs (14.2 ± 1.3 nm) and a subsequently 'click' reaction between the maleimide group on the UCNPs surface and the thiolated porphyrin-jeffamine molecules yielded the porphyrin-UCNPs (2.0% w/w, porphyrin-jeffamine:UCNP) [107]. The energy transfer from the UCNPs to the PS was demonstrated by the quenching of the luminescence emission of the UCNPs in the range of 400–650 nm which matches the absorption band of the PS. In solution, the porphyrin-UCNPs were able to produce 1O2 following irradiation at 980 nm (0.5 W·cm−2) with rates that were slightly higher than those produced by the same concentrations of free porphyrin-jeffamine and by the clinically used hematoporphyrin mono-methylether irradiated directly at 635 nm (0.5 W·cm−2). The efficiency of the porphyrin-UCNPs to generate 1O2 was investigated in an intestinal cancer cell line LS180 using MTT assay. A decrease in cell viability of almost 100% was observed after NIR irradiation (980 nm, 1 W·cm−2, 10 min) of LS180 cells treated with 1.5 mg·ml−1 of porphyrin-UCNPs (36 μM porphyrin-jeffamine), whereas the non-irradiated cells incubated with the same concentration of nanoparticles showed a cell viability ca. 85% [107].
Natural coating strategies including the incorporation of protein layers (e.g. bovine serum albumin (BSA) [92] and apolipoproteins [108]) or cell membranes [109] (e.g. red blood cells and stem cells) have been also employed for loading PS drugs on the surface of UCNPs. Sabri et al reported the covalent functionalisation of RB on the surface of BSA-UCNPs for PDT of cancer [92]. First, RB hexanoic acid was covalently linked to the free amine groups of the lysine units present on the BSA of BSA-NaGdF4:Yb,Er nanoparticles. The energy transfer from the UCNPs to the RB was investigated at different RB concentrations, i.e. 3, 20 and 30 μM; and the maximum energy transfer (68%) was observed for the particles containing 20 μM RB. Higher concentrations of RB (30 μM) led to a decrease in energy transfer due to the self-quenching between the RB molecules. The NIR activated production of 1O2 was evidenced using DPBF and monitoring the decrease in absorption intensity at 415 nm following 980 nm irradiation. Interestingly, the number of molecules of 1O2 produced was estimated to be 5.9 × 1021 molecules, which is above the number of molecules required to produce cell damage (7 × 109) [110]. Confocal microscopy was used to study the RB-BSA-UCNPs cellular uptake and to compare it with that of the free RB hexanoic acid and of BSA-UCNPs. Interestingly, RB was localised in the cytoplasm of human lung cancer A549 cells, BSA-UCNPs did not penetrate the cellular membrane and RB-BSA-UCNPs were localised in the cytosol. MTT assay was used to study the cell viability of the A549 cells treated with the RB-BSA-UCNPs (250 μg·ml−1) following 10 min irradiation at 980 nm (13 mW·cm−2). Although a 36% cell death was determined for the irradiated cells incubated with RB-BSA-UCNPs, an intrinsic cellular toxicity of 15% was observed without light irradiation. Even though the authors showed an efficient covalent RB loading on the surface of the UCNPs, the ability of those nanosystems to induce cell death via PDT could be further improved.
Stem-cells have been used as biomimetic carriers of UCNPs for PDT resulting in a prolonged blood circulation time and enhanced tumour passive targeting. The first example of stem-cell coated UCNPs was reported by Gao et al in 2016 and presented an exhaustive investigation both in vivo and in vitro [111]. NaYF4:Yb,Er nanoparticles were coated with a mesoporous silica layer and two PS drugs, MC540 and ZnPc (overlap the green, ca. 540 nm, and red, ca. 660 nm, UCNP emissions, respectively), were incorporated into it to enhance the PDT effect. Stem-cell-membranes isolated from human and rat bone-marrow-derived mesenchymal stem cells were used to prepare the stem-cell-membrane vesicles via physical extrusion method [111]. Stem-cell-membrane coated MC540-ZnPc-mSiO2-UCNPs were obtained by an extrusion approach in which the particles were mixed with the previously extruded stem-cell-membranes and passed through 200 nm pore-size polycarbonate membrane 11 times. The stem-MC540-ZnPc-mSiO2-UCNPs had a core size of ca. 120 nm and were surrounded by a lipid layer of ca. 10 nm in thickness. The size of the stem-MC540-ZnPc-mSiO2-UCNPs remained stable in solutions containing PBS or foetal bovine serum for up to 2 weeks. The nanocomposites remained stable once taken up by HeLa cells since both the stem-cell-membrane and the UCNPs co-localised in the intracellular environment. The biocompatibility of the stem-MC540-ZnPc-mSiO2-UCNPs was tested by MTT cell viability studies, haemolysis and blood-smear-test; and the three assays confirmed the suitability of the nanoplatform for biological applications. The target-capability of the stem-MC540-ZnPc-mSiO2-UCNPs was confirmed in HeLa cells using confocal microscopy and by comparison with the reduced internalisation of the particles without stem-cell-membrane by the same cell line. These studies were nicely supported by quantitative studies using flow cytometry where an increase in fluorescence intensity was observed for cells treated with a FITC-labelled stem-MC540-ZnPc-mSiO2-UCNPs compared to the increase observed for cells treated with FITC-labelled MC540-ZnPc-mSiO2-UCNPs. To further the applicability of the nanoplatform, in vivo tumour-targeting experiments were performed in mice bearing HeLa tumours using fluorescence imaging and following the labelling of the nanoparticles with a NIR excitable dye, Cy7. These studies confirmed the role that the stem-cell-membranes play in the accumulation of the UCNPs in the tumour and in the reduction of the clearance by the RES. The potential of the stem-MC540-ZnPc-mSiO2-UCNPs as PDT agents was evaluated both in vitro and in vivo. In vitro, HeLa cells were treated with stem-MC540-ZnPc-mSiO2-UCNPs and controls included cells treated with mSiO2-UCNPs, MC540-ZnPc-mSiO2-UCNPs, stem-mSiO2-UCNPs and PBS. Fluorescence cell staining with a living cell fluorophore (calcein AM) and an apoptosis fluorophore (propidium iodide), and quantitative analysis via MTT assay confirmed that only the combination of cells treated with nanoparticles containing the photosensitiser drugs and irradiated at 980 nm (0.35 W·cm−2, 15 min) yielded to reduce cell viability. Interestingly, stem-MC540-ZnPc-mSiO2-UCNPs are twice more effective for PDT than MC540-ZnPc-mSiO2-UCNPs without the stem-cell-membranes thus, confirming the targeting ability of the platform. This complete study concluded with the in vivo validation of the system in HeLa cervical tumour-bearing Balb/c mice. The mice were treated under different conditions including PBS, stem-mSiO2-UCNPs, MC540-ZnPc-mSiO2-UCNPs and stem-MC540-ZnPc-mSiO2-UCNPs, all of them following irradiation with 980 nm laser (0.35 W·cm−1 for 1 h) or without irradiation. 15 d after the treatment, all the mice were euthanised and the tumour sizes were measured, which confirmed the excellent potential of this nanoplatform for in vivo PDT since only those mice that had been treated with stem-MC540-ZnPc-mSiO2-UCNPs following NIR irradiation showed tumour growth inhibition.
Although the majority of the UCNP-based NIR light-triggered PDT systems are designed using organic molecules as PS, some semiconductor materials (such as AgBiS2, ZnO and TiO2) have been also employed as inorganic PS. Incorporation of these shells onto the surface of UCNPs results in an easy PS loading process compared to the incorporation of organic PS drugs. For example, Chu et al reported AgBiS2-coated NaYF4:Yb/Er/Nd@NaYF4:Nd nanoparticles for combined photodynamic and photothermal therapy of cancer in vitro (using 4T1 breast cancer cell) and in vivo (in 4T1 tumor-bearing BALB/c mice) [112]. Dou et al reported a core-shell design with UCNPs (NaYF4:Yb,Tm) as the core and ZnO as the shell (2–3 nm width) and further functionalised with sodium citrate to yield water-dispersible (figure 5) [113]. The energy transfer between the UCNPs and the ZnO occurs upon irradiation at 980 nm yielding to the disappearance of the luminescence emission band that overlaps the absorbance of ZnO. The ROS production was confirmed by fluorescence spectroscopy, using 3'-(p-aminophenyl) fluorescein (ARF) as fluorescent ROS probe [113]. The ZnO-UCNPs were incubated in two breast cancer cell lines (4T1 and MDA-MB-231 cells) showing good biocompatibility at low concentration (10 μg·ml−1). The PDT effect of the ZnO-UCNPs was evaluated in MDA-MB-231 breast cancer cells by MTT assay and a decrease in the cell viability (ca. 50%) was observed 72 h after treatment for cells incubated with ZnO-UCNPs and exposed to NIR light (980 nm) for 30 min. UCNPs have also been functionalised with a TiO2 following different approaches, including the surface coating with a TiO2 shell [114] and the conjugation of TiO2 nanoparticles [115] on the surface of UCNPs. Although the PS-loading of inorganic crystals results in a more straightforward strategy, in most cases, further surface modification is needed to provide hydrophilicity to the systems.
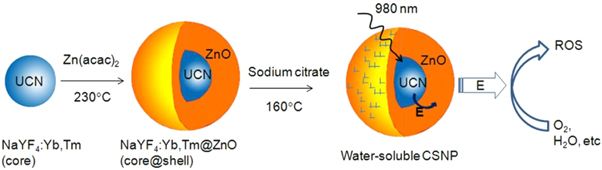
Figure 5. Schematic representation of the ZnO functionalisation of UCNPs and the ROS generation upon irradiation at 980 nm [113]. Reprinted from [113] with permission from Springer Nature.
Download figure:
Standard image High-resolution imageOther photosensitisers such as transition metal-based complexes (e.g. iridium (III) complex [116] and ruthenium (II) complex [117]) have been recently combined with UCNPs for PDT treatment in vitro and in vivo. For example, Meijer et al reported the first example of UCNPs decorated with a metal-based PS drug for NIR-triggered PDT of cancer [117]. NaYF4:Yb,Tm nanoparticles (44.2 ± 2.2 nm) exhibited luminescence at 451, 475, 510, 648, 698, 740 and 803 nm when excited at 969 nm, being 451 and 475 nm the desired wavelengths to activate the PS Ru(II) complex. A lipid film was formed around the UCNPS that contained the Ru(II) complex (5 mol%) and the UCNPs were hydrated using MES/acetate buffer. A 12% energy transfer between the UCNPs and the Ru(II) complex was calculated using steady-state and time-resolved fluorescence spectroscopies [117]. This low energy transfer could be attributed to the long distance between the UCNPs core and the PS or due to a low concentration of PS on the surface of the UCNPs. The Ru(II) complex-UCNPs were able to generate 1O2 following irradiation at 969 nm (2.0 W, 50 W·cm−2, 2 h) as indicated by an increase in the absorption intensity of DHFA [117]. Although water-dispersible Ru(II) complex-UCNPs with an excellent overlap between the absorption of the PS and the emission of the UCNPs were successfully synthesised, the low energy transfer limits the 1O2 production and thus the potential of this nanosystems for PDT of cancer.
Recently, a porphyrin-based MOF has been employed by Shi et al as PS drug for the development of PS-UCNPs for PDT of cancer [118]. The authors stressed the ability of the presented nanoplatform to achieve Type I and Type II PDT via NIR excitation and the reported results addressed this shelling point. A layer of meso-tetra(4- carboxyphenyl)porphine (TCPP)-MOF was grown, through a solvothermal process, on the surface of NaYF4:Tm,Yb nanoparticles (ca. 29 nm) modified with a polyvinylpyrrolidone (PVP) layer [118]. The nanoparticles were then coated with a layer of ultra-small TiO2 nanoparticles which was used as a second PS drug yielding UCNPs-MOF-TiO2 nanoparticles. Upon excitation at 980 nm, the naked UCNPs emitted bands at 291, 344, 360, 450, 475 and 648 nm, which strongly decreased upon addition of the MOF and TiO2 confirming the efficient energy transfer between the UCNPs and the other materials. The ability of the UCNPs-MOF-TiO2 to produce ROS following 980 nm irradiation was determined by monitoring the decrease in the absorbance intensity of DPBF (able to react with several ROS). UCNPs-MOF-TiO2 were slightly superior in the generation of ROS than UCNPs-MOF. In vitro experiments were performed in MCF-7 breast cancer cells. Cells were incubated with UCNPs-MOF-TiO2 or UCNPs-MOF and further incubated with singlet oxygen sensor green (1O2 probe), dihydroethidium (O2 ·− probe) and hydroxyphenyl fluorescein (HO• probe), irradiated at 980 nm (0.72 W·cm−2, 5 min) and imaged using confocal microscopy. UCNPs-MOF were able to produce only 1O2 while UCNPs-MOF-TiO2 were able to produce 1O2, O2 ·− and HO·, thus probing the ability of the UCNPs-MOF-TiO2 to be used for Type I and Type II PDT. The efficacy of the UCNPs-MOF-TiO2 for PDT was further confirmed using MTT assays (and confocal microscopy using propidium iodide staining) reporting a 70% decrease in cell viability only when MCF-7 cells were treated with the nanoparticles and irradiated with NIR light. Furthermore, MTT assays also reported the superior PDT effect of UCNPs-MOF-TiO2 over UCNPs-MOF. The UCNPs-MOF-TiO2 were also evaluated for in vivo PDT using MCF-7 tumour-bearing BALB/c mice. The mice were treated under four different conditions: PBS or UCNPs-MOF-TiO2 (1 mg·ml−1, 100 μl) intravenously injected, and no irradiation or irradiation at 980 nm (0.72 W·cm2, light dose 648 J·cm2, 15 min) following injection. Only those mice treated with the nanoparticles and irradiated with NIR light showed a considerable tumour volume reduction over time with complete disappearance after 90 d and prolonged survival rates for as long as 100 d (the mice in the control groups survived for less than 50 d); thus, confirming the potential of the newly developed platform for PDT of cancer.
3.2. Active targeting
Although a lot of the examples of UCNPs reported for PDT follow passive targeting strategies taking advantage of the EPR effect [59, 119], the efficiency of the treatment can be significantly enhanced using active targeting strategies. There are mainly two types of active targeting, stimuli-responsive targeting and ligand-mediated targeting. In the stimuli-sensitive approach, nanoparticles are tuned to respond to specific changes that occur in the cellular environment of cancer cells but not in healthy cells [93]; therefore, results in a very specific treatment minimising the side effects. This can be achieved by tailoring the surface of the nanoparticles or modifying the PS structure. The ligand-mediated targeting consist on further functionalisation of the PS-UCNPs with units such as folic acid, antibodies, peptides, carbohydrates, among others, that can bind specific receptors only present or overexpressed on tumour cells [93]. Recently, UCNPs have also been explored to achieve cell nucleus-targeted PDT [120]. The use of targeting agents facilitates the uptake of the nanoparticles by cancer cells minimising the undesired side effects [93]. The following sections in this review paper will be devoted to reporting recent papers describing the development of PS-UCNPs that can be used to actively target cancer.
3.2.1. Stimuli-responsive targeting
Examples of external stimulus only present on tumour sites include differences in the pH [121, 122], H2O2 concentration [123] or the presence of specific enzymes [124] in the intracellular microenvironments. Delivery platforms are designed bearing this in mind and thus, the PS drugs are activated only when they reach environments that exhibit these specific conditions. For example, Han and co-workers synthesised UCNPs covalently functionalised with a clinically used prodrug, 5-aminolevulinic acid (ALA), that could be released intracellularly and form the PS protoporphyrin IX only under the acidic environments found in cancer cells [122]. The reported UCNPs consist of cubic phase α-NaYF4 doped with 80% Yb3+ and 20% Er3+ as core and CaF2 as the shell (α-NaYF4:Yb,Er@CaF2, ca. 26 nm). The functionalisation of the UCNPs with ALA was performed though a covalent pH-sensitive hydrazone linked to poly(acrylic acid)-functionalised UCNPs with the aim of the prodrug being released by the lower pH of the endosomes and the subsequent formation of the PS protoporphyrin IX in the mitochondria where it could generate 1O2 upon red light excitation [125]. The formation of protoporphyrin IX and the generation of 1O2 was confirmed in HeLa cells incubated with ALA-UCNPs (100 μg·ml−1) following irradiation at 980 nm (0.5 W·cm−2, 10 min) using a 1O2 fluorescent probe, 2',7'-dichlorofluorescein diacetate (DCFDA). No 1O2 production was evidenced in the control group where cells were treated with UCNPs that did not contain ALA (Hyd-UCNPs) [122]. MTT assays showed a cell viability decrease of ca. 70% after 20 min irradiation at 980 nm that was not observed when the cells were incubated with Hyd-UCNPs or free ALA and irradiated or only irradiated. One of the aims of this research was to increase the deep-tissue treatment by amplifying the red emission of the nanoparticles. To this aim, the authors optimised the content of Yb3+ to 80% in solution which was also confirmed in in vitro experiments where the PDT effect of ALA-functionalised core-shell cubic nanoparticles α-NaYF4:Yb(80%),Er(2%)@CaF2 was compared with that of hexagonal core-shell UCNPs (β-NaYF4:Yb(20%),Er(2%)@β-NaYF4) via MTT assay [122]. With the purpose of studying the deeper tissue efficacy of the NIR-triggered PDT, cells treated with ALA-UCNPs were irradiated with NIR light during 40 min through a piece of pork and the results showed a decrease in cell viability [122]. Additionally, in vivo experiments were performed in female Balb/c mice treated with ALA-UCNPs comparing the PDT effect following 980 nm irradiation or direct irradiation of the PS. A significant reduction in tumour size was observed for both treatments with no statistically significant differences found between them. However, when a 1.2 cm piece of pork was placed between the tumour side and the laser source, a phototherapeutic effect was observed when 980 nm was used as irradiation source, but no tumour size reduction was evidenced when red light excitation was employed. These results further confirm the superiority of using UCNPs in combination with NIR excitation for deep tissue penetration PDT [122].
Another example of pH-induced PDT using UCNPs was reported by Feng et al [121]. The 'off-on' strategy was achieved by the incorporation of a pH-sensitive PEGylated polymer and a fluorescence quencher, black hole quencher (BHQ). The photoactivity of the nanoplatform, 'off' at pH 7.4 in the blood, should be 'turned on' once the nanosystem is internalised in the tumour side and thus reaches environments with lower pH. Core-shell NaYF4:Yb,Er,@NaYF4 nanoparticles (ca. 26 nm) were non-covalently functionalised with RB by mixing the oleate-capped UCNPs with RB hexanoic acid. Next, two PEGylated polymeric micelles containing RB or BHQ were prepared and used as polymeric shells for the RB-UCNPs. The 1O2 generation mediated by pH changes was investigated upon NIR light irradiation using DPBF showing that at pH 5.5 the polymeric shells were released from the particles allowing the recovery of the emission and thus the generation of 1O2. The cleavage of the polymeric shell was also confirmed measuring the hydrodynamic diameter of the nanoparticles, and a decrease in size from 102 nm at pH 7.4 to 68 nm at pH 5.5 was observed which agrees with the singlet oxygen production experiments. MCF-7 breast cancer cells were used to evaluate the in vitro PDT effect (MTT assay) of three types of nanoparticles: 'off-on'-RB-UCNPs which contained PEG polymer ligands with RB and BHQ that were cleaved at acidic pH; 'always-off'-RB-UCNPs in which the PEG polymer used contained both RB and BHQ but did not present a pH-cleavable section (negative control); and 'always-on'-RB-UCNPs in which the polymer used was not modified with the quenchers (positive control). The MCF-7 incubated with 'off-on'-RB-UCNPs and irradiated at 980 nm (0.7 W·cm−2, 15 min) exhibited similar cell viability decrease than those cells incubated with the positive control 'always-on'-RB-UCNPs and irradiated. In comparison, only a small decrease (ca. < 20%) was observed when the cells were incubated with 'always-off'-RB-UCNPs and irradiated. The paper claims that these results support the theory of activation of the 'off-on'-RB-UCNPs when taken up by cancer cells. However, a comparison with the results obtained with healthy cells following the same treatment is essential to support the targeting ability of the particles. The ability of the nanosystem to perform in vitro PDT was also confirmed using flow cytometry and staining of apoptotic and necrotic cells, which confirms a similar behaviour between 'off-on'-RB-UCNPs and 'always-on'-RB-UCNPs. The 'off-on'-RB-UCNPs were used to image tumour sides in MCF-7 tumour bearing mice 4 h after intravenous injection of the nanoparticles; and were able to reduce the relative tumour volume following irradiation with 980 nm NIR laser (15 min at 0.6 W·cm−2) [121].
Cancer cells present higher levels of H2O2 than normal cells, and elevated concentrations of H2O2 have been associated with malignant cell proliferation [126]. Thus H2O2-induced PDT has been investigated for the active targeting of cancer cells [123]. For example, Ding et al reported a lanthanide-based nanoplatform functionalised with MC540 as PS and MnFe2O4 as a Fenton catalyst to generate O2 from intracellular H2O2, thus potentially enhancing the efficiency of the PDT in cancer cells by increasing tissue penetration (with UCNPs) and reducing hypoxia levels (increasing O2 concentration at the tumour side) [123]. Core-shell NaYF4:Yb,Er nanoparticles coated with a mesoporous silica shell via silica sol-gel reaction were functionalised with amine groups using 3-aminopropyltriethoxysilane (ATPES). MnFe2O4 nanocrystals, prepared following a hydrothermal method and modified with 2-bromo-2-methylpropionic acid (BMPA), were covalently bonded to the amine-functionalised UCNPs via nucleophilic substitution reaction. The MnFe2O4-UCNPs were further modified with MC540, which absorption band (540 nm) overlaps one of the luminescence wavelengths of the UCNPs (541 nm). The loading of MC540 on the silica porous was achieved with a 11.52 wt% loading efficiency, with no stability studies reported for potential leaching of the PS drug. The MC540-MnFe2O4-UCNPs were able to generate O2 from H2O2 in solution. In vitro experiments were performed to test the ability of the MC540-MnFe2O4-UCNPs to induce the production of 1O2 in HepG human liver cancer cells under both, normoxic and hypoxic conditions. A ROS probe, DHFA, and fluorescence microscopy were used to detect the intracellular 1O2 formation following treatment of the cells with the MC540-MnFe2O4-UCNPs and irradiation at 980 nm (0.5 W·cm−2, 10 min). A comparative study between MC540-MnFe2O4-UCNPs and MC540-UCNPs in HepG cells confirmed the ability of the former to produce 1O2 under hypoxic conditions (1% O2, 5% CO2 and 94% N2) [123]. Both MC540-MnFe2O4-UCNPs and MC540-UCNPs (400 μg·ml−1) induced significant cell mortality when incubated in HepG cells and irradiated at 980 nm (5 min) in normoxic conditions. However, when the cells were incubated under a hypoxic atmosphere, a notable difference in cell viability was observed, ca. 25% for MC540-MnFe2O4-UCNPs and ca. 90% for MC540-UCNPs. These results confirmed the higher PDT efficiency of MC540-MnFe2O4-UCNPs due to the ability of MnFe2O4 to generate O2 [123]. The incorporation of MnFe2O4 onto the UCNPs results not just in a catalytic generation of O2 but also allows the nanoparticles to be magnetically guided to the tumour sites. The accumulation of the MC540-MnFe2O4-UCNPs, injected through the tail, into malignant regions was investigated in vivo using BALB/c mice to which H22 mouse hepatoma cells had been subcutaneously injected. Magnetic-guided and non-guided accumulation of the nanoparticles in the tumour was determined by measuring the yttrium concentration by ICP-AES resulting in higher levels of Y3+ when magnetic guidance was applied [123]. The in vivo PDT effect of MC540-MnFe2O4-UCNPs was then studied in HepG2 tumour bearing nude mice (average tumour volume of 60 mm3) irradiated at 980 nm (10 min). Tumour growth was observed in control groups (treated with PBS or treated with particles without irradiation) in comparison to the impressive tumour inhibition observed for mice treated with MC540-MnFe2O4-UCNPs and irradiated, which was superior to the observed for mice treated with MC540-UCNPs and irradiated; and thus, may confirm the role that MnFe2O4 plays in the generation of O2 to overcome hypoxia. Additionally, a remarkable in vivo PDT improvement was evidenced when MC540-MnFe2O4-UCNPs were magnetically guided to the tumours furthering the great potential of this platform for in vivo PDT, imaging and magnetic resonance imaging [123]. These ambitious examples have confirmed that stimuli-response strategies could be used to reduce the cytotoxicity effects of PDT in normal cells. However, the non-specific distribution of the nanoparticles in the body is still an issue and further efforts are necessary to deliver the nanoparticles specifically and for them to accumulate in the tumour side. A well-known strategy to achieve this is the incorporation of ligands that can recognise receptors expressed only (or overexpressed) on cancer cells, thus increasing the concentration of PS drugs in the cancer tissue and, therefore, significantly improving the efficiency of the PDT.
3.2.2. Receptor-mediated targeting
The multifunctionalisation capability of UCNPs permits the addition of targeting agents onto the surface of nanoparticles resulting in the specific recognition of the cancerogenic tissue/cells [61, 71–73, 127]. Functionalisation of the nanosystems with targeting agents offers advantages over passive targeting such as high specificity and selectivity, reducing any adverse effects due to non-targeted toxicities and enhancing the cellular uptake. In general, actively targeted PDT has been shown to maximise the accumulation of the PS drugs into malignant tissues and minimise the potential toxicity in normal tissues [56].
To achieve a targeted application, specific targeting agents can be incorporated into the nanoparticle, including monoclonal antibodies [56, 127], peptides [71], aptamers [61], carbohydrates [72, 128] and other receptors as folate [73] and transferrin [129] (figure 6). A summary of the examples of UCNPs reporting receptor-mediated targeting for PDT of cancer can be found in table 1 and these are explained in detail below.

Figure 6. Schematic representation of targeting agents reported in the literature to induce active targeting of cancer cells when UCNPs have been used for PDT of cancer: (a) UCNP functionalised with the different targeting agents and (b) specific recognition of the corresponding receptors on the cell membrane.
Download figure:
Standard image High-resolution imageTable 1. Summary of the UCNPs-based systems reporting receptor-mediated targeting for PDT.
| Targeting ligand | UCNPs | λex (nm) | PS | Conjugated PS to the UCNPs | PDT in vitro | PDT in vivo | References | |
|---|---|---|---|---|---|---|---|---|
| Antibody | Monoclonal antibody | NaYF4:Yb,Er@NaGdF4 | 980 | RB | Mesoporous silica layer | HT-29 human colon adenocarcinoma cells | n/a | Liang et al [130] |
| Trastuzumab | ZnPc-NaYF4:Yb,Er | 975 | ZnPc | Covalent bond through cysteamine | MCF-7 and SK-BR-3 breast cancer cells | n/a | Ramírez-García et al [131] | |
| Affibody® | NaYF4:Yb,Tm | 980 | TiO2 | TiO2 layer | A431 human epidermoid, H596 human lung adenosquamous carcinoma, H460 lung cancer, MCF-7 breast cancer and HepG2 liver cancer cells | Female Balb/c nude mice | Lucky et al [132] | |
| Folic acid | NaYF4:Yb,Er | 980 | MC540 and ZnPc | Mesoporous silica layer | B16-F0 murine skin cancer cells | C57BL/6 mice injected with B16-F0 melanoma cells | Idris et al [133] | |
| NaYF4:Yb,Er | 980 | TPP | Phospholipid | KB cells and REF52 cells | n/a | Thanasekaran et al [134] | ||
| NaYF4:Yb,Er | 980 | ZnPc | Amphiphilic chitosan | Bel-7402 human liver, HELF human embryo lung and MDA-MB-231 breast cancer cells | Mice bearing Bel-7402 tumours | Cui et al [135] | ||
| NaYF4:Yb,Er | 980 | RB | Covalent bonded to AEP surface coating | JAR choriocarcinoma and noncancerous NIH 3T3 fibroblast cells | n/a | Liu et al [104] | ||
| NaYF4:Yb,Er | 980 | ZnPc | Covalent bonded to poly(alylamine) surface coating | HeLa cervical ancer cells and A549 human alveolar adenocarcinoma cells | Hepa1–6 tumour-bearing C57/6J mice | Xia et al [136] | ||
| NaYF4:Yb,Ho@NaYF4:Nd@NaYF4 | 808 | RB | Covalent bonded though amide coupling | HeLa cervical ancer cells | n/a | Wang et al [91] | ||
| NaYF4:Yb,Er | 980 | Hypericin | Mesoporous silica layer | HeLa cervical cancer cells and 293T embryonic kidney cells | n/a | Yang et al [137] | ||
| NaYF4:Yb,Er@NaYF4:Yb,Tm | 980 | C60MA | Covalent bonded to poly(alylamine) surface coating | HeLa cervical ancer cells and A549 human alveolar adenocarcinoma cells | n/a | Liu et al [138] | ||
| NaYF4:Yb,Er@NaYF4:Yb,Tm | 980 | C60MA | Ligand exchange reaction | HeLa cervical cancer cells and A549 human alveolar adenocarcinoma cells | Hepa1–6 tumour-bearing C57/6J mice | Liu et al [139] | ||
| NaYbF4:Nd@NaGdF4:Yb,Er@NaGdF4 | 808 | Ce6 | Covalent bonded to AEP surface coating | KB cells and A549 human alveolar adenocarcinoma cells | n/a | Ai et al [140] | ||
| NaYF4:Yb/Er | 980 | Pha | Covalent linked via acid-labile hydrazone linker | MCF-7 breast cancer cells | n/a | Zhao et al [141] | ||
| NaErF4@NaLuF4 | 808 | TCPP | Covalent bonded via APTES addition | HeLa cervical cancer cells | n/a | Lim et al [142] | ||
| Transferrin | NaYF4:Yb,Tm | 980 | PFSBT and Tc | Physical absorption | MCF-7 breast cancer cells | H22 tumour-bearing ICR mice | Liu et al [143] | |
| NaYF4:Yb,Tm:NaYF4 | 980 | HA | Mesoporous silica shell | HeLa cervical cancer and MCF-7 breast cancer cells | n/a | Zhang et al [144] | ||
| NaYF4:Yb,Er@NaYF4 | 980 | Ce6 | Mesoporous silica shell | HeLa cervical cancer and L02 human liver cells | n/a | Zhang et al [145] | ||
| Peptide | RGD | NaYF4:Yb,Er | 980 | MC540 | Amphiphilic coating | MCF-7 breast cancer cells | n/a | Wang et al [146] |
| RGD | NaYF4:Yb,Er | 980 | ZnPc | Mesoporous silica shell | HeLa cervical cancer cells | n/a | Hou et al [147] | |
| c(RGDyK) | NaYF4:Yb,Er | 980 | Ppa | Covalent bonded to chitosan surface coating | U87-MG brain cancer cells and MCF-7 breast cancer cells | n/a | Zhou et al [148] | |
| TAT | NaYbF4:Nd@NaGdF4:Yb,Er@NaGdF4 | 808 | Ppa | Hydrophobic interactions PMAO-PEG surface coating | HeLa cervical carcinoma cells | n/a | Zhang et al [149] | |
| pHLIP | NaYbF4:Nd@NaGdF4:Yb,Er@NaGdF4 | 808 | Ppa | Hydrophobic interactions PMAO-PEG surface coating | HeLa cervical carcinoma cells and 4T1 mice breast cancer cells | 4T1 tumour-bearing mice | Ai et al [150] | |
| RGD and TAT | NaYF4:Yb,Er | 980 | ZnPc | Lipid micelles | MCF-7 breast cancer cells and B16F1 murine melanoma cells | n/a | Wang et al [151] | |
| cMBP | NaYF4:Yb,Er@NaLuF4 | 980 | ZnPc | Polimerisation | 4T1 mice breast cancer and MDA-MB-231 breast cancer cells | 4T1 tumour-bearing mice | Wang et al [152] | |
| CaB | NaYF4:Gd@NaYF4:Er,Yb@NaYF4:Nd,Yb | 808 | RB | Covalent bond through alendronic acid linker | HeLa cervical carcinoma cells | HeLa tumour-bearing BALB/c nude mice | Li et al [153] | |
| HER-2 | NaYF4:Yb,Er | 975 | MB | Lipid micelles with or without conjugation | SK-BR-3 breast cancer cells | n/a | Panikar et al [154] | |
3.2.2.1. Antibodies
Antibodies are proteins that recognise, highly specifically, other proteins. Some antibodies, such as trastuzumab and cetuximab, have been approved for therapeutics of some types of cancer [155]; and a large number of reports describe the modification of drugs and nanoparticles for the targeted delivery to cancer cells. Antibodies were the first targeting agents used to deliver PS-UCNPs specifically to the cancer cells to perform PDT [85]. In this pioneering work, Zhang et al covalently functionalised the antibody anti-MUC1/episialin onto the surface of silica-capped UCNPs loaded with M-540 to target breast cancer cells [85]. Following this initial paper, a large number of papers have reported antibody-PS-UCNPs conjugates for the targeted delivery of the nanoparticles to cancer cells/tissues. Making use of UCNPs as delivery platforms, antibody-targeted PDT has also been explored in combination with other therapies, for example for the delivery of chemotherapeutic drugs both in vitro and in vivo [156, 157].
Intending to improve the functionalisation of UCNPs with antibodies maintaining the functionality and selectivity of the targeting agent, Liang et al reported a new synthetic strategy in which antibodies were added, in a controlled manner, to the UCNPs through a bifunctional fusion protein [130]. Core-shell NaYF4:Yb,Er@NaGdF4 nanoparticles were synthesised using a solvothermal decomposition method and coated with a silica layer in the presence of RB using a water-in-oil microemulsion method yielding ca. 43 ± 2 nm UCNP@SiO2(RB). The UCNP@SiO2(RB) were further modified with a monoclonal antibody following a novel protocol that would allow the control of the orientation of the antibody and thus, minimise the hindrance of the antigen-binding sides during and after the functionalisation process. First, the UCNPs were modified with a bifunctional fusion protein that contained a silica-specific solid-binding peptide—to bind to the silica on the UCNP@SiO2(RB)—fused to an antibody-binding protein to specifically bind the Fc fragment of the antibody. Once this step was performed, a monoclonal antibody for epithelial cell adhesion molecules (EpCAM; also known as CD326) was added to the nanoparticle conjugate yielding anti-EpCAM-UCNP@SiO2(RB). Anti-EpCAM functionalisation was confirmed by a negative shift of the zeta-potential values and an increase in the hydrodynamic diameter of the nanoparticles. The energy transfer between the UCNPs and the RB was evidenced by a decrease of the upconversion luminescence emission at ca. 545 nm. The ability of the anti-EpCAM-UCNP@SiO2(RB) to produce 1O2 was tested under 980 nm excitation (1.5 W·cm−2) using DPBF with a 30% of the probe consumed after 30 min irradiation. No 1O2 production was observed for non-irradiated anti-EpCAM-UCNP@SiO2(RB) or for irradiated anti-EpCAM-UCNP@SiO2 confirming the importance of having the three elements together, UCNPs + RB + 980 nm excitation, to generate 1O2. The ability of the anti-EpCAM-UCNP@SiO2(RB) to specifically target cancer cells was investigated in vitro using EpCAM-overexpressing human colon adenocarcinoma HT-29 cells and EpCAM-negative murine microglia BV2 cells. After 1 h incubation, the luminescence emission of the UCNPs was observed in the HT-29 but negligible fluorescence was detected in the BV2 cells. UCNP@SiO2(RB) without antibody were also tested in HT-29 cells resulting in lower emission intensity than cells incubated with anti-EpCAM-UCNP@SiO2(RB). Additionally, a control murine monoclonal antibody CRY104 (non-specific to EpCAM) was conjugated to the UCNP@SiO2(RB) and incubated in HT-29 cells resulting in an insignificant green emission [130]. The intracellular ROS production of the anti-EpCAM-UCNP@SiO2(RB) upon 980 nm irradiation was confirmed in HT-29 cells using DCFH-DA (that converts to DCFH intracellularly) as 1O2 probe. A greater increase in the green fluorescence emission intensity due to the formation of DCFH was observed for those cells that had been incubated with anti-EpCAM-UCNP@SiO2(RB) and exposed to 980 nm irradiation compared to the change observed in the cells treated with anti-EpCAM-UCNP@SiO2. The PDT effect was also investigated in vitro using MTT assay resulting in ca. 20% dark cytotoxicity at 200 μg·ml−1 of anti-EpCAM-UCNP@SiO2(RB) and a decrease to ca. 40% cell viability upon irradiation at 980 nm (1.5 W·cm−2, 10 min) [130].
Ramírez-García et al developed ZnPc-NaYF4:Yb,Er (60% Yb3+ and 2% Er3+) nanoparticles bioconjugated with trastuzumab (TRAS) for the specific targeted PDT of HER2-positive breast cancer cells [131]. First, a ligand exchange reaction to remove the oleic acid from the oleate capped UCNPs was performed using cysteamine (Cys), and then ZnPc molecules were covalently incorporated through carbonamide bond formation between the amine group of the Cys and the carboxyl group of the ZnPc. The ZnPc-UCNPs were then bioconjugated with TRAS via EDC/NHS chemistry and the concentration of antibody and reaction time were optimised to avoid aggregation and non-specific orientation. The functionalisation of the UCNPs was confirmed measuring the Fourier transform infrared (FTIR) spectrum. The TRAS-ZnPc-UCNPs showed good colloidal stability and a size of ca. 23 nm. The energy transfer between the luminescence at 659 nm and the absorption at 675 nm (ZnPc) upon 975 nm excitation was calculated to be 84.3% [131]. ZnPc-UCNPs and TRAS-ZnPC-UCNPs were able to generate 1O2 in solution following irradiation at 972 nm (ABMA monitoring) while no obvious production was observed for the control nanoparticles (Cys-UCNPs). Subsequently, the targeting ability of the TRAS-ZnPC-UCNPs was studied in MCF-7 (HER2-negative) and SK-BR-3 (HER2-positive) breast cancer cells using confocal microscopy with 980 nm excitation. Cys-UCNPs were internalised in both cells lines whereas the ZnPC-UCNPs were not observed in the cells, indicating that the uptake of the nanoparticles is diminished when the ZnPc is present on the surface of the UCNPs. Finally, the TRAS-ZnPC-UCNPs were incubated in both cell lines showing only the upconversion luminescence signal on the SK-BR-3 cells. To evaluate The PDT effect of the TRAS-ZnPC-UCNPs, several concentrations of the nanoparticles (0–1000 μg·ml−1) were incubated in both cell lines and the cell viability was measured (XTT assay) after 975 nm irradiation (0.71 W·cm−2, 5 min). Similar cytotoxicity results were obtained for both cell lines when the cells were treated either with Cys-UCNPs or ZnPc-UCNPs. However, higher reductions of the cell viability were observed when SK-BR-3 were incubated with TRAS-ZnPc-UCNPs and irradiated at 975 nm [131]. Therefore, the bioconjugation of PS-UCNPs with trastuzumab enable the targeted PDT of cancer cells that overexpress the HER2 receptor.
Epithelial growth factor receptors (EGFRs) are commonly overexpressed in lung and breast cancers and in glioblastoma, thus different EGFR-based targeting strategies have been developed using antibodies [158]. Zhang and co-workers fabricated UCNPs functionalised with a non-immunoglobulin-derived affinity protein known as Affibody® [132]. TiO2-capped NaYF4:Yb,Tm nanoparticles were bioconjugated with the anti-EGFR Affibody through a PEG chain. To this aim, dimerised anti-EGFR was first reduced using dithiothreitol (DTT) and then reacted to maleimide-PEG-COOH yielding anti-EGFR-PEG-COOH that was grafted to silane and used for the covalent functionalisation of the TiO2-UCNPs yielding anti-EGFR-PEG-TiO2-UCNPs (ca. 50 nm). The antibody functionalisation was confirmed using FTIR. The anti-EGFR-PEG-TiO2-UCNPs showed stability after 6 h stored in water and cell culture medium (10% FBS) but not when stored in PBS. The anti-EGFR-PEG-TiO2-UCNPs were evaluated in vitro in a range of cell lines including EGFR-positive cells such as human epidermoid A431 and human lung adenosquamous carcinoma H596, EGFR wild type cells such as lung cancer H460 cells, and EGFR-negative cells such as MCF-7 and HepG2 cells. The results presented in the paper indicate the selectivity of the designed nanosystem towards EGFR expressing cells. The work was completed with a detailed in vivo evaluation of the anti-EGFR-PEG-TiO2-UCNPs. The nanoparticles were hemocompatible up to a dose of 50 mg·kg−1 and exhibited 100% survival with no obvious toxic side effects in animals up to 120 d. Furthermore, the in vivo PDT effect of the anti-EGFR-PEG-TiO2-UCNPs was investigated confirming a delay in tumour growth and improved survival rates compared to conventional Ce6 PDT (655 nm) [132].
3.2.2.2. Folic acid
Folate receptors (FRs) are overexpressed in many types of cancers including breast, lung, kidney and ovarian. There are 100–300 times more FRs in cancer cells than on healthy cells and they can be divided into three isoforms (FRα, FRβ and FRγ) being FRα the most extensively expressed in cancer cells [159, 160]. Folic acid (FA) presents a high binding affinity to FRs (dissociation constant (KD) of 0.1–1 nM) [159] and, once bound to the FR, FA can be internalised via endocytic transport and accumulates in the cytosol [160]. Thus, the conjugation of FA with organic molecules and nanoparticles has been used as a targeting strategy in cancer diagnosis and therapy. FA (figure 7) has a low molecular weight and high stability; and it contains an acid terminal unit that can be employed to link amino-functionalised molecules or nanoparticles via amide formation reaction [160]. FA-mediated active targeting using FA-UCNPs has been explored for PDT of cancer providing high sensitivity and specificity, thus reducing the systemic side effects [104, 133–140, 161–164].
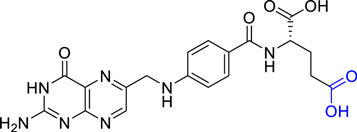
Figure 7. Chemical structure of folic acid; the terminal acid group (blue) of the glutamic acid is commonly used to bind other moieties.
Download figure:
Standard image High-resolution imageThe first example of in vivo targeted PDT using FA-PS-UCNPs was reported by Idris et al in 2012 [133] where mesoporous silica coated NaYF4:Yb,Er nanoparticles were functionalised with two PS drugs, MC540 and ZnPc, and FA as targeting agent. The FA-ZnPc-MC540-UCNPs emitted two main upconverting luminescence emissions upon 980 nm irradiation, at 540 and 660 nm, overlapping with the absorption spectrum of MC540 and ZnPC, respectively. Dual-PS-UCNPs exhibited higher 1O2 generation following 980 nm irradiation (2.5 W·cm−2, during intervals of 20 min) than irradiated single-PS-UCNPs (using ABMA) [133]. The intracellular production of ROS by the dual-PS-UCNPs following 980 nm irradiation (40 min) was confirmed in B16-F0 murine skin cancer cells using DCFDA and confocal microscopy. The control groups, single-PS-UCNPs and void UCNPs, showed lower ROS production. Only a reduction in cell viability of 55% (MTS assay) was obtained when the B16-F0 cells were incubated with the dual-PS-UCNPs and irradiated at 980 nm (40 min) whereas a 25% decrease was observed for the void UCNPs showing the cytotoxicity of the UCNPs [133]. The efficacy of the dual-PS-UCNPs for in vivo PDT was investigated by injecting the nanoparticles under the skin of C57BL/6 mice bearing B16-F0 melanoma cells and then irradiating at 980 nm (415 mW·cm−2, 2 h), which induced inhibition of the tumour growth and an enhancement in the population of apoptotic cells as compared to control experiments. A second approach was used in which the dual-PS-UCNPs were intratumorally injected into C57BL/6 mice bearing melanoma tumours and irradiated at 980 nm for 1 h. This treatment slowed tumour growth during the 11 d period when the mice were monitored. Dual-PS-UCNPs were further functionalised with FA to target the FR overexpressed on B16-F0 cells and PEG to increase the blood circulation time of the UCNPs. Compared to the UCNPs without FA-PEG, the FA-PEG-dual-PS-UCNPs showed a significantly greater reduction in tumour growth when they were intravenously injected in mice bearing B16-F0 melanoma tumours and following NIR irradiation (4 h) [133]. Although better results were obtained after the incorporation of FA and PEG, further optimisation is required since the complete regression of the tumours was not achieved with the reported nanosystem. To this aim, optimisation of the number of encapsulated PS drugs and the porous size of the silica layer can be assessed by determining the maximum energy transfer from the UCNPs to the PSs.
Incorporation of FA in amphiphilic coated UCNPs has also been investigated for active targeted PDT of cancer. Thanasekaran et al reported the encapsulation of PS drugs following phospholipid coating of the surface of UCNPs [134]. Compared to PEGylated strategies, lipid coating results in biocompatible and colloidal stable nanoparticles that are inert to immunological resistance [134]. Additionally, the fatty chain of the phospholipid layer enables the loading of PS drugs through hydrophobic interactions. In this work, oleate-capped NaYF4:Yb,Er UCNPs were coated with zwitterionic phospholipids (L-α-phosphatidylcholine (EggPC) and cholesterol, 1:1 molar ratio) resulting in colloidal stable UCNPs with an average diameter of 40 ± 2 nm [134]. A screening of several PS drugs, which absorption overlaps the upconversion luminescence bands, including RB, meso-tetraphenylporphine (TPP), ZnPc, aluminum phthalocyanine chloride (AlPc), fac-(2,2'-bipyridine)-tricarbonylbromorhenium(I) (Re(bpy)(CO)3Br), perinaphthenone (PN) and MB, was performed and resulted in hydrophobic PS, such as the porphyrin-based PS, encapsulating more efficiently and releasing at a lower rate than the less hydrophobic PS [134]. Determination of 1O2 generation by the PS-phospholipid-UCNPs irradiated at 980 nm was performed using p-nitroso-dimethylaniline (RNO) in the presence of imidazole (RNO + imidazole method). TPP-UCNPs showed the best performance in the 1O2 production compared to the rest of PS drugs and were selected for cellular experiments [134]. The TPP-UCNPs were modified with FA (TPP-UCNP-FA) to assess the targeting ability of the nanosystem. TPP-UCNP-FA nanoparticles were separately incubated in FR-positive KB cells (keratin-forming tumour cell line HeLa) and FR-negative REF52 cells (rat embryonic fibroblast) showing higher receptor-mediated cellular uptake in KB cells. PDT studies performed in KB cells and REF52 cells incubated with TPP-UCNP-FA and irradiated at 980 nm (1 h) resulted in greater cell dead for FR positive than for FR negative cells (cells viability determined staining the cells with propidium iodide). TPP-UCNP-FA were also investigated for in vivo PDT in mice bearing CT-26wt tumours via intratumoral injection and following irradiation at 980 nm (1 W·cm−2, 30 min). The tumour size was monitored over 10 d and the results showed a very similar tumour growth delay for mice injected with TPP-UCNP-FA and TPP-UCNP and irradiated with NIR light [134].
Another example of FA-modified amphiphilic coated UCNPs loaded with PS drugs was published by Cui et al [135]. FA-modified amphiphilic N-succinyl-N'-octyl chitosan (FA-SOC) was first synthesised by amide formation reaction between the carboxyl groups of the FA and the amino groups of the SOC and used to coat previously synthesised oleate-caped NaYF4:Yb,Er nanoparticles (ca. 50 nm) via hydrophobic interactions between the octadecyl groups on the UCNPs and the octyl groups of FA-SOC. ZnPc was trapped into the nanosystem through hydrophobic interactions. The luminescence band of the UCNPs at 660 nm was used to activate the ZnPc to generate 1O2 whereas the second luminescence band (540 nm) was used to image the UCNPs. The highest loading capacity of ZnPc into the amphiphilic chitosan shell was 10%; however, the optimal behaviour was obtained with ca. 6% loading. Releasing studies of the ZnPc were performed at different pHs (7.4, 6.5 and 5.7) showing less than 20% leaching after 50 h at 37 °C in all cases [135]. The FA-SOC-ZnPc-UCNPs were incubated in human liver cancer Bel-7402 cells and imaged using confocal microscopy confirming the intracellular co-localisation of the ZnPc and the UCNPs. Cytotoxicity experiments incubating FA-SOC-ZnPc-UCNPs in human embryo lung HELF and human breast MDA-MB-231 cancer cells showed a negligible decrease in cell viability (using MTT assay) under dark conditions. Several cancer cell lines with different levels of FR expression (Bel-7402, MDA-MB231 and A549) were cultured with the same concentration of nanoparticles showing a higher uptake by cells expressing higher levels of FR. The FA-mediated internalisation was further confirmed with an FR blocking experiment in which before the incubation of the nanoparticles, FR-positive Bel-7402 and MDA-MB-231 cells were treated with free FA. These results demonstrated that the uptake of the FA-SOC-ZnPc-UCNPs in FR-positive cancer cells was via FR-mediated endocytosis [135]. The intracellular 1O2 generation was investigated under 980 nm and 660 nm irradiation for the indirect and direct activation of ZnPc, respectively. To this end, DPBF and DCFDA ROS probes were separately added to Bel-7402 cells incubated with FA-SOC-ZnPc-UCNPs showing, in both cases, similar results. Higher ROS production was observed when the cells were excited at 660 nm (ca. 70%) than when 980 nm laser was used (ca. 55%) using the same power density (0.2 W·cm−2). When the cells were covered with pork tissue (1 cm), higher ROS generation was determined for cells irradiated at 980 nm (ca. 40%) than at 660 nm (ca. 15%). In vivo studies, performed with FA-SOC-UCNPs (without ZnPc), showed high toxicity of the nanoconstruct when administered at high doses (>150 mg·kg−1) and accumulation in the liver, intestines, lungs and kidneys 24 h post-injection. The targeting ability of FA-SOC-UCNPs over SOC-UCNPs was also demonstrated in vivo. Finally, the PDT treatment was investigated in S180 tumour-bearing mice using 980 nm or 660 nm irradiation conditions. 660 nm irradiation was superior when the tumours were directly exposed to the irradiation; however better results were obtained with 980 nm irradiation when a 1 cm pork tissue was placed over the tumour [135].
To avoid the leaching of the PS drug from the nanosystem, Zhang and co-workers described UCNPs covalently functionalised with RB and containing FA as targeting agent [104]. Amine-functionalised NaYF4:Yb,Er nanoparticles were obtained by ligand exchange using 2-aminoethyl dihydrogenphosphate (AEP) replacing the oleylamine ligands obtained from the synthesis of the UCNPs. RB hexanoic acid was covalently bound to the amine-functionalised UCNPs via EDC/NHS chemistry yielding RB-UCNPs (figure 8) [104]. The luminescence spectrum of the UCNPs exhibited a significant decrease of the upconversion luminescence band at 540 nm in the presence of RB while the band at 650 nm remained stable; thus, confirming the effective energy transfer from the UCNPs to the RB. The FRET efficiency was estimated to be ca. 83%, which is higher than for non-covalent RB encapsulation strategies which reported maximum energy transfer efficiencies of ca. 65%–68% [91, 92]. The energy transfer was further confirmed by measuring the luminescence decay lifetimes, showing a decrease in the average decay value recorded at 540 nm [104]. The production of 1O2 by the RB-UCNPs was examined using DPBF, showing a 50% decrease in absorption after 16 min irradiation at 980 nm. To enhance the targeting efficiency of the RB-UCNPs in cancer cells, FA was covalently attached to the surface of the UCNPs through a PEG ligand (NH2-PEG-COOH). FA-RB-UCNPs were incubated at different concentrations in JAR choriocarcinoma and noncancerous NIH 3T3 fibroblast cells and the targeted-PDT treatment following NIR irradiation was investigated using MTT assay [104]. The JAR cells showed a significant decrease in cell viability with an increase in FA-RB-UCNPs concentration, while NIH 3T3 cells did not show changes in cell viability. A negligible decrease in cell viability was also observed for the control non-irradiated cells incubated with FA-RB-UCNPs [104].

Figure 8. Schematic representation of the preparation of the FA-RB-UCNPs: NaYF4:Yb,Er UCNPs functionalised with RB, PEG and FA [104]. Reprinted from [104] with permission from American Chemical Society.
Download figure:
Standard image High-resolution imageAnother strategy to enhance the energy transfer between the UCNPs and the PS drug is to enhance the upconversion luminescence of the UCNPs. To this aim, different strategies have been developed including the construction of core-shell nanoparticles to avoid any energy losses due to surface defects and/or to increase the doping concentration of the lanthanide ions to enhance the upconversion emission from the UCNPs. Using their previous work as starting point [104], Zhang and co-workers designed NaYF4:Yb,Er UCNPs with 5% additional Yb3+ concentration (25% instead of the initial 20%) to increase the upconversion luminescence at 660 nm [136]. The resulting nanoparticles (ca. 30 nm) were coated with poly(allylamine) yielding NH2-UCNPs. ZnPc (containing carboxyl groups) was covalently attached to the NH2-UCNPs via crosslinking reaction. Additionally, PEG succinimidyl carbonate was incorporated onto the surface of the UCNPs enhancing the dispersibility and stability of the nanoparticles in biological media. Different concentrations of ZnPc were loaded onto the UCNPs and a 6% loading was found to be optimal for the generation of 1O2 under 980 nm irradiation (as determined using DPBF). A comparative study between covalent and non-covalent (physical absorption) incorporation of ZnPc onto the UCNPs was performed showing higher 1O2 production when the PS drug was covalently attached to the nanoparticles [136]. To achieve cancer targeting, FA was covalently linked to the ZnPc-PEG-UCNPs via a cross-linking strategy. Subsequently, the energy transfer between the UCNPs and the ZnPc was determined using steady-state upconverting luminescence spectroscopy and decay kinetics resulting in an 80.9% [136], similar to the value obtained for the covalently functionalised RB-UCNPs (83%) [104]. After the confirmation of the outstanding efficiency to generate 1O2 upon 980 nm irradiation in non-aqueous solution (74% DPBF consumption after 4 min irradiation), FA-ZnPc-PEG-UCNPs were incubated in HeLa cells (FR-positive) and human alveolar adenocarcinoma A549 cells (FR-negative). The upconversion fluorescence images of the incubated cells under 980 nm excitation (0.19 W·cm−2) showed the efficient cellular uptake via FR-mediated endocytosis in FR-positive cells whereas negligible upconversion emission was observed for the FR-negative cells. The targeting capability of the FA ligands was further demonstrated with FR blocking experiments in HeLa cells (previous addition of free FA) showing the absence of fluorescence in the incubated cells upon 980 nm excitation [136]. The PDT effect was studied using MTT assay and showed a significant decrease (70%) in cell viability when cells were treated with 200 μg·ml−1 of FA-ZnPc-PEG-UCNPs and irradiated at 980 nm (0.39 W·cm−2, 10 min). The cell viability results showed considerable dark cytotoxicity of the nanoparticles at concentrations above 200 μg·ml−1 whereas a negligible decrease was obtained with lower concentrations. The PDT effects were also investigated in vivo using epa1–6 tumour-bearing C57/6J mice. Two weeks following intratumoral injection of the FA-ZnPc-PEG-UCNPs and irradiation at 980 nm (0.39 W·cm−2, 15 min) a significant reduction in the tumour growth was observed (tumour inhibitory ratio ca. 80.1%). This effect was not observed for control groups treated with NIR irradiation or FA-ZnPc-PEG-UCNPs only. The behaviour of ZnPc-PEG-UCNPs without FA to fully confirm the targeting ability of the nanoparticles was not investigated in this work [136].
Wang et al developed UCNPs (NaYF4:Yb(8%)/Ho(1%)@NaYF4:Nd(20%)@NaYF4 core-shell-shell) functionalised with RB through an amide coupling reaction using EDC/NHS chemistry in which is the first example reported in the literature with UCNPs excitable at 808 nm for PDT treatment [91]. The 1O2 production of the RB-UCNPs was investigated in solution using DPBF. A comparative study of PDT performance of RB-UCNPs, depending on the active layer thickness, was performed using 808 nm irradiation showing the larger production of 1O2 with a shell thickness of 1.6 nm (compared to shells of 0.4, 2.5 and 4.2 nm). HeLa cells were used to confirm the efficiency of the PDT effect in vitro following 808 nm excitation, and minimisation of the overheating effect when using this irradiation wavelength was confirmed following a comparative study with 980 nm irradiation [91].
Hypericin-loaded NaYF4:Yb,Er nanoparticles modified with FA were reported by Yang et al [137]. The oleate-capped UCNPs were first coated with a silica layer and the photosensitiser hypericin was covalently bound to the silica shell. The absorption band of hypericin at 550 nm overlapped the green luminescence emission of the UCNPs ensuring the effective energy transfer. FA was then conjugated to the surface of the hypericin-UCNPs for the receptor-mediated delivery of the UCNPs [137]. The production of 1O2 by the FA-hypericin-UCNPs under 980 nm irradiation was confirmed using ABMA. Two photon laser scanning confocal microscopy was used to study the uptake of the FA-hypericin-UCNPs in FR-overexpressing cancer cells (HeLa cervical cancer cells) and normal cells with lower FR expression (293T embryonic kidney cells), showing higher internalisation of the nanoparticles in HeLa cells. Additionally, the uptake of FA-hypericin-UCNPs and hypericin-UCNPs was compared in HeLa cells resulting in a higher fluorescence emission intensity in the cells when FA-hypericin-UCNPs were incubated [137]. The PDT effect of the FA-hypericin-UCNPs was qualitatively investigated using apoptosis assays, staining treated HeLa cells with Annexin V-FITC/PI after irradiation at 980 nm. The fluorescence images of HeLa cells treated with FA-hypericin-UCNPs and NIR irradiation showed apoptotic cells whereas control groups of cells treated with the nanoparticle but without irradiation did not exhibit any indication of the apoptotic process. Flow cytometry was used to evaluate the PDT effect of FA-hypericin-UCNPs in HeLa cells, showing 36% of early and late apoptosis after 15 min irradiation at 980 nm of treated HeLa cells [137].
Zhang and co-workers reported core-shell NaYF4:Yb,Er@NaYF4:Yb,Tm covalently functionalised with monomalonic fullerene (C60MA) as PS drug, PEGylated ligands to enhance the biocompatibility of the nanoparticles, and FA as targeting agent [138]. To achieve the covalent chemical bonding between the ligands and the nanoparticles, UCNPs were first coated with poly(allylamine) providing amine functionalisation. C60MA was covalently attached via amide bond formation and a PEGylation process was then performed using PEG-succinimidyl carbonate to increase the water-dispersibility of the C60-UCNPs. The resulting PEG-C60-UCNPs presented a hydrodynamic diameter of 64 nm measured using dynamic light scattering (DLS). The UCNPs exhibited several upconverting luminescence emission bands (450, 475, 540, 650 and 808 nm) following 980 nm irradiation, which overlapped (except 808 nm) the broad absorption spectrum of the C60MA. The effective energy transfer between the UCNPs and the C60MA was confirmed using steady state upconversion luminescence measurements and lifetime luminescence decays. Even though the intensities at 450, 475, 540 and 650 nm were all significantly quenched, the maximum energy transfer was calculated to be 79% at 450 nm [138]. The 1O2 production of the PEG-C60-UCNPs was confirmed using fluoresceinyl cypridina luciferin analogue (FCLA) as chemical probe. PEG-C60-UCNPs were further functionalised with FA via covalent amide formation between the carboxyl group of the FA and the free amine groups on the surface of the nanoparticles. The resulting FA-PEG-C60-UCNPs were incubated in FR-positive HeLa cells both, using a folate-free and a folate-supplemented medium, and in FR-negative A549 cells, and the cellular uptake was monitored recording the emission at 808 nm using confocal microscopy. FA-PEG-C60-UCNPs were taken up by HeLa cells cultured in a folate-free medium; however, fewer nanoparticles were internalised by HeLa cells cultured with a folate-supplemented medium and by A549 cells. Unfortunately, cell viability studies (MTT assay) showed that very high irradiation power (limits set for human skin exposure to 980 nm light is 0.72 W·cm−2) and high nanoparticle concentration were needed to induce only a ca. 50% viability decrease in HeLa cells incubated with FA-PEG-C60-UCNPs (500 μg·ml−1) and irradiated at 980 nm (1.37 W·cm−2, 10 min) [138]. The authors claimed that the low PDT efficiency was caused by large distances between the C60 and the core of the UCNPs. To reduce the distance, Zhang and co-workers functionalised core-shell NaYF4:Yb,Er@NaYF4:Yb,Tm nanoparticles with C60 following a non-covalent strategy [139]. A ligand exchange method was used to replace the oleylamine groups on the surface of the UCNPs with C60MA molecules presenting carboxyl groups. Additionally, PEG-block-poly(caprolactone) was incorporated on the C60-UCNPs to enhance the stability of the nanosystems in aqueous solutions. The maximum loading capacity obtained for C60 was found to be 22.5% which was higher than when the covalent strategy was followed (10.5%) [138]. The stability of the PEG-C60-UCNPs was investigated showing a C60 release of 2.5% after 72 h in PBS [138]. An enhanced energy transfer between the UCNPs and the PS was obtained by the ligand exchange strategy compared to the covalent assembly, 98.7% and 79% at 450 nm, respectively. The 1O2 production efficiency following irradiation at 980 nm was confirmed using FCLA but the results cannot be directly compared to those from the covalently assembled C60-UCNPs since different concentrations of C60 were used. The PEG-C60-UCNPs were incubated in HeLa cells and the production of 1O2 was achieved following 980 nm irradiation (0.39 W·cm−2, 5 min) as confirmed using DCFDA and fluorescence images [139]. To achieve targeting, the PEG-C60-UCNPs were further functionalised with FA, directly attached to the PEG units, obtaining a FA loading of 5.1%. The FA-PEG-C60-UCNPs were incubated in HeLa (FR-positive) and A549 (FR-negative) cells and a higher cellular uptake was observed for HeLa cells [139]. Subsequently, the PDT effect of FA-PEG-C60-UCNPs was studied in HeLa cells (MTT assay) and a 35% viability decrease was observed for cells incubated with FA-PEG-C60-UCNPs (500 μg·ml−1) and irradiated at 980 nm (0.39 W·cm−2, 10 min). The dark toxicity of the FA-PEG-C60-UCNPs in HeLa cells was negligible at 500 μg·ml−1 [139]. This study concluded that the ligand exchange design of FA-PEG-C60-UCNPs exhibits superior in vitro therapeutic effect than the covalently conjugated FA-PEG-C60-UCNPs at lower laser power density. In vivo experiments were performed in mice bearing Hepa1-6 tumours (FR-positive) that showed tumour accumulation of tail vein injected FA-PEG-C60-UCNPs 2 h following injection. On the other hand, C60-UCNPs needed 24 h to accumulate in the tumour following injection and due to the EPR effect.
Ai et al reported core-shell-shell NaYbF4:Nd@NaGdF4:Yb,Er@NaGdF4 nanoparticles covalently functionalised with Ce6 and PEGylated modified FA [140]. These UCNPs were excitable at 808 nm and emitted luminescence at 520–560 nm and 650–690 nm, the later overlapping the absorption spectrum of Ce6. The nanoparticles were first modified with AEP via ligand exchange to obtain the amino functionalisation used to covalently attach the Ce6. Finally, the Ce6-UCNPs were capped with FA via NH2-PEG-COOH [140]. The energy transfer between the UCNPs and the Ce6 was confirmed by steady-state upconversion luminescence measurements showing a 70% efficiency and by luminescence lifetime decays. The production of 1O2 by FA-Ce6-UCNPs under 808 nm irradiation increased with increasing Ce6 loading concentrations as observed using DPBF, being 10% (w/w) the most effective concentration and thus, the selected for the biological experiments. Experiments of light penetration depth were conducted in solution adding different thicknesses of pork slices between the sample and the laser sources (808 or 976 nm). A decrease in 1O2 generation was observed with increasing thickness and deeper tissue penetration was achieved with 808 nm light [140]. In vitro experiments using FA-Ce6-UCNPs were performed in KB cells (FR-positive) and A549 cells (FR-negative) and resulted in higher uptake of the nanoparticles by the FR-positive human cancer cells. The intracellular PDT effect of FA-Ce6-UCNPs was investigated and a decrease in cell viability (MTT assay) to 8% was observed for KB cells incubated with the nanoparticles following irradiation at 808 nm. Different concentrations of nanoparticles were tested showing an evident PDT effect after 5 min irradiation when cells were incubated with 100 μg·ml−1 of FA-Ce6-UCNPs (negligible dark toxicities were observed for this nanoparticle concentration) [140].
Zhao et al developed UCNPs with folic acid-polyethylene glycol-poly(aspartic acid-hydrazone)-dihydrolipoic acid (FA-PEAH) polymer chains conjugated to the surface [141]. Further functionalisation with pheophorbide a (Pha) as PS drug via an acid-labile hydrazone linker allowed for pH-responsive release of the Pha within the lysosomal compartments of the cells. The emission band of UCNPs at ca. 670 nm overlapped the main absorption band of Pha (broad band at 690 nm) indicating the potential energy transfer between the UCNPs and the PS drug. The successful activation of the Pha upon excitation of the FA-PEAH-UCNPs-Pha at 980 nm was confirmed in solution using DPBF. Intracellular experiments in MCF-7 cells proved the improved water dispersibility of the Pha when present on the surface of the UCNPs. Furthermore, FA-PEAH-UCNPs-Pha (30 μg·ml−1, 980 nm, 0.1 mW·cm−2, 5 min) showed higher cytotoxicity than the free Pha confirming the improved PDT treatment when using UCNPs. The FA-PEAH-UCNPs-Pha showed also an enhanced PDT effect compared to the PEAH-UCNPs-Pha confirming the targeting ability of the nanosystem [141]. Another example of UCNPs functionalised with FA for targeted PDT was developed by Lim et al in which TCPP was used as PS drug [142]. Core-shell UCNPs were coated with silica and further functionalised via 3-aminopropyltriethoxy-silane (APTED) addition with TCPP-NHS, PEG-NHS, and FA/PEG-NHS. The resulting UCNPs@SiO2-NH2@FA/PEG/TCPP nanoparticles were tested in HeLa cells showing similar toxicity results for the free TCPP irradiated at 660 nm and for the UCNPs@SiO2-NH2@FA/PEG/TCPP irradiated under 808 nm. The cell viability was compared under normoxic and hypoxic conditions showing a significant reduction of the PDT effect in the absence of O2 [142].
3.2.2.3. Transferrin
The receptor for transferrin (known as TfR1 or CD71) is expressed at low levels in most normal human tissues. However, this receptor is expressed at levels many times higher in malignant cells and thus why it is a popular receptor to target for cancer therapy [165].
To target this receptor, Liu et al designed transferrin decorated UCNPs for the targeted PDT of cancer cells upon NIR irradiation [143]. The oleate-capped NaYF4:Yb,Tm nanoparticles containing 20% Yb and 0.5% Tm were first treated to remove the oleic acid ligands via an acid treatment and then functionalised with polyelectrolyte poly[9,9-bis(4'-sulfonatobutyl)fluorine-alt-co-(1,4-benzo-{2, 1', 3}-thiadiazole)] sodium salt (PFSBT) as PS and with transferrin (Tf) as targeting agent. The PFSBT-Tf-UCNPs were then further functionalised with a second PS, titanocene (Tc) able to generate free radicals and with high binding affinity toward the iron-chelating Tf. Irradiation of the UCNPs at 980 nm resulted in emission peaks at 290, 345, 361, 450, 474 and 800 nm, which, apart from 800 nm, overlapped with the absorption of Tc and PFSBT. An 87% energy transfer from the UCNPs to the PS drugs was estimated at 450 nm and lifetime measurements showed a decrease from 594 to 389 μs which also confirmed the energy transfer [143]. The PFSBT-Tf-Tc-UCNPs were incubated in MCF-7 breast cancer cells to examine the PDT effect of the nanostructures upon 980 nm irradiation (0.6 W·cm−2). The photo-cytotoxicity of the PFSBT-Tf-Tc-UCNPs was determined by fluorescence microscopy using calcein acetoxymethylester (Calcein-AM) to stain the live cells and propidium iodide to stain the dead cells. The results obtained at different concentrations of PFSBT polymer on the nanoparticles showed a high number of dead cells when irradiating at 980 nm UCNPs loaded with 10 μg of the PFSBT. Experiments with different concentrations of Tf-Tc on the UCNPs were also performed showing an increased cell death when higher amounts were used. In vivo PDT experiments using PFSBT-Tf-Tc-UCNPs were performed in a mouse model bearing the H22 tumour and showed a significant inhibition of the tumour growth when the animals received the PFSBT-Tf-Tc-UCNPs and irradiation at 980 nm (0.6 W·cm−2, 30 min) [143].
Transferrin has also been used for the targeted delivery of PS-UCNPs containing also the chemotherapeutic drug doxorubocin (DOX)—dual therapy systems [144, 145]. Zhang et al reported NaYF4:Yb, Tm:NaYF4 nanoparticles containing the PS hypocrellin A (HA) incorporated into a mesoporous silica shell that was further functionalised to incorporate DOX via silane coupling reaction and a UV cleavable linker (4-(2-carboxy-ethylsulfanylmethyl)-3-nitro-benzoic acid (CNBA)) that functions as a gate to permit the controlled release of the drugs [144]. Finally, transferring was incorporated into the system to achieve targeted delivery. HA-UCNPs were able to produce 1O2 in solution under irradiation at 980 nm (0.5 W·cm−2) as monitored using DFBF. The CNBA-DOX-HA-UCNPs showed a photoinduced controlled release of the DOX upon irradiation with NIR light (0.5 W·cm−2) ascribed to the UV cleavable CNBA linkers. The PDT effect of the CNBA-DOX-HA-UCNPs was evaluated (MTT assay) in HeLa and MCF-7 cells observing a synergistic effect of the dual treatment following irradiation at 980 nm (0.15 W·cm−2, 20 min). Following these studies, Tf was incorporated into the UCNPs and the targeting ability of the nanosystem was evaluated in HeLa and MCF-7 cancer cells, and 293T normal cells were used as controls. Higher accumulation of Tf-CNBA-DOX-HA-UCNPs was observed compared to CNBA-DOX-HA-UCNPs [144]. Similar results were obtained by the same authors when Ce6 was used as the PS drug and the enzyme and pH-sensitive linker succinic acid-glycine-phenylalanine-leucine-glycine was used as the gate to release DOX [145].
3.2.2.4. Peptides
Peptides have also been used for the targeting of cancer cells since they can enhance the ability to enter the cells (cell-penetrating peptides) or they can specifically target tumour epitopes [166]. In addition, peptides are easy to synthesise, present moderate chemical stability and their small molecular weight allows better tumour penetration. Trans-activating transcriptional (TAT) activator is an example of cell-penetrating peptides that have been successfully used for intracellular delivery of nanoparticles including micelles, liposomes, quantum dots, UCNPs, polymeric, iron oxide, silica and gold nanoparticles [149, 151, 167]. The cancer-associated receptors targeted by peptides include integrins, prostate-specific membrane antigen, transferrin receptor, HER2, aminopeptidase N and CXCR4 [168]. The tripeptide arginine—glycine—aspartic acid (RGD) (figure 9) is commonly used to target cancer cells and preferentially binds to αv β3 integrin receptors which are overexpressed on angiogenic endothelial cells [166].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Chemical structure of RGD peptide.
Download figure:
Standard image High-resolution image{kind=link}
Peptide-decorated UCNPs have been developed for NIR-activated targeted-PDT of cancer [146–154]. For example, Wang et al developed polymer-coated UCNPs functionalised with MC540 and RGD peptide to achieve targeted PDT [146]. First, poly (maleic anhydride-alt-1-octadecene) (PMAO) was grafted with L-α-phosphatidylethanolamine, dioleoyl (DOPE) and RGD peptide. Next, NaYF4:Yb,Er nanoparticles were coated with the RGD-PMAO-DOPE via reverse phase evaporation method resulting in a hydrodynamic diameter of 147 nm. MC540 was trapped into the amphiphilic shell showing a loading efficiency of 9.3%. The 1O2 production of MC540-RGD-PMAO-DOPE-UCNPs was confirmed in solution under 980 nm irradiation using ABMA. MC540-RGD-PMAO-DOPE-UCNPs were taken up by MCF-7 breast cancer cells as confirmed by measuring the upconversion luminescence of the UCNPs. The intracellular ROS production was confirmed upon irradiation of cells incubated with MC540-RGD-PMAO-DOPE-UCNPs at 980 nm (ABMA emission measured intracellularly) [146]. However, the authors did not report controls of, for example, non-irradiated cells treated with the MC540-RGD-PMAO-DOPE UCNPs to confirm that the observed ROS production was a consequence of the 980 nm irradiation. To investigate the intracellular PDT effect of the nanoparticles, MTS assay of treated MCF-7 breast cancer cells was performed. A 60% reduction of the cell viability was obtained after 30 min irradiation of the cells incubated with MC540-RGD-PMAO-DOPE-UCNPs at 980 nm [146]. Although the cell viability decrease obtained for cells treated with MC540-RGD-PMAO-DOPE-UCNPs was higher than for the cells treated with MC540-PMAO-DOPE UCNPs (40%), controls of, for instance, the dark toxicity of the nanoparticles were not performed. Another example, by Hou et al, reports UCNPs decorated with a mesoporous silica shell doped with ZnPc as PS and a cross-linked lipid triple layer formed by PEGylated amphiphilic octadecyl-quaternised polyglutamic acid (OQPGA) which can be further functionalised with cationic RGD peptide [147]. The ZnPc-UCNPs showed an encapsulation efficiency of ca. 65%. A 35% ZnPc leached out after 200 min when no crosslinked lipid layer was present and this was reduced to 12.5% in the presence of the lipid layer [147]. The cellular uptake was investigated in HeLa cells substituting the ZnPc for rhodamine to facilitate fluorescence detection in the microscope. After confirming the co-localisation of the UCNPs and the dye by fluorescence microscopy, RGD peptide was incorporated onto the nanoparticles and a slight increase in cellular uptake was observed. The RGD-ZnPc-UCNPs were able to generate 1O2 under 980 nm irradiation at higher rates than when free ZnPc was tested (ABMA used as probe). The PDT effect of RGD-ZnPc-UCNPs was evaluated in HeLa cells (MTT assay) showing lower cell viability for cells incubated with RGD-ZnPc-UCNPs and ZnPc-UCNPs and irradiated at 980 nm (1.5 W·cm−2, 30 min) than when no irradiation was applied. High irradiation power was needed to achieve these results [147]. Although the authors proved the efficiency of the cross-linked lipid layer on the UCNPs reducing the leaching of the PS drug and enhancing the stability of the nanoparticles, additional experiments are required to demonstrate the targeting activity of the nanoparticles decorated with RGD peptide. For instance, tests of the PDT effect in vivo will validate the selectivity of the UCNPs to reduce the tumour size via PDT.
The aspartyl residue of the linear RGD can suffer a chemical degradation and thus cause a lack of biological activity. The stability against enzymatic degradation of the RGD peptides can be enhanced by using cyclic RGD derivatives [169]. An example of cyclic RGD peptide for targeted PDT using UCNPs was published by Zhou et al and involved NaYF4:Yb,Er nanoparticles modified with o-carboxymethylated chitosan and further functionalised with cyclic Arg-Gly-Asp-(D)-Tyr-Lys (c(RGDyK)) and Ppa as PS drug trough the carboxyl groups and the amine groups of the o-carboxymethylated chitosan, respectively [148]. Ppa was loaded at 2.8 mg Ppa·mg−1 UCNPs and the RGD-Ppa-UCNPs exhibited high stability at different pH (5, 7.4 and 8) for 24 h. The RGD-Ppa-UCNPs were able to produce a higher amount of 1O2 when irradiated at 635 nm (20 mW·cm−2) compared to free Ppa (FCLA used as probe) [148]. In vitro experiments were then performed to study the αv β3 integrin targeting capacity of the RGD-Ppa-UCNPs. Integrin positive cells (U87-MG brain cancer cells) and integrin negative cells (MCF-7 breast cancer cells) were incubated with RGD-Ppa-UCNPs and the intracellular production of ROS was measured using DCFA upon irradiation at 980 nm (0.5 W·cm−2). Higher production of 1O2 was observed for the integrin positive cell line. The αv β3 integrin-mediated endocytosis of the nanoparticles was confirmed with control experiments where the U87-MG cells were pre-treated with an excess of free c(RGDyK). The PDT effect of RGD-Ppa-UCNPs was also investigated using CCK8 cell viability assay with U87-MG and MCF-7 cells, resulting in a significant difference between both cells lines, 60% and 15% reduction, respectively, following irradiation at 980 nm (0.5 W·cm−2) [148]. The reported nanosystem showed higher 1O2 generation than the free PS drug, elevated stability and specific binding to the integrin αv β3-positive tumour cells. In vivo experiments would be relevant to confirm the benefit of NIR activated PDT for deep-seated tumours and to further confirm the targeting activity of the RGD-Ppa-UCNPs.
Another targeting moiety based on peptides is the TAT protein which is a cell-penetrating peptide that can be used as intracellular drug delivery vector. For example, Zhang et al reported core-shell-shell NaYbF4:Nd@NaGdF4:Yb,Er@NaGdF4 nanoparticles functionalised with Ppa as PS drug, PEG and TAT peptide [149]. The oleate-capped UCNPs were first modified with PMAO-PEG-NH2 allowing the covalent functionalisation of the TAT. Then, Ppa was loaded onto the surface of the PMAO-PEG-TAT-UCNPs via hydrophobic interactions resulting in a loading capacity of 0.6 mg Ppa·mg−1 UCNPs. The 1O2 generation upon 808 nm irradiation of the Ppa-UCNPs at different loading concentrations (0.025, 0.05, 0.1 and 0.2 ratios) was confirmed using DPBF as chemical probe with the best results obtained when using a loading amount of Ppa of 0.05 mg Ppa·mg−1 UCNPs. The Ppa-UCNPs were stable in a PBS buffer solution (pH 7.4) for a minimum of 4 h [149]. Subsequently, TAT modified UCNPs exhibited a higher cellular uptake in HeLa cells than UCNPs without TAT—as determined by quantification of Gd concentrations using ICP-OES. Intracellular generation of ROS upon irradiation at 808 nm was observed in HeLa cells incubated with the modified UCNPS using DCFDA. These results agree with the significant decrease in the cell viability following 808 nm irradiation (6 W·cm−2, 5 min) [149]. Co-localisation studies were performed showing the majority of the Ppa accumulated in the same cytoplasmic region of HeLa cells than the UCNPs, verifying the stability of the Ppa-UCNPs after their internalisation by the cells. Pap was localised in the mitochondria as confirmed by co-localisation studies using MitoTracker green. Additionally, JC-1 assay was used to monitor the damage in the mitochondria upon 808 nm irradiation of cells incubated with Ppa-UCNPs, confirming the potential of the nanosystem for mitochondria-targeted PDT [149]. However, although the authors demonstrate the phototherapeutic efficiency of the Ppa-UCNPs and the enhanced cellular uptake when TAT peptide was present, it is important to note that the laser power used in this work is extremely high and not applicable for in vivo experiments. Therefore, the efficiency of the system at lower power densities needs to be determined. In order to enhance the cancer-targeting ability of the UCNPs, the same group developed UCNPs decorated with the pH-low insertion peptide (pHLIP) that is a water-soluble membrane peptide able to specifically cross the cell membrane only at slightly acidic pH [150]. The authors synthesised Ppa-core-shell-shell NaYbF4:Nd@NaGdF4:Yb,Er@NaGdF4 containing pHLIP covalently attached to the UCNPs. A Pap release of 10% was observed for pHLIP-Ppa-UCNPs stored under neutral and acidic pH conditions (PBS buffer of pH 7.4 or 6.5) for 40 h [150]. Subsequently, the targeting ability of the pHLIP-Ppa-UCNPs dissolved in PBS at pH 6.5 and 7.4 was investigated in HeLa cervical carcinoma cells. Using confocal laser microscopy, higher emission intensities were observed upon 808 nm excitation when the cells were incubated with pHLIP-Ppa-UCNPs at pH 6.5 indicating the elevated efficiency of the pHLIP-Ppa-UCNPs to penetrate the cell membrane in an acidic environment. Following the same protocol, a comparative study between pHLIP-Ppa-UCNPs and NH2-Ppa-UCNPs was performed showing higher UCNPs concentration for the pHLIP-Ppa-UCNPs at pH 6.5. These results indicated that the higher efficiency of the pHLIP-Ppa-UCNPs to enter cancer cells at slightly acidic pH is consequence of the pHLIP peptide functionalisation. Then, the intracellular ROS production of the pHLIP-Ppa-UCNPs at both pH values was evaluated under 808 nm irradiation (6 W·cm−2) during 10 min using DCFDA as a probe. The results showed higher ROS generation when HeLa cells were incubated with pHLIP-Ppa-UCNPs at pH 6.5 as expected from the higher uptake at this pH. Measurements of the cell viability were performed to evaluate the photo-cytotoxicity of the pHLIP-Ppa-UCNPs in HeLa cells. A significant decrease of the cell viability was observed when the cells were treated with 50 μg·ml−1 of pHLIP-Ppa-UCNPs at pH 6.5 and irradiated at 808 nm (6 W·cm−2) during 5 min. At this concentration, the cell viability of the non-irradiated cells remained 95.2 ± 11.5%. At neutral pH conditions, a very small reduction of the cell viability was observed even with 200 μg·ml−1 of pHLIP-Ppa-UCNPs indicating that the PDT efficiency of the pHLIP-Ppa-UCNPs increases at slightly acidic microenvironments. Additionally, 4T1 cells were incubated with pHLIP-Ppa-UCNPs at pH 6.5 showing the same behaviour than for HeLa cells [150]. In vivo experiments were then performed, in BALB/c mice bearing 4T1 tumours, confirming the ability of the pHLIP-Ppa-UCNPs to reduce the tumour size via PDT treatment (980 nm, 0.5 W·cm−2, 30 min).
To combine the targeted delivery achieved when using RGD peptide and the ability to cross the cell membrane when functionalising nanoparticles with TAT peptide, Wang et al designed UCNPs modified with RGD/TAT polymeric lipid micelles and functionalised with ZnPc [151]. The authors combined three lipid proteins based on OQPGA including PEG-OQPGA, RGD-OQPGA and TAT-OQPGA. The UCNPs and the ZnPc were encapsulated in the PEG-RGD-TAT-OQPGA lipid micelles prepared using a thin-layer evaporation method obtaining a hydrodynamic size of 82.5 nm. The encapsulation efficiency and the loading efficiency of the ZnPc into the lipid micelles were evaluated showing opposite behaviour, the loading efficiency increased when increasing the volume of solution containing ZnPc whereas the encapsulation efficiency decreased. Additionally, leaching studies were performed showing a 50% ZnPc release after 6 h when then loading efficiency was 1.6%. The ability of the nanoparticles to produce 1O2 upon 980 nm excitation was confirmed using ABMA [151]. Next, the intracellular 1O2 production was investigated in MCF-7 breast cancer cells incubated with PEG-RGD-TAT-OQPGA-ZnPc-UCNPs using DCFDA as ROS marker. The PDT effect of PEG-RGD-TAT-OQPGA-ZnPc-UCNPs was further investigated in MCF-7 cells using MTS assay showing a decrease in cell viability of 85% after 980 nm excitation. A 60% cell viability decrease was obtained for the nanoparticles without RGD/TAT indicating that the modification of the UCNPs with RGD/TAT targeting agents allows for a higher uptake of the nanoparticles. The targeting capability of the nanoparticles was evaluated in B16F1 murine melanoma cells overexpressing αv β3 integrin. Cells incubated with RGD-TAT-OQPGA-ZnPc-UCNPs showed stronger upconversion luminescence upon 980 nm light irradiation compared to the nanoparticles without RGD peptide [151]. Although a higher uptake of the nanoparticles by cancer cells was observed for the RGD-TAT-OQPGA-ZnPc-UCNPs than for the nanoparticles without targeting agents, further experiments are required to verify the actual targeting ability. For instance, blocked cells with free RGD peptide previous incubation with the nanoparticles, uptake comparison experiment with a negative-αv β3 integrin cell line and in vivo tests would confirm the potential of the RGD-TAT-OQPGA-ZnPc-UCNPs as nanosystems for targeted-PDT of cancer.
Other peptides, such as cMBP, have been shown to have targeting properties towards triple-negative breast cancer (TNBC) cells compared to other cancer cells and normal cells. To apply the targeted PDT treatment to the TNBC cells, Wang et al synthesised ZnPc@mPEG-PLGA@UCNPs (ZUPEA) using double emulsion coprecipitation method [152]. The resulting ZUPEA nanoparticles were further functionalised with the cMBP-peptide to recognise TNBC cells. Higher loading of the cMBP-ZUPEA was observed in the TNBC cells (MDA-MB-231 cells) compared to the normal cells (MCF-10A cells) and the MCF-7 breast cancer cell and A549 lung cancer cell, indicating the successful targeting of the nanoparticles to the TNBC cells. The cMBP-ZUPEA exhibited good biocompatibility in normal and cancer cells and showed a great cytotoxicity upon irradiation at 980 nm (1.0 W·cm−2, 10 min,). cMBP-ZUPEA were evaluated in vivo in 4T1 tumour-bearing mice and resulted in a higher PDT effect following 980 nm irradiation than when the mice were treated with ZnPc and irradiated at 650 nm. 12 d following PDT treatment with the UCNPs, significant suppression of the tumour was observed while no abnormalities were observed in the organs of the treated mice [152].
In a recent work, Li et al developed UCNPs functionalised with an antenna molecule 800CW (for luminescence enhancement), RB as PS drug, Cy3 for diagnosis after treatment, and cathepsin B (CaB) substrate peptide labelled with QSY7 quencher for intracellular cathepsin B (CaB)-responsive PDT [153]. The UCNPs were first functionalised with alendronic acid (ADA) to facilitate further functionalisation. Next, the 800CW dye, RB and Cy3 were immobilised on the surface of the UCNPs and the CaB substrate peptide chain GRRGLGC with terminus-labelled QSY7 (Pep-QSY7) was also incorporated. In the final nanosystem, the presence of the QSY7 quenches the emission of the Cy3 and the RB, thus cancelling the production of 1O2 upon irradiation at 808 nm. Upon recognition of the CaB and cleavage of the QSY7 from the UCNPs, the fluorescence emission of the Cy3 is recovered and the RB is activated for the efficient PDT. The PDT effect of the UCNPs in HeLa cells was evaluated by monitoring the formation of ROS with DHR and the cytotoxicity with MTT assay, showing selective and CaB concentration-dependent phototoxicity. The PDT effect of the multifunctional nanosystem was evaluated in vivo using a HeLa tumour xenograft mouse model. Antitumour efficiency was observed for mice intratumorally injected with the nanoparticles (300 μg·ml−1) and irradiated at 808 nm (1.0 W·cm−2, 40 min), which was not observed for control groups (ie. without UCNPs but irradiated under the same conditions or injected with UCNPs but non-irradiated) [153].
4. Conclusions
Based on the number of publications that report the use of PS-UCNPs for the effective treatment of cancer via PDT (both in vivo and in vitro) to date, these nanosystems exhibit the potential to become the next generation of nanodrugs for PDT. Further to their advantage of being NIR excitable and therefore with potential for the treatment of cancers in deep-lying tissue, UCNPs can accumulate in tumour tissues passively via the EPR effect and actively by the incorporation of targeting agents to the nanoparticle surface which results in nanodrugs with reduced side effects. However, despite the large body of research that has shown the viability of UCNPs for PDT as an alternative to the currently used photosensitiser drugs, further improvements are needed to bring this treatment to the clinic. Further efforts are needed to improve their biocompatibility and selectivity which would lead to improvements in therapeutic efficacy.
Acknowledgments
This work was supported by the Faculty of Sciences and School of Chemistry at the University of East Anglia in partnership with Mr and Mrs Whittaker oncology fellowship. The authors have declared that no conflicting interests exist.
Data availability statement
No new data were created or analysed in this study.