
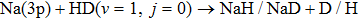
Abstract
Non-adiabatic dynamical calculations are carried out for the  reaction on the diabatic potential energy surfaces of Wang et al. (Sci. Rep. 2018, 8, 17960) by using the time-dependent wave packet method. The state-to-state integral cross sections and differential cross sections of two reaction channels (NaH/NaD+D/H) are calculated for collision energy up to 0.4 eV. The cross section branching ratio indicates that the dominant reaction channel changes from NaD+H to NaH+D when the collision energy is larger than 0.227 eV. The products from two reaction channels both prefer to form in vibrationally cold but rotationally hot states, and they both tend to forward scattering.
reaction on the diabatic potential energy surfaces of Wang et al. (Sci. Rep. 2018, 8, 17960) by using the time-dependent wave packet method. The state-to-state integral cross sections and differential cross sections of two reaction channels (NaH/NaD+D/H) are calculated for collision energy up to 0.4 eV. The cross section branching ratio indicates that the dominant reaction channel changes from NaD+H to NaH+D when the collision energy is larger than 0.227 eV. The products from two reaction channels both prefer to form in vibrationally cold but rotationally hot states, and they both tend to forward scattering.
Export citation and abstract BibTeX RIS
1. Introduction
The reactions of excited-state alkali atoms with hydrogen molecule have received much attention because of their unique advantages for studying non-adiabatic processes in reaction dynamics.[1–17] As the most intriguing characteristic, the alkali-hydrogen reactions are highly endoergic in their ground state. The energy to initiate the reaction is easily achieved via electronic excitation of the alkali atom, therefore the transitions of electronic states necessarily occur when the reaction proceeds from the excited entrance valley to the ground exit valley.[13]
For the  reaction, several excited states (32P, 42S, 42P, 32D, and 62S) of sodium atom were considered in previous studies. Regarding the collision of Na(32P)+H2, both quenching[17–24]
reaction, several excited states (32P, 42S, 42P, 32D, and 62S) of sodium atom were considered in previous studies. Regarding the collision of Na(32P)+H2, both quenching[17–24]  and reactive[15–20]
and reactive[15–20]  processes were investigated. Motzkus et al.[15] demonstrated two-step collision process for the formation of sodium hydride from the collision of Na(3p)+H2, where the vibrationally excited hydrogen molecule from first quenching process plays a significant role. The rate for NaH formation was determined by rate-equation-model based on the two-step reaction model, and other reaction schemes were ruled out. Regarding the
processes were investigated. Motzkus et al.[15] demonstrated two-step collision process for the formation of sodium hydride from the collision of Na(3p)+H2, where the vibrationally excited hydrogen molecule from first quenching process plays a significant role. The rate for NaH formation was determined by rate-equation-model based on the two-step reaction model, and other reaction schemes were ruled out. Regarding the  reaction, Bililign et al.[1,2] observed a bimodal rotational state distribution for the NaH products, which was attributed to the two different reaction pathways. The side-on-attack mechanism leads to highly rotationally excited products, while the end-on-attack mechanism generates products with low-rotational excitation. Motzkus et al.[17] performed comparative studies for the two previous reactions using several nonlinear techniques. Consequently, vibrational state distributions of NaH from two reactions were determined. The time scale of NaH formation revealed that the
reaction, Bililign et al.[1,2] observed a bimodal rotational state distribution for the NaH products, which was attributed to the two different reaction pathways. The side-on-attack mechanism leads to highly rotationally excited products, while the end-on-attack mechanism generates products with low-rotational excitation. Motzkus et al.[17] performed comparative studies for the two previous reactions using several nonlinear techniques. Consequently, vibrational state distributions of NaH from two reactions were determined. The time scale of NaH formation revealed that the  reaction is direct. Chang et al.[4] obtained rotational and vibrational state distributions of NaH from the reactions of Na(42S, 32D and 62S) plus H2. A bimodal rotational feature was found in the 42S and 32D reaction, which is similar to the 42P reaction. However, rotationally cold but vibrationally hot product population was found in the 62S reaction which was explained by the collinear abstraction mechanism. The results conclude that the increasing atomic size of Na may hinder the insertion reaction mechanism.
reaction is direct. Chang et al.[4] obtained rotational and vibrational state distributions of NaH from the reactions of Na(42S, 32D and 62S) plus H2. A bimodal rotational feature was found in the 42S and 32D reaction, which is similar to the 42P reaction. However, rotationally cold but vibrationally hot product population was found in the 62S reaction which was explained by the collinear abstraction mechanism. The results conclude that the increasing atomic size of Na may hinder the insertion reaction mechanism.
In this work, we intend to investigate the dynamics of the  reaction. The reaction involves two coupled potential energy surfaces (PESs), thus diabatic PESs should be considered in the dynamical calculations. Recently, a new set of highly accurate diabatic PESs (called WYYC[16] PESs) of the NaH2 system was developed by Wang et al.[16] and the dynamic studies were carried out for the
reaction. The reaction involves two coupled potential energy surfaces (PESs), thus diabatic PESs should be considered in the dynamical calculations. Recently, a new set of highly accurate diabatic PESs (called WYYC[16] PESs) of the NaH2 system was developed by Wang et al.[16] and the dynamic studies were carried out for the  reaction. For dynamical calculations of state-to-state reactions, the quasi-classical-trajectory[25–28] method and time-dependent wave packet[29–37] (TDWP) method have been used widely. The TDWP method has unique advantage for the dynamical study of non-adiabatic reactions. In the present work, the dynamical calculations of the
reaction. For dynamical calculations of state-to-state reactions, the quasi-classical-trajectory[25–28] method and time-dependent wave packet[29–37] (TDWP) method have been used widely. The TDWP method has unique advantage for the dynamical study of non-adiabatic reactions. In the present work, the dynamical calculations of the  reaction are carried out by using TDWP method based on the WYYC diabatic PESs. The rest of this paper is organized as follows. A brief description regarding TDWP method is presented in Section 2. Dynamic results and detailed discussion are performed in Section 3. Finally, some conclusions are drawn from the present study in Section 4.
reaction are carried out by using TDWP method based on the WYYC diabatic PESs. The rest of this paper is organized as follows. A brief description regarding TDWP method is presented in Section 2. Dynamic results and detailed discussion are performed in Section 3. Finally, some conclusions are drawn from the present study in Section 4.
2. TDWP method
The TDWP method is particularly powerful for studying the dynamics of state-to-state reactions and has been used to study many atom-diatom reactions[29–32] and reactions involving polyatom.[33–37] This method is also effective to study the dynamic of non-adiabatic reactions,[10,16,32] which involve several coupled PESs. The basic principle of the TDWP method is to solve the time-dependent Schrödinger equation. The initial wave packet containing all the information about reactants propagates on the PES, and the dynamic information can be extracted from total wave function after propagating for enough time. The solution of wave function at time t is given by
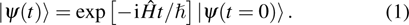
For an atom-diatom reaction of  , the time-independent Hamiltonian operator in the body-fixed reactant Jacobi coordinates R (distance of A from the center of mass of BC), r (bond length of BC) and γ (the angle between R and r) can be written as
, the time-independent Hamiltonian operator in the body-fixed reactant Jacobi coordinates R (distance of A from the center of mass of BC), r (bond length of BC) and γ (the angle between R and r) can be written as
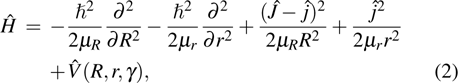
where  is the total angular momentum operator,
is the total angular momentum operator,  is the rotational angular momentum operator of BC,
is the rotational angular momentum operator of BC,  and
and  denote the corresponding reduced mass, and
denote the corresponding reduced mass, and  is the interaction potential of the reaction system.
is the interaction potential of the reaction system.
The initial wave function ψ (t=0) can be written as
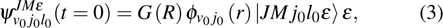
where  is the space-fixed rotational basis,
is the space-fixed rotational basis,  is a Gaussian wave packet, and
is a Gaussian wave packet, and  is the rovibrational eigenfunction of the BC molecule.
is the rovibrational eigenfunction of the BC molecule.
The second order split operator[38] is used to propagate the wave packet. During the propagation of the wave packet, the absorbing potential expressed by the R and r coordinate is used to avoid the reflection of the wave packet from the boundaries. The absorbing potential used in the TDWP calculations is in the following form:
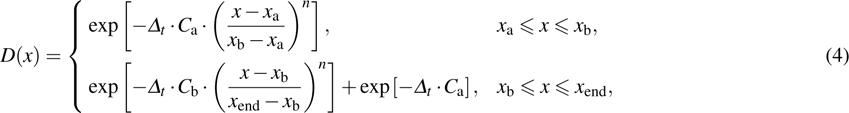
where x represents the R or r coordinate; xa, xb, and xend are the positions of absorbing potentials; Ca, Cb, and n determines the strength of the absorbing potential.
After propagating for enough time, the state-to-state S-matrix  is obtained by using the reactant coordinate-based (RCB)[40] method.
is obtained by using the reactant coordinate-based (RCB)[40] method.
The state-to-state reaction probability is obtained by using
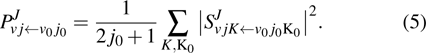
The state-to-state integral cross section (ICS) is calculated from
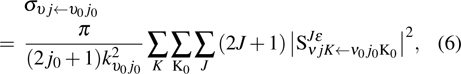
where  is the momentum in the entrance channel. The state-to-state differential cross section (DCS) is obtained from
is the momentum in the entrance channel. The state-to-state differential cross section (DCS) is obtained from
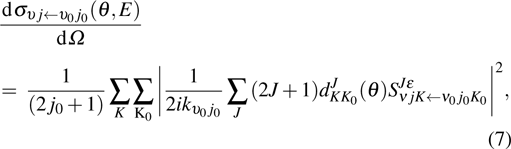
where θ is the scattering angle, and  is the reduced Wigner rotation matrix.
is the reduced Wigner rotation matrix.
For a non-adiabatic reaction correlated with two electronic states, diabatic PESs should be considered in the TDWP calculations. The  in Eq. (2) can be written as a 2 × 2 Hermitian matrix
in Eq. (2) can be written as a 2 × 2 Hermitian matrix
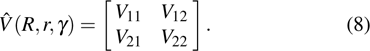
In this work, the WYYC[16] diabatic PESs are employed in the TDWP dynamical calculations for the  reaction. A more detailed description regarding the TDWP method can be found in the relevant literature.[39–42]
reaction. A more detailed description regarding the TDWP method can be found in the relevant literature.[39–42]
3. Results and discussion
In this work, state-to-state quantum dynamical calculations of the  reaction are carried out by using the TDWP method. The initial rotational–vibrational states of reagent are set to be v0 = 1, and j0 = 0. The full Coriolis-coupling is involved in the TDWP calculations. A lot of tests were carried out on the reaction probability of different total angular momentum values to obtain appropriate numerical parameters for the TDWP calculations, which are listed in Table 1. The state-to-state reaction probabilities, ICSs and DCSs are calculated for two reaction channels.
reaction are carried out by using the TDWP method. The initial rotational–vibrational states of reagent are set to be v0 = 1, and j0 = 0. The full Coriolis-coupling is involved in the TDWP calculations. A lot of tests were carried out on the reaction probability of different total angular momentum values to obtain appropriate numerical parameters for the TDWP calculations, which are listed in Table 1. The state-to-state reaction probabilities, ICSs and DCSs are calculated for two reaction channels.
Table 1. Numerical parameters used in TDWP calculations.
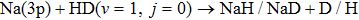
|
|
|---|---|
| Grid/basis range and size | R (bohr) 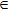 [0.1, 15.0], [0.1, 15.0],  , , 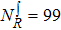
|
r (bohr) 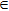 [0.1, 15.0], [0.1, 15.0], 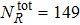 , , 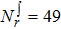
|
|
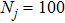
|
|
Initial wave packet ![$\exp \left[-\tfrac{{(R-{R}_{{\rm{c}}})}^{2}}{2{{\rm{\Delta }}}_{R}^{2}}\right]\cos ({k}_{0}R)$](https://content.cld.iop.org/journals/1674-1056/28/6/063401/revision1/cpb_28_6_063401_ieqn35.gif)
|
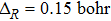
|
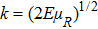 with E = 0.5 eV with E = 0.5 eV |
|
| Total propagation time | 30000 a.u. |
| Time step | 15 a.u. |
| Energy step | 0.001 eV |
| J values | 0 to 60 |
For the non-adiabatic  reaction, electronic states of reactants and products correlate with the first excited and ground electronic states of NaH2 system, respectively. This indicates that the transition between two electronic states occurs along the reaction path. In the present work, the WYYC[16] diabatic PESs are used in the TDWP calculations. Regarding the WYYC[16] PESs, the ab initio single-point energy is calculated by the multi-reference configuration interaction method with large basis sets (cc-pw-CVQZ for Na atom and aug-cc-PVQZ for H atom), and the neural network method is used to fit the PESs. The V22 surface of WYYC[16] PESs connects electronic states of reactant and products of the
reaction, electronic states of reactants and products correlate with the first excited and ground electronic states of NaH2 system, respectively. This indicates that the transition between two electronic states occurs along the reaction path. In the present work, the WYYC[16] diabatic PESs are used in the TDWP calculations. Regarding the WYYC[16] PESs, the ab initio single-point energy is calculated by the multi-reference configuration interaction method with large basis sets (cc-pw-CVQZ for Na atom and aug-cc-PVQZ for H atom), and the neural network method is used to fit the PESs. The V22 surface of WYYC[16] PESs connects electronic states of reactant and products of the  reaction. Therefore, the initial wave packet is constructed on the V22 surface and product information was collected at the V22 surface after propagating for enough time in the TDWP calculations. For a better description of the non-adiabatic dynamics in the
reaction. Therefore, the initial wave packet is constructed on the V22 surface and product information was collected at the V22 surface after propagating for enough time in the TDWP calculations. For a better description of the non-adiabatic dynamics in the  reaction, a schematic energy diagram of the reactants and products is shown in Fig. 1 based on the WYYC[16] PESs. Blue and red lines represent V11 and V22 surface of WYYC[16] diabatic PESs, respectively. The cross of V11 and V22 curves indicates the transition between two electronic states along the reaction path. As seen from product energy diagram, the product channel of NaD+H opens easier than that of NaH+D, because vibrational constant of NaD molecule is smaller than that of NaH. There still exists a threshold in reaction when HD is excited to v = 1 state; however, the threshold disappears when HD is excited above the v = 2 state. Moreover, it should be noted that there is a potential well along the reaction path, which may generate a long-lived complex. A more detailed description regarding WYYC diabatic PESs could be found in Ref. [16].
reaction, a schematic energy diagram of the reactants and products is shown in Fig. 1 based on the WYYC[16] PESs. Blue and red lines represent V11 and V22 surface of WYYC[16] diabatic PESs, respectively. The cross of V11 and V22 curves indicates the transition between two electronic states along the reaction path. As seen from product energy diagram, the product channel of NaD+H opens easier than that of NaH+D, because vibrational constant of NaD molecule is smaller than that of NaH. There still exists a threshold in reaction when HD is excited to v = 1 state; however, the threshold disappears when HD is excited above the v = 2 state. Moreover, it should be noted that there is a potential well along the reaction path, which may generate a long-lived complex. A more detailed description regarding WYYC diabatic PESs could be found in Ref. [16].

Fig. 1. Energy diagram of reactants, products, and the most possible reaction path on WYYC diabatic PESs. Blue and red line represent V11 and V22 elements of WYYC PESs, respectively.
Download figure:
Standard imageThe reaction probabilities of two reaction channels from the  reaction are shown in Fig. 2 each as a function of collision energy at several selected J values. Many oscillation peaks are found in reaction probability curves, which can be attributed to the dynamical resonances. The reaction threshold becomes larger and the values of reaction probabilities decrease as the J value increases, which is attributed to the increasing centrifugal potential energy in the total Hamiltonian. Due to the fact that the increased centrifugal potential energy can reduce the depth of potential well in the reaction path, the oscillations in the reaction probabilities gradually subside as the J value increases. In addition to these similarities, there are also some differences in reaction probability between two reaction channels. The reaction threshold of NaD+H channel (almost 0.186 eV) is lower than that of NaH+D channel (almost 0.206 eV) because of the difference in zero-energy point between NaD (almost 0.057 eV) and NaH (almost 0.077 eV) molecule. This is consistent with the energy diagram as shown in Fig. 1. Moreover, the threshold of the J = 60 partial wave for the NaD+H product channel is approximately 0.45 eV, which is higher than that of NaH+D product channel (approximately 0.4 eV). The convergence of reaction probability for the NaD+H channel is faster than that of NaH+D channel, which may be attributed to the different reduced mass in centrifugal potential at product Jacobi coordinates.
reaction are shown in Fig. 2 each as a function of collision energy at several selected J values. Many oscillation peaks are found in reaction probability curves, which can be attributed to the dynamical resonances. The reaction threshold becomes larger and the values of reaction probabilities decrease as the J value increases, which is attributed to the increasing centrifugal potential energy in the total Hamiltonian. Due to the fact that the increased centrifugal potential energy can reduce the depth of potential well in the reaction path, the oscillations in the reaction probabilities gradually subside as the J value increases. In addition to these similarities, there are also some differences in reaction probability between two reaction channels. The reaction threshold of NaD+H channel (almost 0.186 eV) is lower than that of NaH+D channel (almost 0.206 eV) because of the difference in zero-energy point between NaD (almost 0.057 eV) and NaH (almost 0.077 eV) molecule. This is consistent with the energy diagram as shown in Fig. 1. Moreover, the threshold of the J = 60 partial wave for the NaD+H product channel is approximately 0.45 eV, which is higher than that of NaH+D product channel (approximately 0.4 eV). The convergence of reaction probability for the NaD+H channel is faster than that of NaH+D channel, which may be attributed to the different reduced mass in centrifugal potential at product Jacobi coordinates.

Fig. 2. The reaction probabilities of the two reaction channels for  reaction at several selected J values.
reaction at several selected J values.
Download figure:
Standard imageThe state-to-state ICSs and DCSs are calculated for collision energy up to 0.4 eV based on the convergence of reaction probability at the maximum value of J. The total and product vibrationally state-resolved ICSs of two product channels from the  reaction are shown in Fig. 3. The ICS curves are very smooth and increase monotonically as the collision energy increases. The vibrationally excited products rise with the increase of collision energy. Due to the vibrational constant of NaD being smaller than that of NaH, the opened channels for NaD+H are more than for NaH+D. Thresholds of ICSs are consistent with those shown in the reactant and product energy diagram. From the productʼs vibrational state-resolved ICSs, the NaD and NaH products both prefer to form in the vibrational ground state in the whole range of the calculated collision energy. As seen from total ICSs, the NaD+H reaction channel opens easier than the NaH+D reaction channel. However, the NaH+D channel gradually overtakes the dominant position as collision energy increases. To clarify this competition, the cross-section branching ratio ICS(NaH)/ICS(NaD) is shown in Fig. 4 as a function of collision energy. At low collision energy (
reaction are shown in Fig. 3. The ICS curves are very smooth and increase monotonically as the collision energy increases. The vibrationally excited products rise with the increase of collision energy. Due to the vibrational constant of NaD being smaller than that of NaH, the opened channels for NaD+H are more than for NaH+D. Thresholds of ICSs are consistent with those shown in the reactant and product energy diagram. From the productʼs vibrational state-resolved ICSs, the NaD and NaH products both prefer to form in the vibrational ground state in the whole range of the calculated collision energy. As seen from total ICSs, the NaD+H reaction channel opens easier than the NaH+D reaction channel. However, the NaH+D channel gradually overtakes the dominant position as collision energy increases. To clarify this competition, the cross-section branching ratio ICS(NaH)/ICS(NaD) is shown in Fig. 4 as a function of collision energy. At low collision energy ( ), branching ratio is lower than 1.0 and increases monotonically with collision energy increasing. For high collision energy (
), branching ratio is lower than 1.0 and increases monotonically with collision energy increasing. For high collision energy ( ), branching ratio is larger than 1.0 and fluctuates around 1.3. Therefore, the NaD+H product channel is dominant in the Na(3p)+HD(v = 1) reaction at low collision energy (
), branching ratio is larger than 1.0 and fluctuates around 1.3. Therefore, the NaD+H product channel is dominant in the Na(3p)+HD(v = 1) reaction at low collision energy ( ), and it is surpassed by NaH+D channel as collision energy increases. A similar competition between two reaction channels was found in the Au+HD reaction[43] and it was explained as the fact that the D atom can easily get away from the potential well because of its larger mass than that of the H atom. This explanation is also applicable to the
), and it is surpassed by NaH+D channel as collision energy increases. A similar competition between two reaction channels was found in the Au+HD reaction[43] and it was explained as the fact that the D atom can easily get away from the potential well because of its larger mass than that of the H atom. This explanation is also applicable to the  reaction.
reaction.
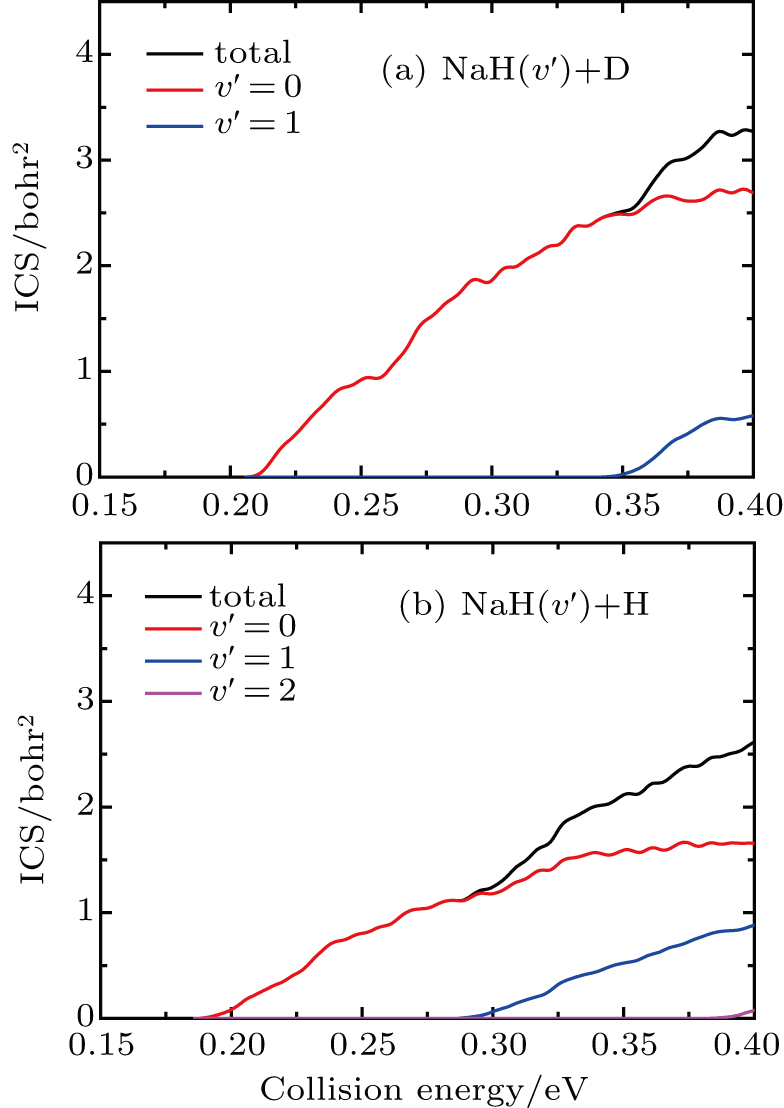
Fig. 3. Total and product vibrational state-resolved ICSs of two reaction channels for  reaction.
reaction.
Download figure:
Standard image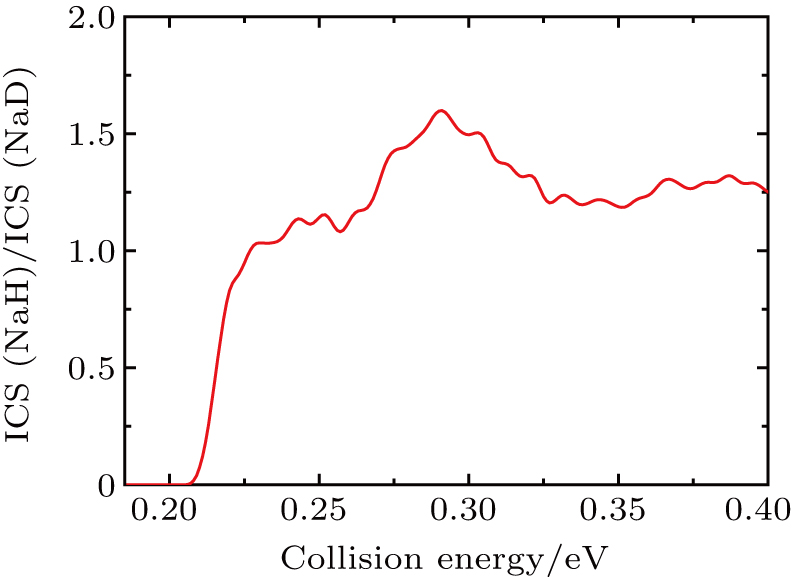
Fig. 4. The cross-section branching ratio ICS(NaH)/ICS(NaD) of the  reaction versus collision energy.
reaction versus collision energy.
Download figure:
Standard imageTo obtain more details regarding energy distribution of products from the Na(3p)+HD(v = 1) reaction, rotational state distributions of products at some values of selected collision energy are shown in Fig. 5. Only vibrational ground state products are depicted in Fig. 5 because products primarily form in the vibrational ground state, as mentioned above. As seen from Fig. 5, products of two reaction channels both prefer to form in rotationally excited states that are different from the vibrationally exited states. The product rotational states' distributions become broader and the maximum populations of  become larger as collision energy increases. The rotational state distribution of NaD is broader than that of NaH and the maximum population of
become larger as collision energy increases. The rotational state distribution of NaD is broader than that of NaH and the maximum population of  in the NaD is larger than that in the NaH at the same collision energy. This is attributed to fact that the rotational constant of NaD is smaller than that of NaH, therefore, the more the channels are opened for NaD, the larger the product rotational state density will be than that of NaH at the same collision energy.
in the NaD is larger than that in the NaH at the same collision energy. This is attributed to fact that the rotational constant of NaD is smaller than that of NaH, therefore, the more the channels are opened for NaD, the larger the product rotational state density will be than that of NaH at the same collision energy.
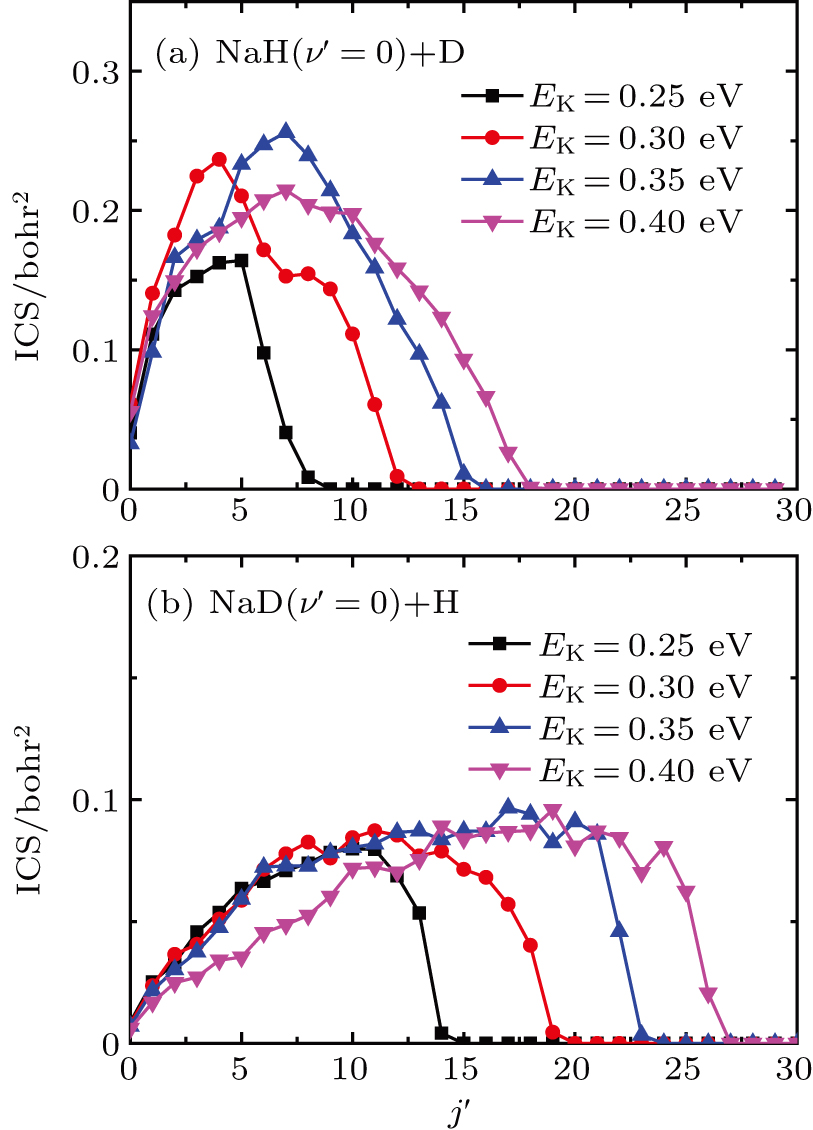
Fig. 5. Rotational state distributions of products from the  reaction respectively at four values of collision energy (0.25, 0.30, 0.35, and 0.40 eV).
reaction respectively at four values of collision energy (0.25, 0.30, 0.35, and 0.40 eV).
Download figure:
Standard imageThe three-dimensional total DCSs of two reaction channels from the  reaction are shown in Fig. 6. There are many peaks at extreme angles 0° and 180° which are corresponding to forward and backward scattering, respectively. The products of two reaction channels both prefer forward scattering, especially at low collision energy. Significant forward scattering peaks reveal that the reaction is dominated by the direct reaction mechanism. Small backward scattering peaks rise as the collision energy increases, which may be attributed to the opening collinear abstraction reaction channel. To understand the information regarding product scattering direction in depth, the product state-resolved angular distributions of two product channels at several values of selected collision energy are shown in Fig. 7. The forward scattering products from the NaH+D channel are mainly at lower rotational states (
reaction are shown in Fig. 6. There are many peaks at extreme angles 0° and 180° which are corresponding to forward and backward scattering, respectively. The products of two reaction channels both prefer forward scattering, especially at low collision energy. Significant forward scattering peaks reveal that the reaction is dominated by the direct reaction mechanism. Small backward scattering peaks rise as the collision energy increases, which may be attributed to the opening collinear abstraction reaction channel. To understand the information regarding product scattering direction in depth, the product state-resolved angular distributions of two product channels at several values of selected collision energy are shown in Fig. 7. The forward scattering products from the NaH+D channel are mainly at lower rotational states ( ). However, the forward scattering products from the NaD+H channel each have a wide rotational state distribution. The forward scattering products from two reaction channels both can be excited to higher rotational excited states as the collision energy increases, which is consistent with the above discussion. Moreover, many oscillations are found along the scattering angle. A similar phenomenon was observed in the
). However, the forward scattering products from the NaD+H channel each have a wide rotational state distribution. The forward scattering products from two reaction channels both can be excited to higher rotational excited states as the collision energy increases, which is consistent with the above discussion. Moreover, many oscillations are found along the scattering angle. A similar phenomenon was observed in the  reaction,[44] and the observed forward angular oscillations were explained by the contribution of partial waves. The period
reaction,[44] and the observed forward angular oscillations were explained by the contribution of partial waves. The period  in the angular oscillation can be used to estimate which J partial wave has primary contribution by using
in the angular oscillation can be used to estimate which J partial wave has primary contribution by using  .[44] As seen from Fig. 7, the period
.[44] As seen from Fig. 7, the period  decreases as the collision energy increases. This indicates that the J value of partial wave contributing to the oscillations increases as the collision energy increases.
decreases as the collision energy increases. This indicates that the J value of partial wave contributing to the oscillations increases as the collision energy increases.
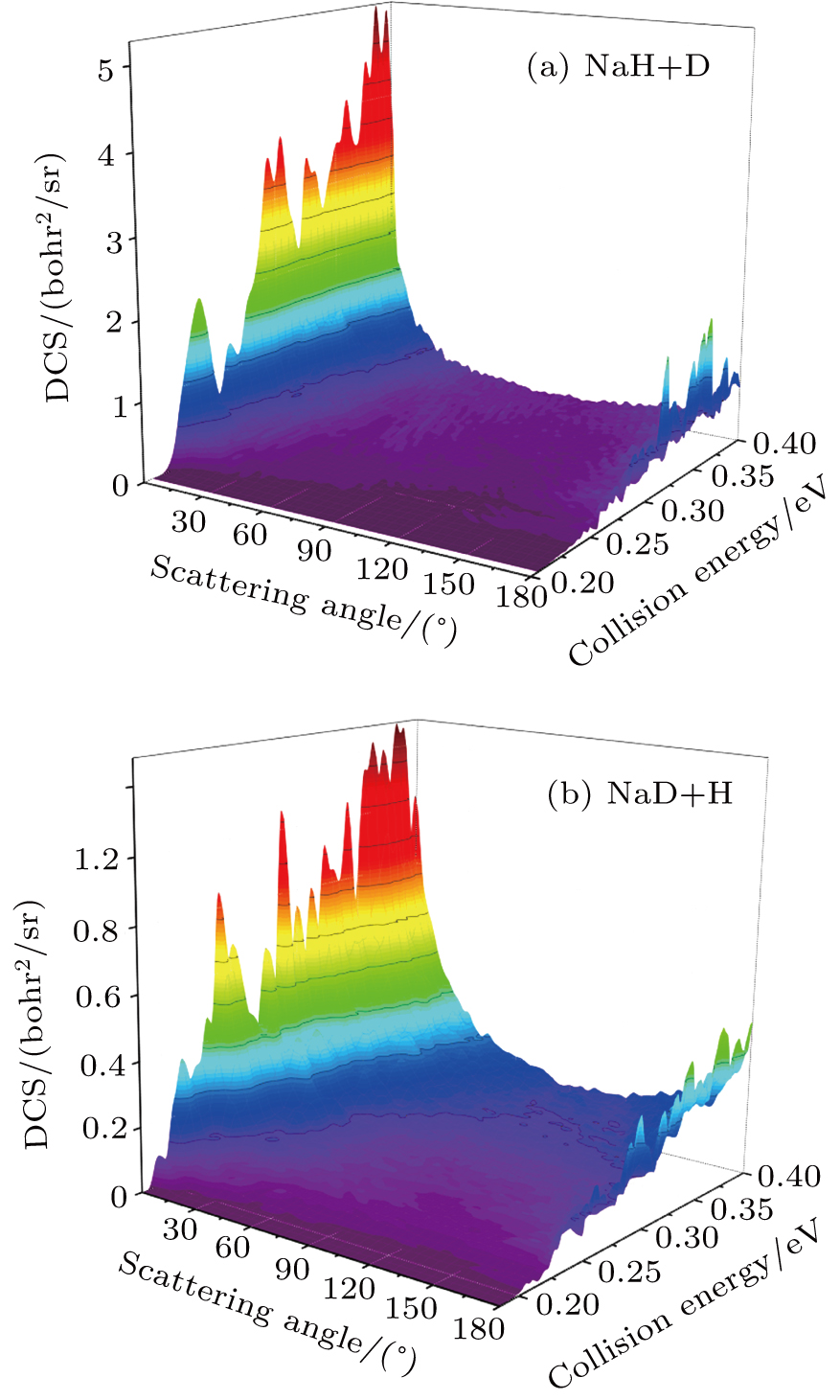
Fig. 6. Three-dimensional DCSs of two reaction channels for  reaction versus collision energy and scattering angle.
reaction versus collision energy and scattering angle.
Download figure:
Standard image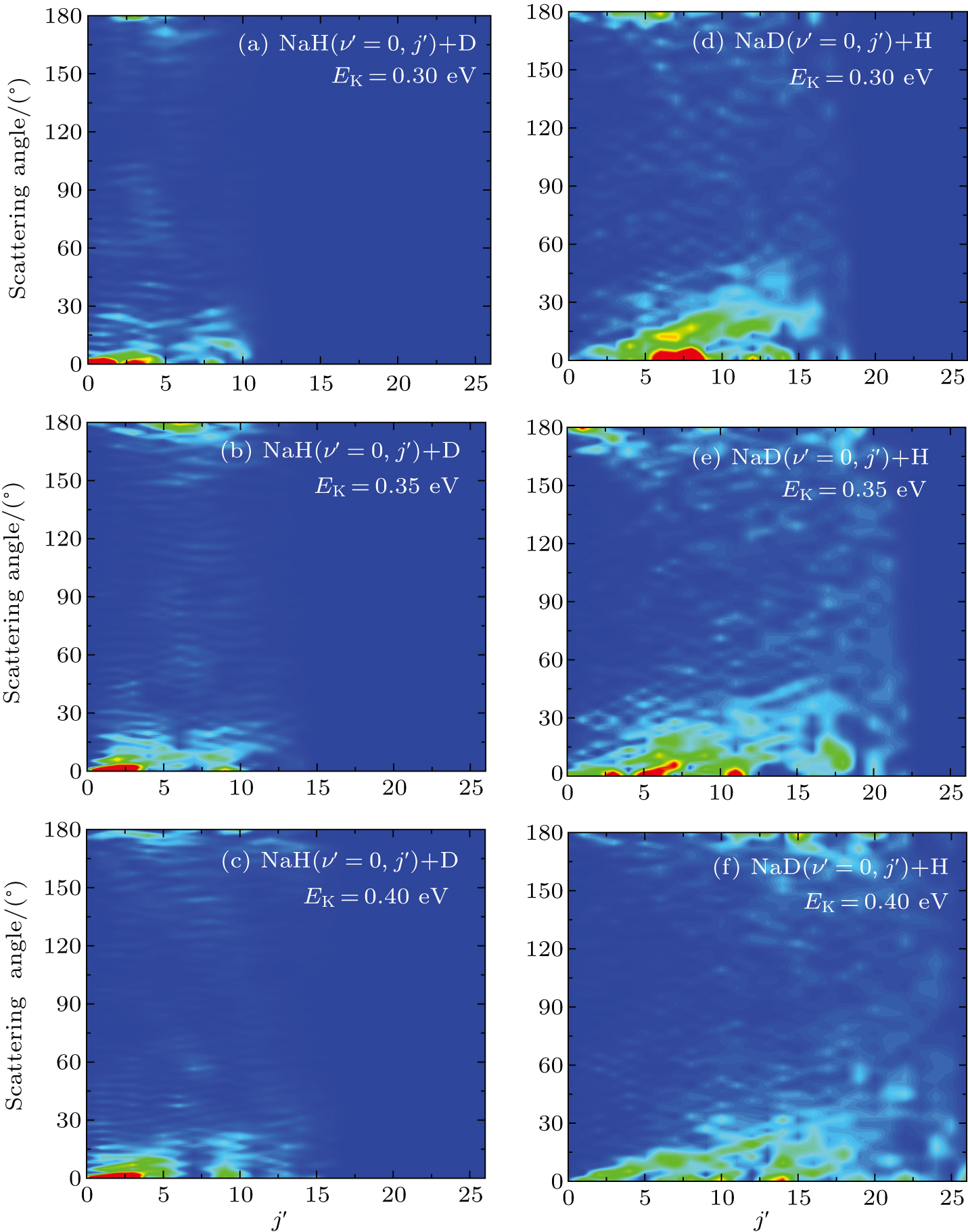
Fig. 7. State-to-state DCSs of two reaction channels for  reaction at three collision energies (0.3, 0.35, and 0.4 eV).
reaction at three collision energies (0.3, 0.35, and 0.4 eV).
Download figure:
Standard image4. Conclusions
In this work, the dynamics of the non-adiabatic  reaction are investigated using the TDWP method. The state-to-state reaction probabilities, ICSs and DCSs of two product channels from the Na(3p)+HD(v = 1, j = 0) reaction are calculated. The threshold of the NaD+H product channel is lower than that of NaH+D because of the difference in zero-point energy between NaD and NaH. The product vibrational-state resolved ICSs show that the products of two reaction channels both prefer to form in vibrational ground state. However, distributions of product rotational states have peaks at excited states. The curves of total ICS indicate that there is a competition between the two product channels with collision energy changing. From the cross-section branching ratio it follows that the NaD+H channel dominates the Na(3p)+HD(v = 1) reaction at the collision energy lower than 0.227 eV, and then the NaH+D channel gradually becomes dominant as the collision energy increases. Total DCSs show that the products of two reaction channels both prefer forward scattering. The forward scattering NaH products mainly populate at lower rotational excited sates, while NaD products have a broad rotational state distribution.
reaction are investigated using the TDWP method. The state-to-state reaction probabilities, ICSs and DCSs of two product channels from the Na(3p)+HD(v = 1, j = 0) reaction are calculated. The threshold of the NaD+H product channel is lower than that of NaH+D because of the difference in zero-point energy between NaD and NaH. The product vibrational-state resolved ICSs show that the products of two reaction channels both prefer to form in vibrational ground state. However, distributions of product rotational states have peaks at excited states. The curves of total ICS indicate that there is a competition between the two product channels with collision energy changing. From the cross-section branching ratio it follows that the NaD+H channel dominates the Na(3p)+HD(v = 1) reaction at the collision energy lower than 0.227 eV, and then the NaH+D channel gradually becomes dominant as the collision energy increases. Total DCSs show that the products of two reaction channels both prefer forward scattering. The forward scattering NaH products mainly populate at lower rotational excited sates, while NaD products have a broad rotational state distribution.
Footnotes
- *
Project supported by the National Natural Science Foundation of China (Grant No. 11774043).