



Abstract
Stabilization of magnetic order in clusters/nanoparticles at elevated temperatures is a fundamentally challenging problem. The magnetic anisotropy energy (MAE) that prevents the thermal fluctuations of the magnetization direction can be around 1–10 K in free transition metal clusters of around a dozen atoms. Here we demonstrate that a graphene support can lead to an order of magnitude enhancement in the anisotropy of supported species. Our studies show that the MAE of supported Co5 and Co13 clusters on graphene increase by factors of 2.6 and 25, respectively. The enhancement is linked to the splitting of selected electronic orbitals that leads to the different orbital contributions along the easy and hard axis. The conductive support enables a magnetic interaction between the deposited species and the nature of the magnetic interaction can be controlled by the separation between supported clusters or by vacancies offering an unprecedented ability to tune characteristics of assemblies.
Export citation and abstract BibTeX RIS
1. Introduction
The principal quantity controlling the applications of magnetic clusters/nanoparticles is the magnetic anisotropy energy (MAE) [1–3] that determines the minimum energy for disorienting the direction of magnetization [4, 5]. For small clusters containing around dozen atoms, the MAE is only of the order of few kelvins and one of the customary approaches to increase the MAE is then to deposit clusters on substrates like Pt marked by strong spin–orbit interaction (SOI) [4, 6]. In this work, we propose an alternate approach that enhances intrinsic anisotropy of individual clusters and further allows assemblies with magnetic interaction between them. These features emerge on a substrate that is neither magnetic nor has significant SOI. We show that Co5 and Co13 clusters, that have a low magnetic anisotropy of 0.9 meV (10.4 K) and 0.12 meV (1.4 K) respectively, undergo substantial enhancement of MAE when supported on graphene. The enhancement is driven by the modest interaction that changes the atomic structure and couples to selected orbitals to stabilize the orientation of the magnetic moment via the intrinsic SOI. The present work complements a recent development where metal-carbon nanoparticles with size ≈8 nm have been found to have blocking temperature (TB) in excess of 570 K [5]. These nanoparticles have layers of Co atoms separated by carbon layers and our studies have shown that the large MAE is driven by the reduced mixing between C and Co states. In fact, our calculated anisotropies were within a few percent of the experimental values.
In this work, we examine Co clusters supported on graphene. Graphene is an ideal substrate as it is strong and non-reactive. In addition, it is conducting so that the deposited species can be accessed in experiment [7, 8]. In general, the bonding with C in metal carbides quenches the magnetic moment of transition metal (TM) atoms [9]. However, since the C atoms in graphene are strongly bonded, the deposited clusters are expected to bind weakly preventing substantial reduction of the moment. The mild interaction, however, could affect MAE as the binding with C-p electrons could provide directionality to the d-states of the TM clusters. Co5 and Co13 clusters were selected as prototype systems to demonstrate the observed effect, since the free clusters have low MAE. We show that the deposited species not only exhibit enhanced anisotropy, the interaction between the deposited species can be modified by separation and by creating defects in the support.
2. Computational method
The magnetic and electronic properties were probed using density functional theory (DFT) within generalized gradient approximation (GGA) using the parameterizations by Perdew et al [10]. We have used the Vienna ab initio simulation package [11, 12] for our calculations which uses the plane wave basis set. For the case of strong correlations, we used a GGA + U approach [13] with a U value of 4.0 eV for Co 3d-states. The projector-augmented wave method [14] was used to represent the electron–ion interaction and the valence states of Co and C were described by [Ar]  , and [He]
, and [He]  , respectively. The energy cutoff for the plane-wave basis was set to be 400 eV. The calculations for free Co5 and Co13 clusters are done using simulation box of size (12×12×12) Å3 and (15×15×15) Å3, respectively, which is sufficiently large to minimize the interaction between the periodic images.
, respectively. The energy cutoff for the plane-wave basis was set to be 400 eV. The calculations for free Co5 and Co13 clusters are done using simulation box of size (12×12×12) Å3 and (15×15×15) Å3, respectively, which is sufficiently large to minimize the interaction between the periodic images.
In order to calculate the MAE, we have performed non-self consistent calculation with the SOI included where the charge density is kept fixed as obtained from the self-consistent scalar relativistic calculation. The details of the calculational procedure of MAE has been published elsewhere [21]. In order to calculate MAE and the distance-dependent exchange energies for deposited clusters, we have used (5 × 3) and (14 × 6) supercells of graphene sheets constructed from an orthorhombic primitive cell consisting of 60 and 336 carbon atoms, respectively. The length of the supercell along the direction perpendicular to graphene plane is set to 23 Å. While the free cluster calculations are performed using only one k-point, i.e., at the Γ-point, a k-mesh of (3 × 3 ×1) was used for graphene-supported clusters within the Mokhorst–Pack scheme to generate the special k-points for constructing the charge density. The structural optimization is done using the conjugate gradient method. The tolerance for the total energy change for structural optimization was set to 10−6 eV. The electronic density of states (DOS) are calculated with Gaussian broadening parameter of 0.05 eV. The local magnetic moments are calculated by the integration of the spin-density over atom-centered spheres with radius 1.302 and 0.863 Å for Co and C atoms, respectively.
Previous studies have shown that gradient corrected functionals [10] used in this work can provide quantitative estimates for several properties of free clusters or their assemblies including nanomagnets where the magnetic clusters are linked by organic linkers [15]. One such example is a Mn12O12 acetate nanomagnet where Pederson and Khanna obtained a MAE of 56 K, close to the experimental value of 55.6 K. The functionals also lead to electronic band gaps in cluster assembled materials within few percent of the experimentally measured values [16]. The success of the functionals in describing cluster properties could be due to the localization of charge in clusters that reduces the delocalization errors known to be one of the principal sources of failure of DFT in extended systems [17]. For supported clusters, Co5 and Co13 were placed on the graphene sheet at various locations and with different orientations, to determine the most stable configuration.
3. Results and discussions
The lowest energy structures for the graphene-supported clusters as well as the free ones along with their magnetic moments are shown in figure 1. Previous studies on free clusters have indicated that Co5 has a triangular bipyramidal structure while the Co13 has an icosahedral shape marked by high symmetry and stability [18].

Figure 1. Geometrically optimized structures of free (a) and (b) and graphene-supported (c) and (d) Co5 and Co13 showing the onsite spin moments (in  ) mi, where 'i' denotes the atom index. The light (orange) and dark (black) balls denote the Co and C atoms, respectively. The blue arrows represent the easy (E) and hard axis (H) for each case.
) mi, where 'i' denotes the atom index. The light (orange) and dark (black) balls denote the Co and C atoms, respectively. The blue arrows represent the easy (E) and hard axis (H) for each case.
Download figure:
Standard image High-resolution imageFor a Co5 cluster, two geometries are examined, namely, the trigonal bipyramid and a square pyramid. A distorted-triangular bipyramid structure for a free Co5 is found to be 0.14 eV lower in energy than the square pyramidal structure which is in agreement with previous studies [19]. Assuming a perfect triangular bipyramid with bulk Co lattice spacing that corresponds to a Co–Co distance ( ) of 2.50 Å, the two apex atoms with three-fold coordination in the trigonal bipyramid structure contracted to a separation from 3.80 Å in the ideal structure to 3.11 Å with an average nearest neighbor
) of 2.50 Å, the two apex atoms with three-fold coordination in the trigonal bipyramid structure contracted to a separation from 3.80 Å in the ideal structure to 3.11 Å with an average nearest neighbor  of 2.18 Å. The remaining three sites (atoms in the basal plane) with four-fold coordination show an expansion of average
of 2.18 Å. The remaining three sites (atoms in the basal plane) with four-fold coordination show an expansion of average  from 2.50 to 2.64 Å. Two
from 2.50 to 2.64 Å. Two  in the basal plane are of length 2.64 Å, while the other
in the basal plane are of length 2.64 Å, while the other  is of length 2.67 Å, which is reminiscent of Jahn–Teller distortion in clusters [20]. The geometry optimization of free Co13 cluster results in a slightly distorted icosahedral structure compared to its ideal structure [21] where the average radial and outer-shell bond lengths increase by 0.43% and 0.81% compared to the corresponding bond lengths of the ideal icosahedron. The key issue is the change in atomic structure as the clusters are supported on graphene. For Co5, the optimized structure is reminiscent of a square pyramid, where the apex Co atom is bonded to the six-fold hollow site of graphene with the average
is of length 2.67 Å, which is reminiscent of Jahn–Teller distortion in clusters [20]. The geometry optimization of free Co13 cluster results in a slightly distorted icosahedral structure compared to its ideal structure [21] where the average radial and outer-shell bond lengths increase by 0.43% and 0.81% compared to the corresponding bond lengths of the ideal icosahedron. The key issue is the change in atomic structure as the clusters are supported on graphene. For Co5, the optimized structure is reminiscent of a square pyramid, where the apex Co atom is bonded to the six-fold hollow site of graphene with the average  of 2.25 Å. While the
of 2.25 Å. While the  for atoms in the square plane is 2.16 Å, the
for atoms in the square plane is 2.16 Å, the  from apex to basal plane is elongated to 2.40 Å. For supported Co13, the icosahedral symmetry of the cluster is substantially reduced. The most stable structure has two of the Co atoms of cluster bonded to the bridge site of the graphene sheet. The radial
from apex to basal plane is elongated to 2.40 Å. For supported Co13, the icosahedral symmetry of the cluster is substantially reduced. The most stable structure has two of the Co atoms of cluster bonded to the bridge site of the graphene sheet. The radial  in the adsorbed Co13 vary from (2.30 to 2.50) Å whereas the outer shell
in the adsorbed Co13 vary from (2.30 to 2.50) Å whereas the outer shell  shows a variation from (2.31 to 2.78) Å. Such large variations in
shows a variation from (2.31 to 2.78) Å. Such large variations in  w.r.t. the free Co13 cluster indicate a strong structural distortion.
w.r.t. the free Co13 cluster indicate a strong structural distortion.
The strength of the interaction between the cluster and graphene determines the stability of cluster. Therefore, we calculated the binding energy (BE) via the energy difference:
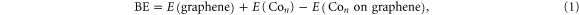
where, n is the number of Co atoms in the cluster. E(graphene), E(Con), and E(Con on graphene) are the total energies of the graphene sheet, free Con cluster, and that of the supported species, respectively. We find that Co5 and Co13 are bound to graphene with a BE of 1.02 and 2.24 eV, respectively, which are lower than the BE of a free Co–C dimer (3.20 eV). This indicates that the clusters are only moderately bound to graphene but enough to prevent the detachment and migration of clusters on the surface under ambient temperature. The binding reduces the spin moments from 13  (free Co5) to 11
(free Co5) to 11  and from 31
and from 31  (free Co13) to 21
(free Co13) to 21  for the deposited species. The reduction in the magnetic moment is non-uniform across the cluster and the maximum reduction occurs at sites linked to graphene as the bonding of the Co-d states with C- sp states reduces the magnetic moment. Figures 1(c) and (d) quantifies this reduction for various sites in Co5 and Co13, respectively.
for the deposited species. The reduction in the magnetic moment is non-uniform across the cluster and the maximum reduction occurs at sites linked to graphene as the bonding of the Co-d states with C- sp states reduces the magnetic moment. Figures 1(c) and (d) quantifies this reduction for various sites in Co5 and Co13, respectively.
The change in geometry due to binding affects the electronic structure that determines the MAE. In small clusters, the main contribution to MAE comes from the SOI which can be calculated by perturbation approach using the charge density obtained from scalar-relativistic treatment. The MAE was obtained by using the magnetic force theorem [23], where we calculated the difference between the maximum and minimum values of the ( )-dependent total energy, i.e., MAE
)-dependent total energy, i.e., MAE ![$=[{{E}_{{\rm max} }}(\theta ,\phi )-{{E}_{{\rm min} }}(\theta ,\phi )]$](https://content.cld.iop.org/journals/1367-2630/17/5/053052/revision1/njp514134ieqn20.gif) [21]. The SO contribution to energy was calculated by constraining the magnetic moment along different directions of azimuthal angle ϕ in steps of 15°. For each ϕ, the polar angle θ was varied in steps of 10°. For free Co5 and Co13, we obtain MAE values of 0.9 and 0.12 meV, respectively. The calculated value of Co13 is found to be larger compared to the previous study [21], as the clusters in the present work are fully optimized without any symmetry constraints, whereas in the previous work, the structure was transformed through a path characterized by two structural parameters and a series of single point calculations were performed. It is known that the presence of structural symmetry reduced the MAE due to cancellation of symmetry of the even order terms. When the symmetry is reduced due to the complete structural optimization associated with local distortions, the lower even order terms contribute to the MAE.
[21]. The SO contribution to energy was calculated by constraining the magnetic moment along different directions of azimuthal angle ϕ in steps of 15°. For each ϕ, the polar angle θ was varied in steps of 10°. For free Co5 and Co13, we obtain MAE values of 0.9 and 0.12 meV, respectively. The calculated value of Co13 is found to be larger compared to the previous study [21], as the clusters in the present work are fully optimized without any symmetry constraints, whereas in the previous work, the structure was transformed through a path characterized by two structural parameters and a series of single point calculations were performed. It is known that the presence of structural symmetry reduced the MAE due to cancellation of symmetry of the even order terms. When the symmetry is reduced due to the complete structural optimization associated with local distortions, the lower even order terms contribute to the MAE.
In the present calculations the MAE of supported Co5 and Co13 are found to be 2.31 meV (27 K) and 2.98 meV (35 K), respectively. These values translate into an increase by factors of 2.6 and 25 compared to the free clusters for Co5 and Co13, respectively. The change in MAE is accompanied by a change in the direction of easy (E) and hard (H) axes as shown in figure 1. We found that while the spin moments remain collinear for all cases, there exists a degree of non-collinearity in orbital moments. This is in agreement with a previous study [22]. The changes in the E and H axes as well as the increase in MAE are ultimately linked to the evolution of the electronic structure. One effect of adsorption is the charge exchange with the support. A Bader analysis [24] of the resulting charges indicates a net charge transfer from the cluster to the graphene of 0.38 e and 1.16 e for Co5 and Co13, respectively, where e is the electronic charge. The numerical values of the local charge on each atom are listed in table 1.
Table 1.
The absolute values of the local spin  and local orbital moments
and local orbital moments  for free (within paranthesis) and supported Co clusters obtained from the scalar-relativistic calculations with SOI included.
for free (within paranthesis) and supported Co clusters obtained from the scalar-relativistic calculations with SOI included.  and
and  denote the orbital moments along the easy and hard axes, respectively. Δ
denote the orbital moments along the easy and hard axes, respectively. Δ  is the change in Bader charge on each atom of the cluster. The Bader charge is calculated for Co atoms with 9 valence electrons. The atom indices are shown in figure 1 for each case.
is the change in Bader charge on each atom of the cluster. The Bader charge is calculated for Co atoms with 9 valence electrons. The atom indices are shown in figure 1 for each case.
| System | Atom index |
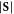
|
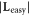
|
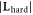
|
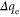
|
|---|---|---|---|---|---|
| Co5 | 1 | 1.258 (2.265) | 0.051 (0.173) | 0.061 (0.134) |
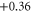
|
| 2 | 2.093 (2.265) | 0.097 (0.172) | 0.092 (0.134) |
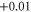
|
|
| 3 | 2.097 (2.261) | 0.098 (0.172) | 0.092 (0.182) |
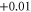
|
|
| 4 | 2.095 (2.198) | 0.091 (0.116) | 0.092 (0.107) |
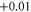
|
|
| 5 | 2.103 (2.198) | 0.092 (0.116) | 0.091 (0.107) | −0.01 | |
| Co13 | 1 | 1.776 (2.106) | 0.096 (0.108) | 0.123 (0.116) |
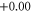
|
| 2 | 1.824 (2.106) | 0.121 (0.108) | 0.141 (0.152) | −0.03 | |
| 3 | 1.820 (2.111) | 0.108 (0.137) | 0.109 (0.112) |
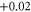
|
|
| 4 | 1.820 (2.111) | 0.115 (0.137) | 0.114 (0.149) | −0.03 | |
| 5 | 1.815 (2.114) | 0.152 (0.149) | 0.092 (0.104) |
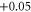
|
|
| 6 | 1.825 (2.112) | 0.104 (0.149) | 0.129 (0.151) |
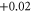
|
|
| 7 | 1.818 (2.114) | 0.166 (0.149) | 0.157 (0.151) |
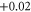
|
|
| 8 | 1.828 (2.113) | 0.114 (0.149) | 0.109 (0.105) |
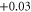
|
|
| 9 | 1.588 (2.111) | 0.114 (0.137) | 0.075 (0.149) |
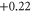
|
|
| 10 | 1.614 (2.112) | 0.130 (0.137) | 0.076 (0.111) |
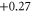
|
|
| 11 | 1.076 (2.106) | 0.065 (0.108) | 0.049 (0.152) |
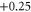
|
|
| 12 | 0.874 (2.100) | 0.058 (0.108) | 0.039 (0.115) |
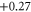
|
|
| 13 | 2.103 (1.920) | 0.025 (0.047) | 0.032 (0.046) |
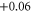
|
The significant charge transfer occurs from the Co atoms bonded to the C-atoms of graphene. This evoked the suspicion that the graphene–cluster interface might be playing an important role in determining the MAE. To gain a physical insight we used the perturbative approach as the change is small [15]. To a second order in perturbation, the change in energy,  , with the quantization axis can be expressed as
, with the quantization axis can be expressed as
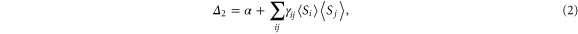
where,
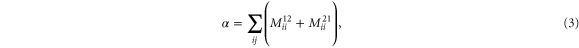
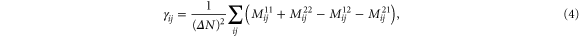
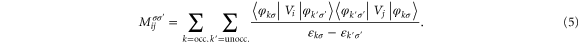
Here,
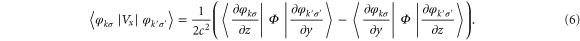
 is the Coulomb potential, Si's are the
is the Coulomb potential, Si's are the  (
( ) component of the total spin of the system along the quantization axis,
) component of the total spin of the system along the quantization axis,  is the single particle energy of the
is the single particle energy of the  state with spin σ (assigned 1 and 2) and
state with spin σ (assigned 1 and 2) and  is the number of unpaired electrons. Since the tensor
is the number of unpaired electrons. Since the tensor  (equation (4)) depends on the orbital character of the system, anisotropy depends on both orbital and spin character of the single particle levels. The matrix elements Mij involve a transition from occupied (k) to unoccupied states (k') and hence the anisotropy is sensitive to the details of the electronic structure. There are four terms coupling the occupied and unoccupied states. Two of these couple spin-up (1) or -down (2) occupied states with unoccupied states of similar spin while the remaining terms involve a spin flip between occupied and unoccupied states. The denominator in these terms involves the energy difference between occupied and unoccupied states and it is reasonable to assume that the main contribution comes from levels close to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels. The minimum energy difference corresponds to the HOMO–LUMO gap which plays a significant role (the values are listed in table 2). In order to show these competing effects, we also show the spin-polarized DOS in figure 2 for the free and deposited Co5 and Co13, respectively. For free clusters, the discrete energy levels have been broadened to fascilitate the comparison with the deposited species. The electronic structures of free Co5 and Co13 clusters lead to a grouping of levels resulting in a larger HOMO–LUMO gap of 0.43 and 0.19 eV, respectively, and the lower values for MAE as 0.9 meV (10.44 K) for Co5 and 0.12 meV (1.4 K) for Co13 clusters. As the clusters are supported on graphene, the electronic states are spread out leading to a lowering of the HOMO–LUMO gap of 0.1 eV (deposited Co5), 0.04 eV (deposited Co13) and a larger number of states close to the Fermi level.
(equation (4)) depends on the orbital character of the system, anisotropy depends on both orbital and spin character of the single particle levels. The matrix elements Mij involve a transition from occupied (k) to unoccupied states (k') and hence the anisotropy is sensitive to the details of the electronic structure. There are four terms coupling the occupied and unoccupied states. Two of these couple spin-up (1) or -down (2) occupied states with unoccupied states of similar spin while the remaining terms involve a spin flip between occupied and unoccupied states. The denominator in these terms involves the energy difference between occupied and unoccupied states and it is reasonable to assume that the main contribution comes from levels close to the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels. The minimum energy difference corresponds to the HOMO–LUMO gap which plays a significant role (the values are listed in table 2). In order to show these competing effects, we also show the spin-polarized DOS in figure 2 for the free and deposited Co5 and Co13, respectively. For free clusters, the discrete energy levels have been broadened to fascilitate the comparison with the deposited species. The electronic structures of free Co5 and Co13 clusters lead to a grouping of levels resulting in a larger HOMO–LUMO gap of 0.43 and 0.19 eV, respectively, and the lower values for MAE as 0.9 meV (10.44 K) for Co5 and 0.12 meV (1.4 K) for Co13 clusters. As the clusters are supported on graphene, the electronic states are spread out leading to a lowering of the HOMO–LUMO gap of 0.1 eV (deposited Co5), 0.04 eV (deposited Co13) and a larger number of states close to the Fermi level.
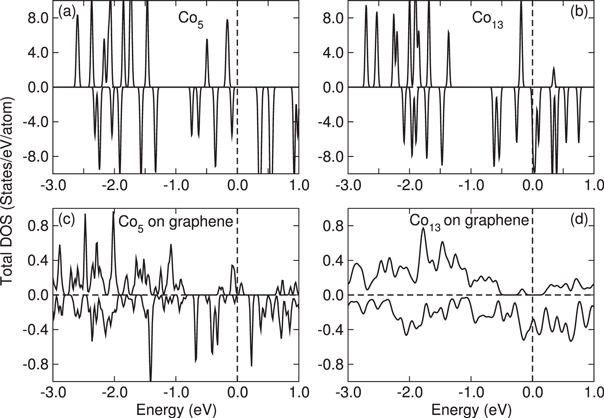
Figure 2. The spin-polarized scalar-relativistic density of states (DOS) for free Co5 and Co13 clusters shown in (a) and (b) and deposited Co5 and Co13 clusters (c) and (d). The dashed vertical line indicates the Fermi level which is set to zero.
Download figure:
Standard image High-resolution imageTable 2.
The MAE (meV), total spin moment ( ) and the HOMO–LUMO gap (eV) for free and graphene-supported Co clusters based on GGA and GGA + U calculations with U = 4 eV for Co-d orbitals.
) and the HOMO–LUMO gap (eV) for free and graphene-supported Co clusters based on GGA and GGA + U calculations with U = 4 eV for Co-d orbitals.
| System | MAE (meV) | Magnetic moment ( ) ) |
HOMO–LUMO gap (eV) | |||
|---|---|---|---|---|---|---|
| GGA | GGA + U | GGA | GGA + U | GGA | GGA + U | |
| Co5 | 0.90 | 1.54 | 13 | 13 | 0.43 | 0.52 |
| Co13 | 0.12 | 0.23 | 31 | 31 | 0.19 | 0.42 |
| Co5 on graphene | 2.31 | 0.48 | 11 | 11 | 0.10 | 0.05 |
| Co13 on graphene | 2.98 | 2.14 | 21 | 26 | 0.04 | 0.003 |
We observed that the HOMO and LUMO for free and deposited clusters are primarily composed of Co-d states. For deposited Co5, the HOMO and LUMO are composed of the states from the Co atom close to the graphene whereas, for deposited Co13, the HOMO is occupied by d-states of Co atoms that are away from graphene and the LUMO is occupied by the d states of the Co atoms close to the graphene. Such difference in the occupancies of the energy levels are probably responsible for the strong enhancement of MAE for the deposited Co13 relative to its free cluster by a factor of 25 unlike that of the adsorbed Co5 which is enhanced by a factor of 3. In addition, the levels around HOMO or LUMO have both spin-up and spin-down character and all four terms contribute to the anisotropy. For Co13 that shows the larger increase in MAE compared to free cluster, the levels in the spin-down for the supported cluster are densely populated around the HOMO and LUMO and contribute to the enhancement of MAE.
Within a second order perturbation theory, anisotropy of a system can be related to the orbital moments by the Bruno formula [25]
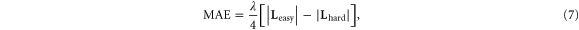
where,  and
and  are the orbital moments along the E and H axes, respectively, and λ is the SO coupling constant, which for 3d elements is about 50 meV [26]. We have calculated the projected orbital angular momentum at the various sites along the E and H axes and these are listed in table 2. For Co5, the maximum difference between
are the orbital moments along the E and H axes, respectively, and λ is the SO coupling constant, which for 3d elements is about 50 meV [26]. We have calculated the projected orbital angular momentum at the various sites along the E and H axes and these are listed in table 2. For Co5, the maximum difference between  and
and  comes from the site 1 that is coupled to the graphene sheet. A similar scenario occurs for Co13 that has multiple sites close to graphene. This would suggest that the coupling of the d-states of the cluster to the p-states of the graphene does stabilize orientation of the orbital moments along the easy axis.
comes from the site 1 that is coupled to the graphene sheet. A similar scenario occurs for Co13 that has multiple sites close to graphene. This would suggest that the coupling of the d-states of the cluster to the p-states of the graphene does stabilize orientation of the orbital moments along the easy axis.
As mentioned earlier, previous studies on Mn12O12-acetate and other nanomagnets indicate that the gradient corrected functionals can provide a reasonably accurate values for the MAE. However, we did investigate the change in MAE if an additional Hubbard term U were included in the functionals. We used a GGA + U functional with a U = 4.0 eV typical for TM systems. The effect of U is generally to shift the energy of occupied orbitals by  and those of the unoccupied orbitals by
and those of the unoccupied orbitals by  . This is the reason that the inclusion of U leads to better values for the band gaps. For MAE that is governed by the transitions from occupied to unoccupied states, the opening of the gaps might be expected to generally reduce MAE. Such a trend has been observed for the free clusters where inclusion of U leads to increase of HOMO–LUMO gap of Co5 and Co13 clusters from 0.43 eV and 0.19 eV to 0.52 eV and 0.42 eV, respectively (refer table 2). Further, the effect of U is to make the d-states more localized that can be expected to reduce the amount of quenching of the moments by the substrate and could enhance the anisotropy. For free clusters, the magnetic moment was the same as in GGA calculations, i.e., 13
. This is the reason that the inclusion of U leads to better values for the band gaps. For MAE that is governed by the transitions from occupied to unoccupied states, the opening of the gaps might be expected to generally reduce MAE. Such a trend has been observed for the free clusters where inclusion of U leads to increase of HOMO–LUMO gap of Co5 and Co13 clusters from 0.43 eV and 0.19 eV to 0.52 eV and 0.42 eV, respectively (refer table 2). Further, the effect of U is to make the d-states more localized that can be expected to reduce the amount of quenching of the moments by the substrate and could enhance the anisotropy. For free clusters, the magnetic moment was the same as in GGA calculations, i.e., 13  for Co5 and 31
for Co5 and 31  for Co13, whereas the anisotropy are calculated as 1.54 and 0.23 meV for Co5 and Co13, respectively. The reduced interaction to the substrate leads to a triangular bipyramid structure for Co5 upon deposition as in a free cluster. Furthermore, the magnetic moment of the deposited Co5 and Co13 are found to be 11
for Co13, whereas the anisotropy are calculated as 1.54 and 0.23 meV for Co5 and Co13, respectively. The reduced interaction to the substrate leads to a triangular bipyramid structure for Co5 upon deposition as in a free cluster. Furthermore, the magnetic moment of the deposited Co5 and Co13 are found to be 11  and 26
and 26  , respectively, indicating that the magnetic moment of Co13 is closer to the free cluster value than in the absence of U. The MAE for deposited Co5 and Co13 were calculated to be 0.48 and 2.14 meV, respectively. Table 2 shows a comparison of the above quantities obtained from GGA and GGA + U calculations. The MAE of Co13 is still enhanced by approximately a factor of 9 from the free cluster but the MAE of Co5 is slightly reduced indicating that the reduced interaction of the cluster from the substrate and the stabilization of the more symmetric geometry adversely affects its MAE.
, respectively, indicating that the magnetic moment of Co13 is closer to the free cluster value than in the absence of U. The MAE for deposited Co5 and Co13 were calculated to be 0.48 and 2.14 meV, respectively. Table 2 shows a comparison of the above quantities obtained from GGA and GGA + U calculations. The MAE of Co13 is still enhanced by approximately a factor of 9 from the free cluster but the MAE of Co5 is slightly reduced indicating that the reduced interaction of the cluster from the substrate and the stabilization of the more symmetric geometry adversely affects its MAE.
While a graphene surface can stabilize orientation of moments, can it also mediate interaction between the supported species? In particular, the possibility of RKKY exchange coupling between magnetic moments of defects has attracted considerable attention. It has been suggested that while the Fermi momentum in a graphene plane is zero and excitations are gapless, the inclusion of spin orbit interactions opens an energy gap at the Fermi level leading to an absence of electrons at the Fermi level. Here we consider a different situation where the cobalt clusters that interact reasonably with the graphene substrate have d-states at the Fermi energy. A Co5 cluster was positioned on the surface and a Co atom was placed on the hollow site of the sheet at increasing separations from Co5 cluster. The position of Co adatom is marked as 1–5 in figure 3(a). At each separation, we examined the stability of magnetic order by calculating the total energy difference between the ferromagnetic (FM) state and the antiferromagnetic (AFM) state where the magnetic moment of the adatom was aligned opposite to that of the magnetic moment on the Co5 cluster as
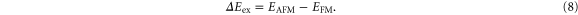
Regions where the exchange energy  is positive (negative) correspond to the situation where the FM (AFM) alignment is more stable. Figure 3(a) shows
is positive (negative) correspond to the situation where the FM (AFM) alignment is more stable. Figure 3(a) shows  as a function of separation between the center of mass of the Co5 cluster and Co adatom. It is observed that
as a function of separation between the center of mass of the Co5 cluster and Co adatom. It is observed that  changes as a function of separation, and become AFM at ≈12 Å. Thereafter it reverts back to FM.
changes as a function of separation, and become AFM at ≈12 Å. Thereafter it reverts back to FM.
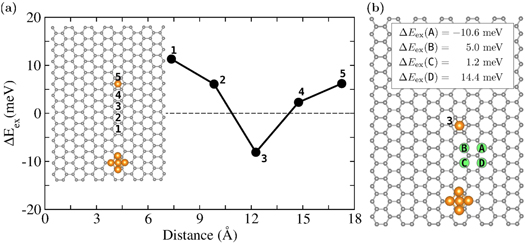
Figure 3. (a)  (defined in equation (8)) in units of meV as a function of separation between the adsorbed species. 1–5 denote the position of single Co atom w.r.t. Co5. (b) The position of vacancies (green balls) are denoted as A-D. The gray and orange balls denote the C and Co atoms, respectively.
(defined in equation (8)) in units of meV as a function of separation between the adsorbed species. 1–5 denote the position of single Co atom w.r.t. Co5. (b) The position of vacancies (green balls) are denoted as A-D. The gray and orange balls denote the C and Co atoms, respectively.
Download figure:
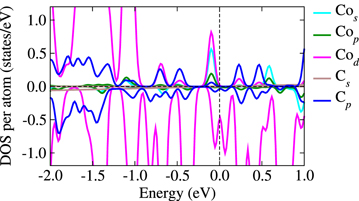
Standard image High-resolution imageTo further probe the origin of such interaction, figure 4 shows the DOS of the system with a cluster supported on graphene. It is interesting to note that there is appreciable mixing between the C p-states and the Co d-states. In fact the C p-states are polarized and mediate the magnetic interaction by coupling to d-states.
{kind=link}
{kind=link}
{kind=link}
Figure 4. The partial and site-projected DOS for the FM interaction of the adsorbed species where the Co atom is placed at a distance of 7 Å from Co5 marked as position '1' in figure 3(a). The vertical line denotes the Fermi level and is shifted to zero. The cyan, green, magenta, brown and blue curves indicate the Co-s, Co-p, Co-d, C-s and C-p states, respectively. The C states are multiplied by a factor 10.
Download figure:
Standard image High-resolution image{kind=link}
Although pristine graphene is nonmagnetic it is well known that the presence of vacancy makes graphene magnetic [27]. Therefore, we have examined if these vacancies could magnetically interact the deposited species and if the nature of interaction and its strength could be altered by the position of vacancy. To examine this, for Co5 and Co, we introduced a carbon monovacancy at four different locations marked as A–D in figure 3(b). After relaxing the structures in each case,  was calculated using equation (8) and the resulting values are shown in figure 3(b). The interaction at 12 Å is AFM without vacancy. The presence of vacancy alters the ground state from AFM to FM for most cases. For the FM case, while the presence of vacancy enhances the total magnetic moment of the system by 1
was calculated using equation (8) and the resulting values are shown in figure 3(b). The interaction at 12 Å is AFM without vacancy. The presence of vacancy alters the ground state from AFM to FM for most cases. For the FM case, while the presence of vacancy enhances the total magnetic moment of the system by 1  compared to the pristine case (12
compared to the pristine case (12  ), the total magnetic moment, however, remains unaffected for the AFM case (10
), the total magnetic moment, however, remains unaffected for the AFM case (10  ). The magnitude of onsite magnetic moment around the vacancy is calculated to be about 1
). The magnitude of onsite magnetic moment around the vacancy is calculated to be about 1  where one of the C atoms close to the vacancy exhibits the largest contribution of 0.5
where one of the C atoms close to the vacancy exhibits the largest contribution of 0.5  . This is in agreement with previous studies [27]. Since the nature of interaction and its strength can be changed by just changing the position of vacancy, therefore for an assembly of clusters the choice of separation and vacancies can be used to enhance the overall
. This is in agreement with previous studies [27]. Since the nature of interaction and its strength can be changed by just changing the position of vacancy, therefore for an assembly of clusters the choice of separation and vacancies can be used to enhance the overall  , which could help to stabilize the magnetic order against thermal fluctuation.
, which could help to stabilize the magnetic order against thermal fluctuation.
4. Summary
The present work emphasizes how a nonmagnetic support having no significant SOI can enhance the MAE in supported clusters and also allow a magnetic interaction that varies with the separation between species as well as can be controlled by defects and their location. The great advantage of graphene is that it is conducting and hence allows easy access to the deposited species. The physical origin of the observed effects is the mild hybridization between the C p-states with the Co d-states that orients the magnetization as well as acts to mediate the interaction between the supported clusters. It will be interesting to see if these effects could be also observed on similar supports including BN sheets and could be extended to other TM clusters.
Acknowledgments
The authors gratefully acknowledge support from the US Department of Energy (DOE) through grant DE-SC0006420 for this work.