


Abstract
The combination of modern scientific computing with electronic structure theory can lead to an unprecedented amount of data amenable to intelligent data analysis for the identification of meaningful, novel and predictive structure–property relationships. Such relationships enable high-throughput screening for relevant properties in an exponentially growing pool of virtual compounds that are synthetically accessible. Here, we present a machine learning model, trained on a database of ab initio calculation results for thousands of organic molecules, that simultaneously predicts multiple electronic ground- and excited-state properties. The properties include atomization energy, polarizability, frontier orbital eigenvalues, ionization potential, electron affinity and excitation energies. The machine learning model is based on a deep multi-task artificial neural network, exploiting the underlying correlations between various molecular properties. The input is identical to ab initio methods, i.e. nuclear charges and Cartesian coordinates of all atoms. For small organic molecules, the accuracy of such a 'quantum machine' is similar, and sometimes superior, to modern quantum-chemical methods—at negligible computational cost.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The societal need for novel computational tools and data treatment that serve the accelerated discovery of improved and novel materials has gained considerable momentum in the form of the materials genome initiative8. Modern electronic structure theory and computer hardware have progressed to the point where electronic properties of virtual compounds can be routinely calculated with satisfactory accuracy. For example, using quantum chemistry and distributed computing, the members of the widely advertised Harvard Clean Energy Project endeavor to calculate relevant electronic properties for millions of chromophores [1]. A more fundamental challenge persists, however: it is not obvious how to distill from the resulting data the crucial insights that relate structure to property in a predictive and quantitative manner. How are we to systematically construct robust models of electronic structure properties that properly reflect the information already obtained for thousands to millions of different chemical compounds?
With increasing numbers of data and available computational resources, increasingly sophisticated statistical data analysis, or machine learning (ML) methods, have already been applied for predicting not only outcomes of experimental measurements but also outcomes of computationally demanding high-level electronic structure calculations. In close analogy to the quantitative structure property relationships (QSPRs) prevalent in cheminformatics and bioinformatics, QSPRs can also be constructed for electronic structure properties. Examples include QSPRs for exchange-correlation potentials using neural networks (NNs) [2, 3], basis-set effects using support vector machines [4, 5] or molecular reorganization energies affecting charge transfer rates [6, 7], or for solid ternary oxides [8]. Ordinarily, these applications rely on association, using regression methods that create statistically optimized relationships between the so-called descriptor variables and the electronic property of interest. Not surprisingly, the heuristic ad hoc identification and formatting of appropriate descriptors represents a crucial and challenging aspect of any QSPR, and is to be repeated for every property and class of chemicals.
We make use of an alternative ML approach, recently introduced by some of us for the modeling of molecular atomization energies [9]. This approach is based on a strict first principles view on chemical compound space [11]. Specifically, solutions to Schrödinger's equation (SE) are inferred for organic query molecules using the same variables that also enter the electronic Hamiltonian H, i.e. nuclear charges ZI and positions RI,9 and that are mapped to the corresponding total potential energy,  [11, 12]. Unlike the aforementioned QSPRs this ML model is free of any heuristics: it exactly encodes the supervised learning problem posed by SE, i.e. instead of finding the wavefunction Ψ which maps the system's Hamiltonian to its energy, it directly maps the system to energy (based on examples given for training),
[11, 12]. Unlike the aforementioned QSPRs this ML model is free of any heuristics: it exactly encodes the supervised learning problem posed by SE, i.e. instead of finding the wavefunction Ψ which maps the system's Hamiltonian to its energy, it directly maps the system to energy (based on examples given for training),  . The employed descriptor, dubbed the 'Coulomb'-matrix, is directly obtained from {ZI,RI}. As such this constitutes a well-defined supervised-learning problem and in the limit of the converged number of training examples the ML model becomes a formally exact inductive equivalent to the deductive solution of SE. It is advantageous that the training data can come from experiment just as well as from numerical evaluation of the corresponding quantum mechanical observable using approximate wavefunctions (separated nuclear and electronic wavefunctions, Slater determinant expansions, etc), Hamiltonians (such as the Hückel or any exchange-correlation potential), and self-consistent field procedures. Building on our previously introduced work [9], we present here a more mature ML model developed to accomplish the following two additional tasks: (i) simultaneously predict a variety of different electronic properties for a single query and (ii) reach an accuracy comparable with the employed reference method used for generating the training set. The presented ML model is based on a multi-task deep artificial NN approach that captures correlations between seemingly related and unrelated properties and levels of theory. A remarkable predictive accuracy for 'out-of-sample' molecules (i.e. molecules that were not part of the training set) has been obtained through the use of random Coulomb matrices that introduce invariance with respect to atom indexing. For training, we generated a quantum chemical database containing nearly 105 entries for over 7000 stable organic molecules, made of up to seven atoms from main-group elements, consisting of C, N, O, S and Cl, saturated with hydrogen to satisfy valence rules [13, 14]. For each molecule, the atomization energy, static polarizabilities, frontier orbital eigenvalues and excitation energies and intensities have been calculated by a variety of widely used electronic structure methods, including state-of-the-art first principles methods, such as hybrid density-functional theory and the many-body single particle Green's function and screened Coulomb interaction (GW) approach (see section 2)10. Figure 1 illustrates the complete property database and how it has been used in model training and prediction.
. The employed descriptor, dubbed the 'Coulomb'-matrix, is directly obtained from {ZI,RI}. As such this constitutes a well-defined supervised-learning problem and in the limit of the converged number of training examples the ML model becomes a formally exact inductive equivalent to the deductive solution of SE. It is advantageous that the training data can come from experiment just as well as from numerical evaluation of the corresponding quantum mechanical observable using approximate wavefunctions (separated nuclear and electronic wavefunctions, Slater determinant expansions, etc), Hamiltonians (such as the Hückel or any exchange-correlation potential), and self-consistent field procedures. Building on our previously introduced work [9], we present here a more mature ML model developed to accomplish the following two additional tasks: (i) simultaneously predict a variety of different electronic properties for a single query and (ii) reach an accuracy comparable with the employed reference method used for generating the training set. The presented ML model is based on a multi-task deep artificial NN approach that captures correlations between seemingly related and unrelated properties and levels of theory. A remarkable predictive accuracy for 'out-of-sample' molecules (i.e. molecules that were not part of the training set) has been obtained through the use of random Coulomb matrices that introduce invariance with respect to atom indexing. For training, we generated a quantum chemical database containing nearly 105 entries for over 7000 stable organic molecules, made of up to seven atoms from main-group elements, consisting of C, N, O, S and Cl, saturated with hydrogen to satisfy valence rules [13, 14]. For each molecule, the atomization energy, static polarizabilities, frontier orbital eigenvalues and excitation energies and intensities have been calculated by a variety of widely used electronic structure methods, including state-of-the-art first principles methods, such as hybrid density-functional theory and the many-body single particle Green's function and screened Coulomb interaction (GW) approach (see section 2)10. Figure 1 illustrates the complete property database and how it has been used in model training and prediction.

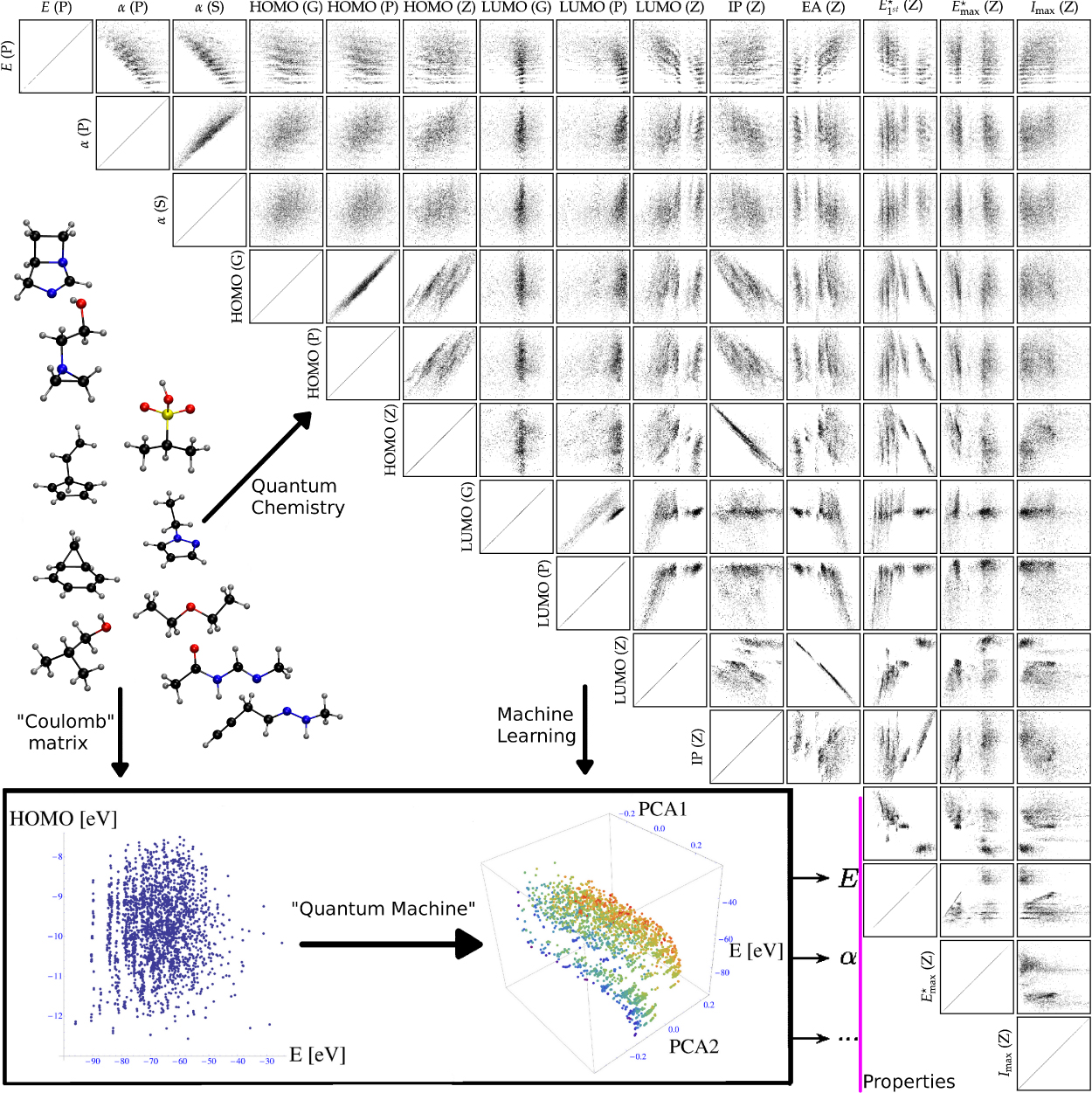
Figure 1. Overview of the calculated database used in training and testing the ML model. The quantum chemistry results for 14 properties of 7211 molecules are displayed. All the properties and level of theory, GW (G), PBE0 (P) and Zerner's intermediate neglect of differential overlap (ZINDO; Z) are defined in section 2.3. Cartoons of ten exemplary molecules from the database are shown; they are used as the input for quantum chemistry, for learning or for prediction. Relying on the input in the 'Coulomb' matrix form, the concept of a 'quantum machine' (QM) is illustrated for two seemingly uncorrelated properties, atomization energy E and HOMO eigenvalue, which are decoded in terms of the largest two principal components (PCA1, PCA2) of the last NN layer for 2k molecules not part of the training. The color-coding corresponds to the HOMO eigenvalues.
Download figure:
Standard image High-resolution image2. Methods
2.1. Molecular structures (input)
While the present ML model approach is generally applicable, for the purpose of this study we restrict ourselves to the chemical space of small organic molecules. For all the cross-validated training and out-of-sample model performance testing, we rely on a controlled test bed of molecules, namely a subset of the general molecular data base (GDB)-13 database [13, 14] consisting of all 7211 small organic molecules that have up to seven second and third row atoms consisting of C, N, O, S or Cl, saturated with hydrogen. The entire GDB-13 database represents an exhaustive list of the ∼ 0.97 B organic molecules that can be constructed from up to 13 such 'heavy' atoms. All GDB molecules are stable and synthetically accessible according to organic chemistry rules [15]. Molecular features such as functional groups or signatures include single, double and triple bonds; (hetero-) cycles, carboxy, cyanide, amide, amine, alcohol, epoxy, sulfide, ether, ester, chloride, aliphatic and aromatic groups. For each of the many possible stoichiometries, many constitutional isomers are considered, each being represented only by a single conformational isomer.
Based on the string representation (SMILES [16, 17]) of molecules in the database, we used the universal force field [18] to generate reasonable Cartesian molecular geometries, as implemented in OpenBabel [19]. The resulting geometries were relaxed using the PBE approximation [20] to Kohn-Sham density functional theory (DFT) [21] in a converged numerical basis, as implemented in the FHI-aims code [22] (tight settings/tier2 basis set). All the geometries are provided in the supplementary material (available from stacks.iop.org/NJP/15/095003/mmedia).
2.2. Molecular representation (descriptor)
One of the most important aspects for creating a functional ML model is the choice of an appropriate data representation (descriptor) that reflects important constraints and properties due to the underlying physics, SE in our case. While there is a wide variety of descriptors used in chem- and bio-informatics applications [23–27], they are conventionally based on prior knowledge about chemical binding, electronic configuration or other quantum mechanical observables. Instead, we derive our representation without any pre-conceived knowledge, i.e. exclusively from stoichiometry and configurational information, from that generated according to the previous subsection. As such, the molecular representation is in complete analogy to the electronic Hamiltonian used in ab initio methods.
For this study, we use a randomized variant of the recently introduced 'Coulomb matrix', M [9, 10]. The Coulomb matrix is an inverse atom-distance matrix representation that is unique (i.e. no two molecules will have the same Coulomb matrix unless they are identical or enantiomers) and retains invariance with respect to molecular translation and rotation by construction:

Off-diagonal elements encode the Coulomb repulsion between nuclear charges of atoms I and J, while diagonal elements represent the stoichiometry through an exponential fit in Z to the free atoms' potential energy. We have enforced invariance with respect to atom indexing by representing each molecule as a probability distribution over Coulomb matrices p(M) generated by different atom indexing of the same molecule. Details for producing such random Coulomb matrices can be found in the supplementary material (available from stacks.iop.org/NJP/15/095003/mmedia).
2.3. Molecular electronic properties (output)
The reference values necessary for learning and testing consist of various electronic ground- and excited-state properties of molecules in their PBE geometry minimum. Specifically, we consider the atomization energies E, static polarizabilities (trace of tensor) α, frontier orbital eigenvalues HOMO and LUMO, ionization potential IP and electron affinity EA. Furthermore, from optical spectrum simulations (10–700 nm), we consider the first excitation energy E*1st, excitation of maximal optimal absorption E*max and its corresponding intensity Imax. Data ranges of properties for the molecular structures and for various levels of theory are given in footnote 9; property mean values in the data set also feature in table 1.
Table 1. Mean absolute errors (MAEs) and root mean square errors (RMSE) for out-of-sample predictions by the ML model, together with typical error estimates of the corresponding reference level of theory. Errors are reported for all 14 molecular properties, and are based on out-of-sample predictions for 2211 molecules using a multi-task multi-layered NN ML model obtained by cross-validated training on 5000 molecules. The corresponding true versus predicted scatter plots feature in figure 3. Property labels refer to the level of theory and molecular property, i.e. atomization energy (Eref), averaged molecular polarizability (α), HOMO and LUMO eigenvalues, ionization potential (IP), electron affinity (EA), first excitation energy (E*1st), excitation frequency of maximal absorption (E*max) and the corresponding maximal absorption intensity (Imax). To guide the reader, the mean value of the property across all 7211 molecules in the database is shown in the second column. Energies, polarizabilities and intensity are in eV, Å3 and arbitrary units, respectively.
| Property | Mean | MAE | RMSE | Reference MAE |
|---|---|---|---|---|
| E (PBE0) | −67.79 | 0.16 | 0.36 | 0.15a, 0.23b, 0.09−0.22c |
| α (PBE0) | 11.11 | 0.11 | 0.18 | 0.05−0.27d, 0.04−0.14e |
| α (SCS) | 11.87 | 0.08 | 0.12 | 0.05−0.27d, 0.04−0.14e |
| HOMO (GW) | −9.09 | 0.16 | 0.22 | – |
| HOMO (PBE0) | −7.01 | 0.15 | 0.21 | 2.08f |
| HOMO (ZINDO) | −9.81 | 0.15 | 0.22 | 0.79g |
| LUMO (GW) | 0.78 | 0.13 | 0.21 | – |
| LUMO (PBE0) | −0.52 | 0.12 | 0.20 | 1.30g |
| LUMO (ZINDO) | 1.05 | 0.11 | 0.18 | 0.93g |
| IP (ZINDO) | 9.27 | 0.17 | 0.26 | 0.20, 0.15d |
| EA (ZINDO) | 0.55 | 0.11 | 0.18 | 0.16h, 0.11d |
| E*1st (ZINDO) | 5.58 | 0.13 | 0.31 | 0.18h, 0.21i |
| E*max (ZINDO) | 8.82 | 1.06 | 1.76 | – |
| Imax (ZINDO) | 0.33 | 0.07 | 0.12 | – |
aPBE0, MAE of formation enthalpy for the G3/99 set [54, 55]. bPBE0, MAE of atomization energy for six small molecules [56, 57]. cB3LYP, MAE of atomization energy from various studies [52]. dB3LYP, MAE from various studies [52]. eMP2, MAE from various studies [52]. fMAE from GW values. gZINDO, MAE for a set of 17 retinal analogues [58]. hPBE0, MAE for the G3/99 set [54, 55]. iTD-DFT(PBE0), MAE for a set of 17 retinal analogues [58].
To also gauge the impact of the reference method's level of theory on the ML model, polarizabilities and frontier orbital eigenvalues were evaluated with more than one method. Static polarizability has been calculated using self-consistent screening (SCS) [28] as well as hybrid density functional theory (PBE0) [29, 30]. PBE0 has also been used to calculate atomization energies and frontier orbital eigenvalues. The electron affinity, ionization potential, excitation energies and maximal absorption intensity have been obtained from ZINDO [31–33]. Hedin's GW approximation [34] has also been used to evaluate frontier orbital eigenvalues. GW is a quasi-particle ab initio many-body perturbation theory, known to accurately account for electronic excitations that describe electron addition and removal processes [34]. The SCS, PBE0 and GW calculations have been performed using FHI-aims; [22, 35], ZINDO/s calculations are based on the ORCA code [36]. ZINDO/s is an extension of the INDO/s semiempirical method with parameters to accurately reproduce single excitation spectra of organic compounds and complexes with rare-earth elements. The INDO Hamiltonian neglects some two-center two-electron integrals in order to simplify the calculation of time-dependent Hartree–Fock equations. While the ZINDO results are usually not as accurate as highly correlated methodologies, the semiempirical Hamiltonian reproduces the most important features of the absorption spectra of many small molecules and complexes, particularly characterizing their most intense bands on the UV–vis spectra. All properties are provided in the supplementary material (available from stacks.iop.org/NJP/15/095003/mmedia).
Similar conclusions hold for the selected levels of theory: the employed methods can be considered to represent a reasonable compromise between computational cost and predictive accuracy. It should be mentioned that ML methods can, in principle, be applied to any method or level of approximation.
2.4. Training the model
Our model consists of a deep and multi-task NN [37, 38] that is trained on molecule–properties pairs. It learns to map Coulomb matrices to all 14 properties of the corresponding molecule simultaneously. NNs are well established for learning functional relationships between the input and the output. They have successfully been applied to different tasks such as object recognition [39] and speech recognition [40]. Given a sufficiently large NN, its universal approximation capabilities [41] and the existence of the underlying noise-free SE, an NN solution can be expected to exist that satisfyingly relates molecules to their properties. Specifically, a deep NN will properly unfold, layer after layer, a complex input into a simple representation of molecular properties. Finding the true relationship unfolding among those that fit the training data can be challenging because there is typically a manifold of solutions. The multi-task setup forces the NN to predict multiple properties simultaneously. This is conceptually appealing because these additional constraints narrow down the search for the 'true model' [42], as the set of models that fit all properties simultaneously is smaller. Details of the NN training procedure can be found in the supplementary material (available from stacks.iop.org/NJP/15/095003/mmedia).
3. Results and discussion
Before reporting and discussing our results, we note the long history of statistical learning of the potential energy hyper surface for molecular dynamics applications. It includes, for example, the modeling of potential energy surfaces with artificial NNs starting with the work of Sumpter and Noid in 1992 [43–49] or Gaussian processes [50, 51]. Our work aims to go beyond single molecular systems and learn to generalize to unseen compounds. This extension is not trivial, as the input representation must deal with molecules of diverse sizes and compositions in the absence of one-to-one mapping between atoms of different molecules.
3.1. Database
Scatter plots among all properties for all the molecules are shown in figure 1. Visual inspection confirms the expected relationships between various properties: Koopman's theorem relating ionization potential to the HOMO eigenvalue [52], the hard soft acid base principle linking polarizability to stability [53] or electron affinity correlating with the first excitation energy. Correlations of identical properties at different levels of theory reveal more subtle differences. Polarizabilities, calculated using PBE0 or with the more approximate SCS model [28], are strongly correlated. Also, less well-known relationships can be extracted from these data. One can obtain to a very decent degree, for example, the GW HOMO eigenvalues by subtracting 1.5 eV from the corresponding PBE0 HOMO values.
Some properties, such as atomization and HOMO energies, exhibit very little correlation in their scatter plot. The inset of figure 1 illustrates how our QM (i.e. the NN-based ML model) extracts and exploits hidden correlations for these properties despite the fact that they cannot be recognized by visual inspection. Similar conclusions hold for atomization energy versus first excitation energy or polarizability versus HOMO energy.
3.2. Accuracy versus training set size
It is an important feature of any ML model that the error can be controlled systematically as the training set size is varied. We have investigated this dependence for our ML model. Figure 2 shows a typical decay of the ML model's mean absolute error (MAE) for predicting the properties of 'out-of-sample' molecules as the number of molecules in the training set increases logarithmically from 500 to 5000. For all the investigated properties, the improvement of error suggests that the MAE could still be lowered even further through the addition of more molecules. However, since the reference method's 'precision' (i.e. the estimated accuracy of the employed level of theory) is reached for almost all properties already using 5000 examples, adding further examples does not make sense. For the atomization energy the decay is particularly dramatic: a tenfold increase in the number of molecules (500 → 5000) reduces the error by 70%, from 0.55 to 0.16 eV. But also for the HOMO/LUMO eigenvalues, the error reduces substantially. We find that the expected error decay law of  is only recovered for the atomization energy; for other properties the error decays more slowly. Figure 2 also features the statistical error bars for the MAEs—a measure of outliers. The error bar is only slightly larger than symbol size, and hardly varies as the training set increases and the testing set decreases.
is only recovered for the atomization energy; for other properties the error decays more slowly. Figure 2 also features the statistical error bars for the MAEs—a measure of outliers. The error bar is only slightly larger than symbol size, and hardly varies as the training set increases and the testing set decreases.

Figure 2. Error decay of the ML model with increasing number of molecules in the training set (shown on a logarithmic scale). The MAE and its error bar are shown for atomization energy (E), polarizability (α), frontier orbital eigenvalues (HOMO, LUMO) and the first excitation energy (E*1st).
Download figure:
Standard image High-resolution image3.3. The final machine learning model
After cross-validated training on the largest training set with 5000 randomly selected molecules, 2211 predictions have been made for the remaining 'out-of-sample' molecules, yielding at once all 14 quantum chemical properties per molecule. The corresponding true versus predicted scatter plots feature in figure 3. The corresponding mean absolute and root-mean square errors (RMSE) are shown in table 1, together with the literature estimates of errors typical of the corresponding level of theory. Errors of all properties range in the single digit percent of the mean property. Remarkably, when compared to published typical errors for the corresponding level of theory, i.e. used as a reference method for training, similar accuracy is obtained—the sole exception being the most intense absorption and its associated excitation energy. This, however, is not too surprising: extracting the information about a particular excitation energy and the associated absorption intensity requires sorting the entire optical spectrum—thus encoding significant knowledge that was entirely absent from the information employed for training. For all other properties, however, our results suggest that the presented ML model makes 'out-of-sample' predictions with an accuracy competitive with the employed reference methods. These methods include some of the more costly state-of-the-art electronic structure calculations, such as GW results for HOMO/LUMO eigenvalues and hybrid DFT calculations for atomization energies and polarizabilities. Work is in progress to extend our ML approach to other properties, such as the prediction of ionic forces or the full optical spectrum. We note, however, that for the purpose of this study any level of theory and any set of geometries could have been used.

Figure 3. Scatter plot of the true value versus the ML model value for all properties. The red line indicates the identity mapping. All units correspond to the entries shown in table 1.
Download figure:
Standard image High-resolution imageThe remarkable predictive power of the ML model can be rationalized by (i) the deep layered nature of the NN model that permits us to progressively extract the relevant problem subspace from the input representation and gain predictive accuracy [59, 60]; (ii) inclusion of random Coulomb matrices for training, effectively imposing invariance of property with respect to atom indexing, clearly benefits the model's accuracy: additional tests suggest that using random, instead of sorted or diagonalized [9], Coulomb matrices also improves the accuracy of kernel ridge regression models to similar degrees; and (iii) the multi-task nature of the NN accounts for the strong and weak correlations between seemingly unrelated properties and different levels of theory. Aspects (i) and (iii) are also illustrated in figure 4.

Figure 4. Principal component analysis (PCA) on the multiple layers of the deep NN. Each point (molecule) is colored according to the rule: E and HOMO large → red; E large and HOMO small → blue; E small and HOMO large → green; E and HOMO small → black. We can observe that the NN extracts, layer after layer, a representation of the chemical space that better captures the multiple properties of the molecule.
Download figure:
Standard image High-resolution imageWe reiterate that evaluation of all 14 properties at the said level of accuracy for an out- of-sample molecule requires only milliseconds using the ML model, as opposed to several CPU hours using the reference methods used for training. The downside of such accuracy, of course, is the limit in transferability. All ML model predictions are strictly limited to out-of-sample molecules that interpolate. More specifically, the 5000 training molecules must resemble the query molecule in a similar fashion as they resemble the 2211 test molecules. For compounds that bear no resemblance to the training set, the ML model must not be expected to yield accurate predictions. This limited transferability might one day become moot through a more intelligent choice and construction of molecular training sets tailored to cover all of a pre-defined chemical compound space, i.e. all of the relevant geometries and elemental compositions, up to a certain number of atoms.
4. Conclusion
We have introduced a ML model for predicting the electronic properties of molecules based on training deep multi-task artificial NNs in chemical space. Advantages of such a QM (conceptually speaking, as illustrated in figure 1) are the following: (i) multiple dimensions: a single QM execution simultaneously yields multiple properties at multiple levels of theory; (ii) a systematic reduction of error: by increasing the training set size the QM's accuracy can be converged to a degree that outperforms modern quantum chemistry methods, hybrid density-functional theory and the GW method in particular; (iii) a dramatic reduction in computational cost: the QM makes virtually instantaneous property predictions; (iv) user-friendly character: training and the use of the QM do not require knowledge about the electronic structure or even about the existence of the chemical bond; (v) arbitrary reference: the QM can learn from data corresponding to any level of theory, and even experimental results. The main limitation of the QM is the empirical nature inherent in any statistical learning method used for inferring solutions, namely that meaningful predictions for new molecules can only be made if they fall in the regime of interpolation.
We believe our results to be encouraging numerical evidence that ML models can systematically infer highly predictive structure–property relationships from high-quality databases generated via first-principles atomistic simulations or experiments. In this study, we have demonstrated the QM's performance for a rather small subset of chemical space, namely for small organic molecules with only up to seven atoms (not counting hydrogen) as defined by the GDB. Due to its inherent first principles setup, we expect the overall approach to be equally applicable to molecules or materials of arbitrary size, configurations and composition—without any major modification. We note, however, that in order to apply the QM to other regions in chemical space with a similar accuracy differing amounts of training data might be necessary.
We conclude that combining reliable databases with ML promises to be an important step toward the general goal of exploring chemical compound space for the computational bottom-up design of novel and improved compounds.
Acknowledgments
This research utilized the resources of the Argonne Leadership Computing Facility at Argonne National Laboratory, which is supported by the Office of Science of the US DOE under contract no. DE-AC02-06CH11357. KRM acknowledges partial support from DFG, Einstein Foundation and EU. MR acknowledges support from the FP7 program of the European Community (Marie Curie IEF 273039). This work was also supported by the World Class University Program through the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology, under grant no. R31-10008.
Appendix A.: Details of random Coulomb matrices
Random Coulomb matrices define a probability distribution over the set of Coulomb matrices and account for different atom indexing of the same molecule. The following four-step procedure randomly draws Coulomb matrices from the distribution p(M): (i) take an arbitrary valid Coulomb matrix M of the molecule, (ii) compute the norm of each row of this Coulomb matrix: n = (∥M1∥,...,∥M23∥), (iii) draw a zero-mean unit-variance noise vector ε of the same size as n and (iv) permute the rows and columns of M with the same permutation that sorts n + ε. An important feature of random Coulomb matrices is that the probability distributions over Coulomb matrices of two different molecules are completely disjoint. This implies that the randomized representation is not introducing any noise into the prediction problem. Invariance to atom indexing proves to be crucial for obtaining models with high predictive accuracy. The idea of encoding known invariances through such data extension has previously been used to improve prediction accuracy on image classification and handwritten digit recognition data sets [61].
Appendix B.: Details of training the neural network
The ML model and the NN perform a sequence of transformations on the input that are illustrated in figure B.1. The Coulomb matrix is first converted to a binary representation before being processed by the NN. The rationale for this binarization is that continuous quantities such as Coulomb repulsion energies encoded in the Coulomb matrix are best processed when their information content is distributed across many dimensions of low information content. Such binary expansion can be obtained by applying the transformation

where ![$\phi : \mathbb {R} \rightarrow [0,1]^\infty $](https://content.cld.iop.org/journals/1367-2630/15/9/095003/revision1/nj459085ieqn4.gif) , the parameter θ controls the granularity of the transformation and sigm(x) = ex/(1 + ex) is a sigmoid function. Transforming Coulomb matrices M of size 23 × 23 with a granularity θ = 1 yields three-dimensional tensors of size [∞ × 23 × 23] of quasi-binary values, approximately 2000 dimensions of which are non-constant. Transforming vectors P of 14 properties with a granularity 0.25 of the same units as in table 1 yields matrices of size [∞ × 14], approximately 1000 components of which are non-constant.
, the parameter θ controls the granularity of the transformation and sigm(x) = ex/(1 + ex) is a sigmoid function. Transforming Coulomb matrices M of size 23 × 23 with a granularity θ = 1 yields three-dimensional tensors of size [∞ × 23 × 23] of quasi-binary values, approximately 2000 dimensions of which are non-constant. Transforming vectors P of 14 properties with a granularity 0.25 of the same units as in table 1 yields matrices of size [∞ × 14], approximately 1000 components of which are non-constant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure B.1. Predicting the properties for a new molecule: (a) enter the Cartesian coordinates and nuclear charges, (b) form a Coulomb matrix, (c) binarize the representation, (d) propagate into a trained NN and (e) scale outputs back to property units.
Download figure:
Standard image High-resolution image{kind=link}
We construct a four-layer NN with 2000, 800, 800 and 1000 nodes at each layer. The network implements the function  , where functions f1, f2 and f3 between each layer correspond to a linear transformation learned from data followed by a sigmoid nonlinearity. The NN is trained to minimize the MAE of each property using the stochastic gradient descent algorithm (SGD) [62]. Errors are back-propagated [63] from the top layer back to the inputs in order to update all parameters of the model. We run 250 000 iterations of the SGD and present at each iteration 25 training samples. During training, each molecule–property pair is presented in total 1250 times to the NN, but each time with different atom indexing. A moving average of the model parameters is maintained throughout training in order to attenuate the noise of the stochastic learning algorithm [64]. The moving average is set to remember the last 10% of the training history and is used for the prediction of out- of-sample molecules. Training the NN on a CPU takes ∼ 24 h. Once the NN has been trained, the typical CPU time for predicting all 14 properties of a new out-of-sample molecule is ∼ 100 ms. Prediction of an out-of-sample molecule is obtained by propagating ten different realizations of p(M) and averaging outputs. Prediction of multiple molecules can be easily parallelized by replicating the trained NN on multiple machines. For more details on training neural networks, the reader is referred to [65, 66]
, where functions f1, f2 and f3 between each layer correspond to a linear transformation learned from data followed by a sigmoid nonlinearity. The NN is trained to minimize the MAE of each property using the stochastic gradient descent algorithm (SGD) [62]. Errors are back-propagated [63] from the top layer back to the inputs in order to update all parameters of the model. We run 250 000 iterations of the SGD and present at each iteration 25 training samples. During training, each molecule–property pair is presented in total 1250 times to the NN, but each time with different atom indexing. A moving average of the model parameters is maintained throughout training in order to attenuate the noise of the stochastic learning algorithm [64]. The moving average is set to remember the last 10% of the training history and is used for the prediction of out- of-sample molecules. Training the NN on a CPU takes ∼ 24 h. Once the NN has been trained, the typical CPU time for predicting all 14 properties of a new out-of-sample molecule is ∼ 100 ms. Prediction of an out-of-sample molecule is obtained by propagating ten different realizations of p(M) and averaging outputs. Prediction of multiple molecules can be easily parallelized by replicating the trained NN on multiple machines. For more details on training neural networks, the reader is referred to [65, 66]
Footnotes
- 8
Based on the Materials Genome Initiative www.whitehouse.gov/mgi announced by the US President Obama in June 2011, four federal science and research agencies (National Science Foundation, Department of Energy, Air Force Research Laboratory and Office of Naval Research) recently published their support: www.whitehouse.gov/blog/2011/10/26/four-new-federal-programs-support-materials-genome-initiative
- 9
The number of electrons is implicitly encoded by imposing charge neutrality.
- 10
Electronic properties considered include PBE0 atomization energies ranging from −800 to −2000 kcal mol−1; the first excitation energies (1.52–36.77 eV), as well as maximal absorption intensities (oscillator strengths (〈j|r|0〉) ranging from 0.05 to 3.35 arbitrary units) and the corresponding excitation energies (3.37–39.69 eV) at the ZINDO level of theory; HOMO and LUMO values calculated at the ZINDO/s, PBE0 and GW level of theory (HOMOPBE0: −10.95 to −5.12; HOMOGW: −14.13 to −6.98 eV; LUMOPBE0: −3.81 to 0.41 eV; LUMOGW: −1.84 to 1.96 eV) (the corresponding gap ranging from 6.9 to 20.2 and from 7.3 to 15.2 eV, respectively); PBE0 and self-consistent screening [28] molecular polarizabilities (2.5–10 Å3); electron affinity (− 3.99 to 2.91 eV) and ionization potentials (6.93–15.73 eV) at the ZINDO/s level of theory.