

Abstract
Density-functional based tight-binding (DFTB) is an efficient quantum mechanical method that can describe a variety of systems, going from organic and inorganic compounds to metallic and hybrid materials. The present topical review addresses the ability and performance of DFTB to investigate energetic, structural, spectroscopic and dynamical properties of gold and silver materials. After a brief overview of the theoretical basis of DFTB, its parametrization and its transferability, we report its past and recent applications to gold and silver systems, including small clusters, nanoparticles, bulk and surfaces, bare and interacting with various organic and inorganic compounds. The range of applications covered by those studies goes from plasmonics and molecular electronics, to energy conversion and surface chemistry. Finally, perspectives of DFTB in the field of gold and silver surfaces and NPs are outlined.
Export citation and abstract BibTeX RIS
1. General introduction
Nanoparticles (NPs) and surfaces made of group 11 noble metals, which includes gold and silver, have deserved a lot of attention motivated by their unique properties that make them materials of choice in a large number of applications [1, 2]. Indeed, silver and gold NPs are well known for their surface plasmon resonance (SPR) effect which originates from coherent oscillations of conduction electrons in response to external time-varying electric fields. SPR confers unique optical properties to gold and silver NPs. For instance, they display high optical absorption over a wide range of both visible and UV light [3] and can also act as photocatalyst to promote chemical reactions [2–5]. This feature can be enhanced or tuned by associating Ag or Au NPs with an organic or inorganic compound as for instance: (i) an insulating support such as ZrO2 or SiO2 to form Au/ZrO2, Ag/ZrO2 or Ag/SiO2 [3]; (ii) a semiconductor support to form NPs-semiconductor composites, for instance Au/TiO2 [3, 6]; (iii) molecular species, in particular dye molecules such as methylene blue or rhodamine 6G [7]. In those devices, light absorption by the NPs generates hot charge carriers that can migrate into the semiconductor or the attached molecules and facilitate photochemical transformations [3, 7]. In addition to solar-to-chemical energy conversion, this charge-carrier transfer is also used for solar-to-electrical energy conversion by improving absorption in photovoltaic devices which allows for the development of more efficient solar-cells [8–10]. One can also take advantage of the SPR effect to produce intense local electromagnetic fields that further enable surface-enhanced Raman scattering (SERS) [11] and metal-enhanced fluorescence [12, 13].
From a purely chemical point of view, gold NPs, when dispersed on specific oxide surfaces, are also a good catalyst for the oxidation of carbon monoxide into CO2 at room temperature [14, 15]. It is worth mentioning that this specific property only appears for particle sizes smaller than ∼8 nm and is not observed for bulk gold which is chemically inert. This is one among a variety of examples highlighting the peculiar character of the nanometer scale.
Biological applications have greatly benefited from the development of Ag and Au NPs. Indeed, silver NPs are well recognized for their antibacterial properties [16–18], and their incorporation in bio-compatible materials has allowed for the development of promising multifunctional biomedical devices [19]. Gold NPs also display interesting biomedical applications due to their biocompatibility and optical properties. For instance, once properly functionalized to improve stability, biocompatibility and functionality, they can be used for molecular and biosensing as well as bioimaging [20–23]. In addition to analytical applications, gold NPs can serve as nanocarriers for gene [24] and drug [25] delivery in the context of cancer therapy [20]. Laser-induced heating of Au NPs can also be used, among others, in photothermal therapy or selective protein denaturation [26].
Developing devices integrating gold or silver NPs that efficiently promote the aforementioned applications is a complex task which requires a deep understanding of several mutually interplaying parameters. Indeed, characteristics of the NPs such as size [1, 27], morphology [1, 27, 28], chemical or dielectric environment [27], as well as the nature and properties of their support [29] can affect their optical and reactive properties and, subsequently, their range of applications. While numerous experimental investigations of the fundamental properties of NPs have been carried out, experiments alone cannot provide a detailed picture of all those parameters, and modeling tools have to be introduced as recently highlighted by Jin and co-workers: 'It is highly desirable to develop efficient methods for computing large nanoclusters such as Au130(SR)50 and Au133(SR)52' [2]. A large variety of computational methods have thus been introduced to tackle the diversity of questions and the complexity arising from gold or silver particles: structure of bare NPs or NPs interacting with molecules or surfaces, dynamics and finite-temperature effects, reactivity, behavior in the excited-states and explicit description of hot carriers generated by SPR. Those methods range from highly accurate ab initio formalisms applicable for a limited number of atoms, to empirical force fields and potentials suitable for very large systems but much less accurate a priori. In between, a variety of intermediate approaches exists, for instance the density functional theory (DFT), that display strengths and weaknesses either considering the properties they are able to describe, their computational efficiency and accuracy, or the size range they can handle. Considering only the size challenge, metal NPs displaying sizes larger than ∼1.6 nm, which corresponds approximately to 150 atoms, can still be challenging to describe at the DFT level when finite-temperature effects need to be included or global optimization studies have to be conducted for extensive exploration of energy landscapes. Nevertheless, some DFT electronic structure calculations have now been published for transition metal NPs containing up to one thousand and even around ten thousand atoms, see for instance some recent publications [30–32]. In contrast, the use of empirical potentials breaks down whenever an explicit treatment of the electronic structure is required. Consequently, approximate quantum mechanical methods have emerged.
Among them, the self-consistent-charge density-functional based tight-binding (SCC-DFTB) approach has known an increasing interest over the last decade for the description of gold and silver materials. SCC-DFTB is derived from the DFT formalism and belongs to the semi-empirical class of methods. It allows for an explicit treatment of the electronic structure while reducing the computational cost thanks to the use of parametrized integrals and a minimal valence basis set. As a consequence, SCC-DFTB can tackle problems requiring an explicit description of the electronic structure even for large systems consisting of several hundreds of atoms.
In the present review, we intend to provide a general panorama of the various applications of the SCC-DFTB approach to the description of gold and silver materials. First, we provide in section 2 a brief overview of the methodologies used to model gold and silver materials. This section is not intended to report exhaustively all the applications of all methods to gold and silver clusters and nanoparticles, much too numerous for this short overview. The interested reader might refer to previous textbooks and feature articles [33–45]. Then, in section 3.1, we summarize the theoretical basis of the SCC-DFTB formalism and discuss in section 3.2 the main issues existing in its parametrization in the case of gold and silver. We present in section 4 the application of SCC-DFTB to efficiently explore potential energy surfaces of gold and silver clusters and NPs in order to characterize their structure, dynamics, thermodynamics and plasmonics. Performances of SCC-DFTB towards the modeling of bulk properties are also discussed in this section. In section 5, we review the various SCC-DFTB studies focusing on gold and silver clusters, NPs and surfaces in interaction with a chemical environment or functionalized. We finally highlight in the conclusion the main perspectives in the application of SCC-DFTB to the description of gold, silver and other metallic systems.
2. A short overview about gold and silver materials modeling
As for other metallic and non-metallic systems, the theoretical description of gold and silver clusters, NPs and surfaces can proceed from a hierarchy of methodologies depending on the size and on the properties of interest. In a bottom-up approach (e.g. from small systems to large ones), one may distinguish (somewhat arbitrarily) the microcluster range from three atoms to a few tens, the cluster range between a few tens and typically one hundred and the NP regime between one or several hundreds and a few ten thousands or hundred thousands of atoms (namely NPs with a diameter larger than 1.6 nm). Obviously, such frontiers are fuzzy and this division in three regimes is only indicative. Its main significance is that in the microcluster and cluster ranges, 'each atom counts' and strong size effects are observed, while in the NP regime, scaling laws are expected to be more relevant (note that the distinction between clusters and microclusters is not always made). The evolution with size obviously depends on the considered property. Furthermore, it should be emphasized that in the case of metals, both electronic and geometric factors contribute to the stability. One may observe magic sizes governed by electronic- or by geometric-shell closings. In the case of 3D monovalent metal clusters, electronic-shell closings in the spherical jellium model are expected for a number of electrons equal to 2, 8, 18, 20, 34, 40, etc... (while a 2D jellium model is relevant for planar microclusters). One should remark that due to hybridization of the atom-localized s electrons with the d shell, both silver and gold can depart from the simple monovalent character. Geometric magic sizes correspond to completion of shells of atoms which leads to highly symmetric structures such as icosahedrons, cuboctahedrons, decahedrons, tetrahedrons or truncated octahedrons. For instance, both the icosahedron and the cuboctahedron correspond to the well-known geometric magic sizes n = 13, 55, 147, 309, 561, etc.
The most accurate theoretical methods involve first-principles descriptions of the electronic structure, i.e. quantum mechanics, and most often a classical treatment of the nuclei in the Born–Oppenheimer framework. Ab initio theories based on the determination of the wavefunction (WFT), such as configuration-interaction or coupled-cluster formalisms [46–48], aim at the exact resolution of the electronic many-body problem and have been used for silver and gold aggregates in the microcluster and cluster regime [34, 39, 49–59]. Alternatively, DFT relies on the determination of the electronic density and has become the most popular first-principles approach for reasonably large systems [60–63]. DFT has been extensively applied over the microcluster and cluster range for silver [59, 64–69] and gold [70–93] and even up to the NP regime with a few hundreds atoms [94–102]. DFT is also the preferred method to investigate the properties of gold and silver surfaces in interaction with chemical species [103–111].
Very large NPs cannot, at the present time, be addressed neither by WFT and hardly by DFT and the only practicable models for structure, dynamics and thermodynamics are those in which the electrons are no longer explicitly treated, the interactions between nuclei being described via potentials or sometimes parametrized densities. In the case of metals and in particular noble-metal materials, many-body potentials, such as the embedded atom model (EAM) [112], also known as the Gupta potential or the second moment approximation (SMA) scheme, have been widely used. Other examples of interaction potentials for metals are the Sutton–Chen potential [113, 114] or the Glue potential based on atomic density ansatz [115]. All those schemes incorporate N-body terms through simplified expressions, some of them deriving from approximations of the tight-binding scheme. Such potentials are able to cover the size domain from one hundred to several tens of thousands atoms [116–129]. The account of many-body effects in these potentials is certainly oversimplified and their form tends to favour high atomic coordinations. They are therefore ususally not relevant in the small size regime where they fail to account for planar structures. They are also unable to describe specific quantum-mechanical effects such as Jahn–Teller shape distorsions and more generally properties directly associated with the electronic structure of the NPs. In between fully first-principles methods and model potentials, semi-empirical schemes can still provide a quantum treatment of the electronic structure while keeping a limited computational cost such as CNDO, NDDO, AMx or PMx approaches [130–133]. For noble or transition metal compounds, various adaptations of the tight-binding schemes [134, 135] have been developed [136–140].
Aside from describing the interaction between metallic atoms, the determination of the structure of NPs is a formidable problem in itself above a few ten atoms. Indeed, the microcluster and the cluster regimes are characterized by a large number of low-energy isomers which often display very different topologies. Highly symmetric configurations are not necessarily the global minima, even for the geometric magic sizes mentioned above, and generally not for generic sizes, even though some of their building patterns can be present. Consequently, global optimization schemes are required to determine the low-energy isomers with the least possible a priori assumptions. The most popular of these algorithms are: (i) genetic algorithms (GA) [141], (ii) algorithms based on molecular dynamics (MD) or Monte Carlo (MC) schemes completed by quenches of selected configurations along the trajectories [142], (iii) basin-hopping algorithms [143, 144], and (iv) swarm algorithms [145]. All these schemes require a huge number (generally more than 106) of energy calculations and possibly energy gradient. Therefore, such algorithms cannot be combined with WFT based methods, and even the direct use of DFT becomes prohibitive above ∼30-40 noble metal atoms as illustrated by the study of Assadollahzadeh and Schwerdtfeger on gold clusters using a genetic algorithm and limited to sizes smaller than 20 atoms [86]. To circumvent this limitation, efficient approaches in the 20-80 size range consist in using pre-screening schemes: a simple model potential (EAM or Glue for instance) allows to generate a panel of low-energy configurations which are further optimized using a higher-level method such as DFT. Besides the lowest-energy isomer question, large NPs also present size transitions between various structural stabilities: icosahedron, cuboctahedron, decahedron, and truncated octahedron which have been largely investigated via model potentials [121, 122, 124, 126]. It should be noted that cluster structures have become experimentally accessible via various techniques such as infra-red spectroscopy [81], photoemission spectroscopy [79, 146, 147], ion mobility measurements [148, 149], trapped ion electron diffraction [83, 149, 150], x-ray diffraction [122, 124, 151], extended x-ray absorption fine structure [152] or scanning transmission electron microscopy [99, 153–156]. Some of those techniques do concern neutral systems while others concern mostly ions that can be mass-selected via mass spectrometry techniques. Consequently, synergetic computational and theoretical approaches can be conducted to accurately determine the structure of gold and silver aggregates.
MD and MC simulations can be used for global structural searches as mentioned above. Furthermore, they can also be used to simulate real-time processes such as isomerisation, reactivity, nucleation, fragmentation or finite temperature behaviour. At low temperature, clusters with many low-lying and shallow energy minima can behave as flexible systems, even possibly liquid at very low temperature. In gold, a particularly interesting zone is the n = 8–12 size range where there is a strong competition between 2D planar structures and 3D geometries [157]. Isomerization of small clusters, and in particular gold cage clusters was also investigated [158]. Thermodynamics of clusters and NPs is another issue that requires even larger scale extensive MD or MC exploration of the energy landscape [158–162]. Heat capacities were determined via DFT for gold clusters up to 20 atoms [159, 160]. Temperature and entropy driven features obviously affect larger NPs and have been considered mostly via the use of model potentials [163–169] and enhanced metastability could be observed for instance in gold [156].
The theoretical description of optical properties and plasmonics has been abundantly documented for noble metal particles [170]. The simplest models are based on the Mie theory of spherical or non spherical (ellipsoidal) metal particles. A number of developments have also been achieved within the jellium model of metal spheres in combination with time-dependent DFT [171, 172]. The most realistic of those jellium-based models may depend on a number of phenomenological parameters, such as different effective dielectric constants used to describe the motion of either s and d electrons or the jellium model itself which can be abrupt or smooth and can also take into account surface granularity. Although the Mie theory or the jellium model are able to predict plasmon widths or transition energies for large NPs (∼50 nm and higher for gold and silver) [173], they are limited when a detailed description of the atomic structure of the particles is required. Explicit electronic excitations in a first-principles framework can be introduced either in WFT approaches such as the equation-of-motion coupled-cluster theory within the linear response approximation [34, 48, 174], or in the time-dependent density-functional theory (TDDFT) [175] in the linear-response formulation of Casida [63]. The latter has become of common use for organic compounds and can be applied to systems containing up to ∼400 atoms, i.e. ∼2000 electrons, for vertical TDDFT calculations and about two times less for geometry optimizations [176]. In the more specific field of metal clusters and nanoparticles, TDDFT has also been largely applied and is still an active area of current research [69, 177–187]. Despite the theoretical formulation of DFT is based on first-principles and is exact for the ground state, an important problem is the delicate choice of the exchange-correlation functional. A forest of possibilities is now available, from local, gradient-corrected and hybrid functionals to double-hybrid and long-range corrected functionals. This choice determines how well DFT and TDDFT behave to account for correlation, van der Waals forces, minimization of the self-interaction error, accuracy in the description of dissociative properties and electronic excited-states, and spectroscopy. For instance, in the case of optical properties, several studies have stressed the importance of using long-range-corrected density functionals to properly describe the absorption spectra of large gold and silver clusters [180, 188, 189].
Increasing computational efficiency (either via algorithms ensuring linear scaling or via the use of massively parallel computing architectures) has become an extraordinary challenge for all first-principles methods including both WFT or DFT methods. Of course, it is of primary importance in the investigation of gold and silver NPs at various scales. At present, only DFT can be viewed as able, from the computational point of view, to bridge the gap between the cluster regime and the large NP regime, including the bulk. However, its effective use in simulations involving extensive sampling for more than a few tens of atoms is computationally very demanding and still difficult to conduct. This is why the SCC-DFTB method which is an efficient alternative to DFT has known a fast development and a growing success for the past twenty years. In the next section, we present the theoretical basis of this method and its parametrization in the general case and in the specific cases of gold and silver systems.
3. The density-functional tight-binding approach
3.1. Theoretical basis
The density-functional based tight-binding method is an approximate DFT scheme in the Kohn–Sham orbital based formalism. The method is briefly presented below, as detailed description of the DFTB basics can be found in the literature, either in the original founding papers [190–192] or in more recent reviews [193–196]. It relies on the use of an atomic minimal basis set  to express the molecular orbitals
to express the molecular orbitals  (MOs):
(MOs):

where  denotes the expansion coefficients.
denotes the expansion coefficients.
Following the idea of Foulkes and Haydock [197], the DFT energy is expanded around a reference density  . When the expansion is limited at first order, the expression of a tight-binding model is recovered. This is often referred to as the zeroth-order DFTB [190, 191] as well as DFTB1. This original approach was further refined by introducing a second-order term [192] in the Taylor expansion. This is referred to as second-order DFTB, self-consistent-charge DFTB (SCC-DFTB) or DFTB2 which corresponds to the following energy expression:
. When the expansion is limited at first order, the expression of a tight-binding model is recovered. This is often referred to as the zeroth-order DFTB [190, 191] as well as DFTB1. This original approach was further refined by introducing a second-order term [192] in the Taylor expansion. This is referred to as second-order DFTB, self-consistent-charge DFTB (SCC-DFTB) or DFTB2 which corresponds to the following energy expression:
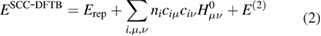
which consists of three terms, namely a repulsive contribution, a so-called band energy term and a second-order term. In this expression, the sum of the first two terms contains the zeroth- and first-order terms of the Taylor expansion, i.e. correspond to zeroth-order DFTB. All the studies performed on gold and silver that are discussed in the present review were performed at the SCC-DFTB level. However, in the following, in order to simplify the notation we will simply refer to DFTB.
In the second term (band energy term) of equation (2), ni is the occupation of orbital i and  is the DFT Kohn–Sham operator matrix element computed at the reference density
is the DFT Kohn–Sham operator matrix element computed at the reference density  and expressed in a localized atomic orbital basis (LCAO). The reference density is taken as the superposition of the isolated atomic densities. Neglecting both three- and four-center integrals and pseudo-potential contributions to two-center terms [193] leads to the following expressions for matrix elements between μ and ν atomic orbitals belonging respectively to atoms α and β:
and expressed in a localized atomic orbital basis (LCAO). The reference density is taken as the superposition of the isolated atomic densities. Neglecting both three- and four-center integrals and pseudo-potential contributions to two-center terms [193] leads to the following expressions for matrix elements between μ and ν atomic orbitals belonging respectively to atoms α and β:
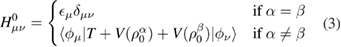
which corresponds to the potential superposition formulation although a density additivity scheme can also be followed [192].
In equation (3),  is the atomic orbital energy of the isolated atom. The two-center term, which is a function of the inter-atomic distance, is determined and tabulated from DFT calculations on the atomic pair. In practice, the values of the two-center terms and of the overlap integrals are stored in the so-called Slater–Koster tables [198] for each atomic pair and over a range of diatomic distances. The implementation of the Slater–Koster tables for a given chemical system is often referred to as the parametrization of the DFTB potential. It may be subject to some empirical modifications as discussed in the following section.
is the atomic orbital energy of the isolated atom. The two-center term, which is a function of the inter-atomic distance, is determined and tabulated from DFT calculations on the atomic pair. In practice, the values of the two-center terms and of the overlap integrals are stored in the so-called Slater–Koster tables [198] for each atomic pair and over a range of diatomic distances. The implementation of the Slater–Koster tables for a given chemical system is often referred to as the parametrization of the DFTB potential. It may be subject to some empirical modifications as discussed in the following section.
The second-order term in equation (3) is expressed as a function of atomic charges  as follows:
as follows:
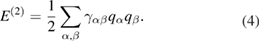
The onsite term  should be identified with the Hubbard parameter or chemical hardness. The two-center term
should be identified with the Hubbard parameter or chemical hardness. The two-center term  mostly contains the
mostly contains the  Coulomb interaction screened at short distances by a contribution of the exchange-correlation second-order derivative. Its complete expression is presented in the seminal paper by Elstner et al [192]. In the standard formulation, atomic charges are computed from the MO coefficients through the Mulliken scheme. It is worth mentioning that the second-order contribution introduced in the DFTB Kohn–Sham operator depends on the MO coefficients. This leads to a variational problem which is solved self-consistently.
Coulomb interaction screened at short distances by a contribution of the exchange-correlation second-order derivative. Its complete expression is presented in the seminal paper by Elstner et al [192]. In the standard formulation, atomic charges are computed from the MO coefficients through the Mulliken scheme. It is worth mentioning that the second-order contribution introduced in the DFTB Kohn–Sham operator depends on the MO coefficients. This leads to a variational problem which is solved self-consistently.
Finally, all the zeroth-order remaining terms are collected in a repulsive contribution Erep which is expressed as a sum of pair potentials  . Technically, the repulsive part is usually parametrized by fitting the difference between the contributions of the other two terms (band energy and second-order terms) and total energy reference calculations.
. Technically, the repulsive part is usually parametrized by fitting the difference between the contributions of the other two terms (band energy and second-order terms) and total energy reference calculations.
Since the derivation of these equations, there have been a number of developments devoted to the improvement and extension of the applications of DFTB. Some consisted in transferring DFT improvements to the DFTB scheme, others are more specific to the DFTB approximations. In connection with the electronic structure description, significant advances are the unrestricted formalism [199], the implementation within periodic boundary conditions and band structure calculations [194], the refinement of atomic charges to go beyond the Mulliken definition [200, 201], inclusion of dispersion contribution [202–204] corrections within the phenomenological Grimme scheme [205], the extension of DFTB to third-order (referred to as DFTB3) in the Taylor expansion of the energy [206] which introduces a charge dependence of the Hubbard parameter, the development of a hybrid short-range/long-range scheme allowing for long-range Hartree–Fock contribution corrections [207], the combination with configuration-interaction methods [208] and the time-dependent DFTB (TDDFTB) approach to treat excited states [209]. Hybrid quantum-mechanical/molecular-mechanical (QM/MM) as well as QM/QM formalisms to incorporate the effect of a chemical environment at low computational cost have also been developed [210, 211]. In the field of dynamics and finite-temperature approaches, one may cite extensive molecular dynamics techniques such as the Car–Parrinello scheme [212, 213] and the coupling of DFTB with global exploration schemes such as genetic algorithms [214], parallel-tempering molecular dynamics (PTMD) [215–219] or basin-hopping techniques [144, 220, 221] or the determination of minimum-energy paths via the nudged elastic band (NEB) algorithm [222]. Calculation of free energies via metadynamics [223–227] or determination of heat capacities with the multiple-histogram method [162, 219, 228, 229] are also available. Techniques to deal with electron dynamics and its possible coupling with nuclei have also been implemented within various schemes such as the Liouville formalism for plasmonics [230], non-adiabatic excited-states dynamics [231, 232], dynamics of electronic transport and its coupling with phonons [233, 234].
3.2. Parametrization and specific issues for gold and silver
The strength of DFTB is to provide both an ab initio justification to parametrized tight-binding schemes and a well-defined strategy to derive the Slater–Koster tables. However, their implementation contains a certain amount of freedom. Indeed, the atomic basis used to compute the two-center matrix elements consists of contracted orbitals, obtained from DFT atomic calculations, and defined by a confinement potential. The choice of this confinement potential can differ between different parameter sets. Recently, it has even been used as a free parameter to improve DFTB properties, for instance electronic energy bands in materials [235]. Different DFT functionals can also be used to achieve the parametrization. Furthermore, DFTB parameters can be further processed and adjusted to improve results with different amounts of empirical intuition, for instance by shifting orbital energies [236] or correcting the short range behavior of matrix elements due to the use of confined orbitals [237, 238]. Such procedures differ from the canonical derivation of the DFTB parameters, but, in a pragmatic way, it can be done based on chemical intuition to correct, for instance, the effects of a too much contracted basis. Such arbitrary adjustments may appear as a disregard of the DFTB theoretical footings and its ab initio character. However one should stress that: (i) the use of approximate DFT functionals or the potential superposition approximation are already a strong deviation from a pure ab initio formalism; (ii) from a pragmatic view, other tight-binding schemes (parametrized empirically) are even less related to first-principles methods.
Another strength of DFTB over other tight-binding schemes is its transferability. Indeed, its theoretical footings make the Slater–Koster table of a given atomic pair a priori transferable from one system to another. Although this is true in essence, this would require heavy fitting of the repulsive part for different combinations of atoms [239], and the parameters themselves are only transferable to other systems if all the necessary Slater–Koster tables are developed in concert with one another. In some specific cases, it is even not possible to develop a DFTB potential that is transferable to chemically different phases of the same element. For instance, Wahiduzzaman et al pointed out the difficulty to implement a carbon-carbon potential accurate enough for both diamond and other carbon phases [235].
Turning now to the specific case of parametrization for silver and gold, the first question relies on the choice of the minimal atomic basis to be used, which of course presents similarities as the two atoms belong to the same group in the periodic table. While the valence atomic orbitals of silver (resp. gold) are the 5s and 4d (resp. 6s and 5d) orbitals, it is relevant to also include 5p (resp. 6p) orbitals, as their radial extension overlaps that of the atomic valence ones. Accounting for relativistic effects is necessary for heavy atoms like gold. For instance, important differences observed for gold and silver clusters involve the critical size for the 2D/3D transition, which occurs at larger sizes in the case of gold as compared to silver. This has been attributed to relativistic effects [74, 240] which strongly modify the valence atomic energies and the hybridization patterns of gold versus silver: dominant s-p mixing in silver favors 3D structures, whereas s-d hybridization in gold favors planar patterns in small clusters. This point is further discussed in section 4.1. At the DFTB level, the parametrized quantities, i.e. atomic orbital energies and two-center Slater–Koster integrals [241], are obtained from scalar-relativistic DFT calculations [242]. Scalar-relativistic effects are thus incorporated in an effective way within the the DFTB matrix elements of equation (2) without resorting to explicit scalar-relativistic operators.
For gold, two parameter sets were derived ten years ago. The first set was proposed by Koskinen et al [241] who showed the importance of relativistic corrections for DFTB to be able to reproduce the correct 2D/3D transition. The role of relativistic effects was also investigated recently in the context of liquid gold membranes [243] by comparing this parameter set with another one derived through the non-relativistic standard procedure [194]. The relativistic parametrization set has been refined later on to achieve better representation of bulk properties through a new definition of the repulsive contribution [244]. The second set was derived by Dong and Springborg [214] in order to conduct extensive exploration of the potential energy surfaces of neutral gold clusters [245]. More recently, in the context of gold interaction with biomolecules, Fihey et al [246] derived a third set of parameters for gold ('auorg set', available at www.dftb.org). These parameters were derived with the aim of accurately modeling both gold bulk and surfaces. They were latter shown to reproduce adsorption energies of biomolecules on gold in good agreement with DFT calculations [247].
For silver, a set of parameters has been derived by Szűcs et al [248] with the initial focus of investigating the adsorption of PTCDA on a silver surface. This parameter set was referred to as 'hyb-0-1 set' at www.dftb.org and has latter been replaced by its extension, 'hyb-0-2 set' (available from www.dftb.org), that can be used for TDDFTB calculations (the two sets of parameters are otherwise identical). These parameters have been used to investigate Si-doped Ag clusters [249], plasmon phenomena on naked and capped silver clusters of different sizes [230, 250–252] and very recently optical properties of silver nanorods, in particular Ag55 nanorod dimers [253]. Sun et al [254], who correlated the Fermi energy and electronic gaps of large clusters with size and geometrical considerations, also benchmarked these parameters with respect to DFT calculations for large clusters.
Oliveira et al recently investigated the quality of some of the aforementioned parameter sets for silver and gold clusters and bulk [255]. In the case of gold, a small modification of the 'auorg set' [246] was found to strongly improve the description of homogeneous small clusters properties while yielding a good description of the gold bulk. The modification consisted in a slight shifting of the energy value  to increase the gap with
to increase the gap with  and
and  and reduce the hybridization of s/d with p orbitals. This highlights that the onsite energies
and reduce the hybridization of s/d with p orbitals. This highlights that the onsite energies  (see equation (3)) of unoccupied atomic orbitals are much less straightforward to define than the onsite energies of the occupied atomic orbitals. Concerning the silver parameters ('hyb-0-2 set' [248]), a slight modification (a scaling of the interatomic Hamiltonian matrix elements by a factor of 0.9) was shown to correct the too early 2D–3D transition in clusters and the overestimation of binding energies and bulk lattice parameter [255].
(see equation (3)) of unoccupied atomic orbitals are much less straightforward to define than the onsite energies of the occupied atomic orbitals. Concerning the silver parameters ('hyb-0-2 set' [248]), a slight modification (a scaling of the interatomic Hamiltonian matrix elements by a factor of 0.9) was shown to correct the too early 2D–3D transition in clusters and the overestimation of binding energies and bulk lattice parameter [255].
In addition to the previously mentioned parametrization works, which aimed at deriving parameters from fitting DFT matrix elements and energies of diatomics, some groups have also tried to obtain parameters for a wide variety of atoms, including silver and gold, via automatized procedures aiming at reproducing particular properties. For instance, Chou et al [256] used a particle swarm optimization technique [145] to derive parameters able to reproduce specific properties from reference data. In another approach, Wahiduzzaman et al [235] developed a strategy based on the optimization of the atomic basis confinement potential to reproduce reference band structure calculations. At the moment, electronic terms, i.e. band energy terms, have been derived for silver and gold but not the repulsive ones [239].
4. From clusters to nanoparticles and bulk
4.1. Small clusters
As mentioned above, there are strong structural differences between silver and gold clusters. An interesting challenge for DFTB in the field of small clusters was thus to check if it was able to reproduce such differential features. The first DFTB investigations of the structural properties of negative gold clusters Au were reported by Koskinen et al [241], showing that DFTB with such parametrization yields planar structures up to size n = 14. They also reported vertical ionization potentials and electron affinities. Dong and Springborg [214] used an optimization scheme based on genetic algorithms to determine the structure and stabilities of clusters in the range 3–58. However, they found the occurrence of 3D-structures already for Au7, which seems much too early with respect to WFT and DFT calculations [41, 91]. We have however to keep in mind that some WFT methods and DFT functionals can also lead to discrepancies as compared to highly accurate WFT based methods such as the coupled cluster scheme as illustrated by Götz and co-workers in the case of Au10 [257]. Dong and Springborg also obtained for Au20 a shape different from the highly symmetric pyramidal shape obtained in DFT and evidenced experimentally [81]. Using an original version of DFTB with a non self-consistent perturbative approach of the on-site charge fluctuations, Rincon et al [258] investigated the structure and energetics of neutral Aun clusters in the range 3–20, quenching a variety of crystal type configurations. Their structures were in quite good agreement with previous DFT results, in particular the onset of 3D structure at size 12, empty cage-like structures for Au16 and Au17 and a pyramidal twentymer Au20 with high Td symmetry, as a subpiece of the fcc (face-centered cubic) bulk lattice for gold. Very recently, Oliveira et al [255] published a systematic work about silver and gold clusters in the range 3–20, conducted with a global minimization scheme based on PTMD. Neutrals, cations and anions were systematically analyzed in order to check the ability of DFTB to cover the different charge situations and the structural modifications resulting from ionization or electron addition in the small cluster range. This work showed several positive features of DFTB and in particular its ability to behave very differently (and satisfactorily) for silver and gold: (i) while the 2D–3D transition occurs at n = 7 in silver clusters whatever the charge, it occurs in the range n = 8–12 for gold clusters (see figure 1), with an increasing transition size from n = 8 for the cations, n = 10 for the neutrals, up to n = 12 for the anions. Thus DFTB correctly accounts for the charge effects in gold clusters, providing structures and energies in agreement with first-principles methods; (ii) DFTB correctly accounts for the preferential trend of silver clusters to build up around five-fold motifs and icosahedral patterns while fcc motifs and the trend to planarity have a major importance for gold clusters. The investigation of Tarrat et al [259] thus recovered pyramidal structures for all three clusters Au20,
were reported by Koskinen et al [241], showing that DFTB with such parametrization yields planar structures up to size n = 14. They also reported vertical ionization potentials and electron affinities. Dong and Springborg [214] used an optimization scheme based on genetic algorithms to determine the structure and stabilities of clusters in the range 3–58. However, they found the occurrence of 3D-structures already for Au7, which seems much too early with respect to WFT and DFT calculations [41, 91]. We have however to keep in mind that some WFT methods and DFT functionals can also lead to discrepancies as compared to highly accurate WFT based methods such as the coupled cluster scheme as illustrated by Götz and co-workers in the case of Au10 [257]. Dong and Springborg also obtained for Au20 a shape different from the highly symmetric pyramidal shape obtained in DFT and evidenced experimentally [81]. Using an original version of DFTB with a non self-consistent perturbative approach of the on-site charge fluctuations, Rincon et al [258] investigated the structure and energetics of neutral Aun clusters in the range 3–20, quenching a variety of crystal type configurations. Their structures were in quite good agreement with previous DFT results, in particular the onset of 3D structure at size 12, empty cage-like structures for Au16 and Au17 and a pyramidal twentymer Au20 with high Td symmetry, as a subpiece of the fcc (face-centered cubic) bulk lattice for gold. Very recently, Oliveira et al [255] published a systematic work about silver and gold clusters in the range 3–20, conducted with a global minimization scheme based on PTMD. Neutrals, cations and anions were systematically analyzed in order to check the ability of DFTB to cover the different charge situations and the structural modifications resulting from ionization or electron addition in the small cluster range. This work showed several positive features of DFTB and in particular its ability to behave very differently (and satisfactorily) for silver and gold: (i) while the 2D–3D transition occurs at n = 7 in silver clusters whatever the charge, it occurs in the range n = 8–12 for gold clusters (see figure 1), with an increasing transition size from n = 8 for the cations, n = 10 for the neutrals, up to n = 12 for the anions. Thus DFTB correctly accounts for the charge effects in gold clusters, providing structures and energies in agreement with first-principles methods; (ii) DFTB correctly accounts for the preferential trend of silver clusters to build up around five-fold motifs and icosahedral patterns while fcc motifs and the trend to planarity have a major importance for gold clusters. The investigation of Tarrat et al [259] thus recovered pyramidal structures for all three clusters Au20,  and
and  , with some Jahn–Teller distortions for the ions, in consistency with the DFT work of Furche et al [148]; (iii) The same investigation determined some electronic properties of gold and silver clusters, in particular the ionization potentials and the electron affinities (see also the early work of Koskinen et al [241]). A good quantitative agreement between DFTB and DFT was obtained for the vertical detachment energies (VDEs) from the gold and silver anions. The vertical ionization potentials (VIPs) of the neutral silver clusters were also found to be quite consistent with the available experimental data, while the agreement with experiment for the VIPs of gold clusters was more disappointing, the DFTB VIPs decreasing somewhat too quickly with increasing sizes in the range n = 3–14. Figures 2 and 3 show the adiabatic IP and EA values for silver and gold respectively, determined from relaxed structure of all charge configurations (neutral, cationic and anionic clusters) in the range n = 3–561. Those curves are in good qualitative agreement with the experimental data presented by Wesendrup et al [260]. In particular, the higher IP and EA of gold over silver clusters is rather well reproduced by DFTB up to n = 20. Finally another interesting feature was the isomerization gap in Au20, which was found to be much larger than in its ionized analogues, while the various charge states of Ag20 exhibit a quasi-degeneracy behaviour with a large number of isomers within a small energy range 0–0.3 eV (even narrower for the anion and the cation). Finally, one may cite a very recent work [221] combining basin-hopping search with DFTB followed by DFT and finding results in essential agreement with those of Tarrat et al [259] for neutral gold clusters.
, with some Jahn–Teller distortions for the ions, in consistency with the DFT work of Furche et al [148]; (iii) The same investigation determined some electronic properties of gold and silver clusters, in particular the ionization potentials and the electron affinities (see also the early work of Koskinen et al [241]). A good quantitative agreement between DFTB and DFT was obtained for the vertical detachment energies (VDEs) from the gold and silver anions. The vertical ionization potentials (VIPs) of the neutral silver clusters were also found to be quite consistent with the available experimental data, while the agreement with experiment for the VIPs of gold clusters was more disappointing, the DFTB VIPs decreasing somewhat too quickly with increasing sizes in the range n = 3–14. Figures 2 and 3 show the adiabatic IP and EA values for silver and gold respectively, determined from relaxed structure of all charge configurations (neutral, cationic and anionic clusters) in the range n = 3–561. Those curves are in good qualitative agreement with the experimental data presented by Wesendrup et al [260]. In particular, the higher IP and EA of gold over silver clusters is rather well reproduced by DFTB up to n = 20. Finally another interesting feature was the isomerization gap in Au20, which was found to be much larger than in its ionized analogues, while the various charge states of Ag20 exhibit a quasi-degeneracy behaviour with a large number of isomers within a small energy range 0–0.3 eV (even narrower for the anion and the cation). Finally, one may cite a very recent work [221] combining basin-hopping search with DFTB followed by DFT and finding results in essential agreement with those of Tarrat et al [259] for neutral gold clusters.

Figure 1. Evolution of the DFTB binding energy of neutral gold (a) and silver (b) clusters as a function of the cluster size for the most stable 2D and 3D structures. Thick lines: 3D clusters. Dashed lines: 2D clusters. Reprinted with permission from [255]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution image
Figure 2. Adiabatic ionization potentials (blue squares) and electron affinities (red circles) of Agn clusters and nanoparticles in the range n = 3–20 and n = 55, 147, 309 and 561. Above n = 20, the joining lines are only indicative. Clusters with sizes n = 55, 147, 309 and 561 are icosahedrons relaxed for each charge state. The dotted line is the experimental Ag(1 0 0) surface work function [261]. Reprinted with permission from [255]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution image
Figure 3. Adiabatic ionization potentials (blue squares) and electron affinities (red circles) of Aun clusters and nanoparticles in the range n = 3–20 and n = 55, 147, 309 and 561. Above n = 20, the joining lines are only indicative. Clusters with sizes n = 55, 147, 309 and 561 are cuboctahedrons relaxed for each charge state. The overlapping dotted lines are the experimental Au(1 0 0) and Au(1 1 0) surface work functions [262, 263]. Although possibly lower in energy (see text), amorphous structures have not been considered in this plot. Reprinted with permission from [255]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution image4.2. Nanoparticles
For larger species encountered in the nanoparticle size range, an important issue explored in the recent years has been the ability of Au NPs to exhibit or not regular structures as a function of their size. Dong and Springborg [214] were the first to provide a detailed description of the structural patterns and suggested the ability of gold to form low coordinated non-regular systems, such as planar and shell like substructures. Yen et al used a modified basin-hopping optimization algorithm [220] and predicted that for Au40 a twisted pyramidal structure (C1 symmetry) should be more stable than a symmetrical tetrahedral structure (C3 symmetry) [221]. From global optimization performed using the PTMD algorithm, Tarrat et al [259] pointed out that disordered Au55 structures are much more stable than highly symmetric isomers such as the icosahedron, the cuboctahedron and the decahedron, confirming the EAM work of Garzon et al [165]. They also demonstrated the presence of planar patterns at the clusters surfaces as well as inner cavities (figure 4). These results were confirmed at the DFT level and are qualitatively consistent with the STEM observations of Wang and Palmer [154]. Concerning larger Au NPs, the same approach was used to investigate the competition between ordered and disordered Au147 isomers (figure 5) and evidenced that the amorphous ones are relevant low-energy candidates expected to contribute in finite-temperature dynamics [101].

Figure 4. Perspective views of the cavity observed inside a low-energy isomer of  [259]. (a) The complete structure of the cluster. (b) The blue atom is cleared to allow for easier observation of the cavity.
[259]. (a) The complete structure of the cluster. (b) The blue atom is cleared to allow for easier observation of the cavity.
Download figure:
Standard image High-resolution image
Figure 5. Perspective view of Au147 clusters [101]. (a) cuboctahedron, (b) icosahedron, (c) amorphous.
Download figure:
Standard image High-resolution imageThe structure of Ag NPs has been much less studied at the DFTB level than the gold one. DFTB has been used to investigate the structure of Ag55, which remains nearly icosahedral for cations, anions and neutrals [259]. This difference in the symmetric nature of silver versus gold NPs was found in line with experiments which suggest highly symmetric structures for silver and more disordered ones for gold [79, 150, 154]. Very recently, DFTB has also been used in the area of machine learning to establish the relationships between the structure and the morphology of silver NPs (with diameters up to 4.9 nm) and their electron transfer properties [254].
Finally, as DFTB is able to optimize particles with a few hundreds of atoms or more, it can be used to examine the consistent convergence of the cohesive energy of gold and silver NPs with increasing sizes towards the bulk value. A two-parameter analytical extrapolation of the cohesive energy [255] was proposed as:
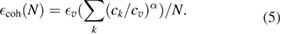
This formula assigns specific cohesion energies  to the atomic sites, depending on their coordination ck, possibly smaller than the coordination
to the atomic sites, depending on their coordination ck, possibly smaller than the coordination  of the volume atoms characterized by a cohesive energy
of the volume atoms characterized by a cohesive energy  . DFTB calculations of cuboctahedral particles with 55, 147, 309 and 561 atoms (compatible with the fcc bulk crystal structures) were used to fit α and
. DFTB calculations of cuboctahedral particles with 55, 147, 309 and 561 atoms (compatible with the fcc bulk crystal structures) were used to fit α and  and hence determine the surface, edge and apex energies (table 1). Regular clusters offer a variety of situations and can provide simultaneous extrapolated information about site energies of facets with various surface symmetries (1 0 0/1 1 1) but also possibly about surface defects with less coordinated surface atoms.
and hence determine the surface, edge and apex energies (table 1). Regular clusters offer a variety of situations and can provide simultaneous extrapolated information about site energies of facets with various surface symmetries (1 0 0/1 1 1) but also possibly about surface defects with less coordinated surface atoms.
Table 1. Coordination (ck) and site cohesive energies  (eV) of cuboctahedral structures of gold and silver clusters derived from the interpolation of large clusters binding energies. Reprinted with permission from [255]. Copyright (2016) American Chemical Society.
(eV) of cuboctahedral structures of gold and silver clusters derived from the interpolation of large clusters binding energies. Reprinted with permission from [255]. Copyright (2016) American Chemical Society.
| Element | Site | ck | 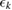 |
|---|---|---|---|
| Ag cubo | |||
| Volume | 12 | 3.022 | |
| Surface(1 1 1) | 9 | 2.600 | |
| Surface(1 0 0) | 8 | 2.445 | |
| Edge | 7 | 2.280 | |
| Apex | 5 | 1.913 | |
| Au cubo | |||
| Volume | 12 | 3.427 | |
| Surface(1 1 1) | 9 | 3.254 | |
| Surface(1 0 0) | 8 | 3.185 | |
| Edge | 7 | 3.110 | |
| Apex | 5 | 2.926 | |
4.3. Dynamics and thermodynamics
As mentioned above, DFTB is well suited for dynamical applications. The analytical gradient is available and makes on-the fly simulations extremely efficient. It allows extensive sampling of initial conditions and rather long trajectories, providing statistically meaningful results in the exploration of dynamical processes such as isomerization, fragmentation or nucleation. Temperature effects may be particularly significant in the case of clusters where many low-energy isomers are in competition. Extensive sampling is also needed to investigate thermodynamical properties and achieve significant thermal averages. DFTB highlighted several very specific dynamical features, connected with finite-size effects and/or the propensity of gold clusters for planar architectures and shapes. Koskinen et al [157] investigated the dynamical coexistence and conversion of 2D and 3D negatively charged liquid clusters Au in the range n = 11–14 around temperature T = 750 K. They demonstrated that although the 2D structures in this size range are lower, the systems exhibit coexistence. This coexistence is related to the fact that the barriers between 2D and 3D minima of the landscape are relatively low, while the barriers between 2D isomers are much higher. Moreover, they also showed that the entropy of the 3D phase is larger and that supercooling may drive the clusters (
in the range n = 11–14 around temperature T = 750 K. They demonstrated that although the 2D structures in this size range are lower, the systems exhibit coexistence. This coexistence is related to the fact that the barriers between 2D and 3D minima of the landscape are relatively low, while the barriers between 2D isomers are much higher. Moreover, they also showed that the entropy of the 3D phase is larger and that supercooling may drive the clusters ( and
and  ) into metastable energetically higher minima of the 3D phase. Such entropic effect is likely to influence the 2D–3D transition thresholds in mobility experiments.
) into metastable energetically higher minima of the 3D phase. Such entropic effect is likely to influence the 2D–3D transition thresholds in mobility experiments.
DFTB was also used to address the thermodynamics of clusters. From the harmonic spectra frequencies of gold clusters, Dong and Springborg calculated the low temperature heat capacities and investigated the possible occurrence of a Boson peak [214] in such small clusters. In a very recent work, Rapacioli et al [162] investigated the thermodynamical properties of the charge states of Au20 from canonical PTMD simulations and the multiple histogram method to determine the heat capacities. They demonstrated that, despite the fact that the ion, the cation and the neutral all have pyramidal and very similar geometries, the canonical heat capacity curves can significantly differ from one another, the melting temperatures of the ions being shifted towards smaller values with respect to the neutral. For the ions, the melting process was shown to take place on a temperature range broader than for the neutral, demonstrating for the magic gold twentymer the dependence of melting upon charge. The low energy landscape of figure 6, which reports the isomerization paths between the lowest energy minima, illustrates the early displacements in the melting processes of the cationic gold cluster  . In the case of Au20, the DFTB simulation also showed the possible occurrence of a convex intruder in the entropy and a negative microcanonical heat capacity within a narrow temperature range, typical of the thermodynamics of finite systems [228, 264, 265].
. In the case of Au20, the DFTB simulation also showed the possible occurrence of a convex intruder in the entropy and a negative microcanonical heat capacity within a narrow temperature range, typical of the thermodynamics of finite systems [228, 264, 265].

Figure 6. DFTB isomerisation path between characteristic structures of  .
.
Download figure:
Standard image High-resolution imageIn addition to those fundamental studies, one may also cite the finite-temperature rearrangement of the electrolyte in the vicinity of gold electrodes that has been studied through a model consisting of a gold cluster with 55 units surrounded by three water layers (640 molecules). A dependence of the orientation of the water molecules was observed as a function of the gold cluster surface charge, this polarisation being limited to the first solvation shell [266].
4.4. Towards plasmonics
One very elegant and promising application of DFTB to gold and silver NPs concerns the modeling of their plasmonic properties. Indeed, as stated in introduction, the field enhancement occurring around a NP due to the localized SPR effect is a subject of considerable ongoing studies. To address this question from a theoretical point of view, a variety of methods have emerged [170]. Among the quantum mechanical methods, TDDFT in the linear-response formulation of Casida [63] has become a very popular tool to compute optical properties of small metal NPs (see section 2). However, it becomes computationally very demanding for systems containing hundreds of metal atoms when electron dynamics, i.e. time evolution of the electrons, has to be considered. To overcome this limitation, the linear response to an electric field of large NPs has been studied by the group of Sánchez through the combination of (i) DFTB to determine the initial single electron density matrix of the considered systems and (ii) the Liouville–Von Neumann equation of motion to describe the time evolution of the density matrices after the application of an external field [250]. As a first application, the field enhancements around two Agn NPs (n = 309 and 489) and a dimer of two Ag201 NPs obtained with this approach and a classical model were calculated. Both formalisms are in qualitative agreement. Nevertheless, the real-time excited-state DFTB scheme provides atomistic information that would be of key importance to further understand the interaction of a molecule with the enhanced field. The performance of this approach for studying the temporal near-field response of the Ag NPs to an arbitrarily shaped electric field was also demonstrated. Very recently, the same methodology allowed Sánchez and co-workers to demonstrate the existence of sub-picosecond breathing-like radial oscillations in silver NPs containing up to 309 atoms [230]. Those oscillations, which start immediately after laser pulse excitation, occur without electron–phonon scattering that takes place at a longer time scale.
A similar methodology combining an initial evaluation of the electronic ground state at the SCC-DFTB level and a real-time propagation of the density matrix using the Liouville–Von Neumann equation of motion was conducted by Ilawe and co-workers to characterize the excitation energy transfer (EET) mechanism along chains of Ag NPs [252] (a similar study was also performed by the same group on a system composed of four Na55 icosahedral NPs [267]). This transfer is made possible by local SPR effects and is of interest to achieve energy and information transfer at high speeds. Thank to the computational efficiency provided by the SCC-DFTB approach, the authors probed the energy transfer along chains of icosahedral Ag55 NPs as a function of interparticle distance (see figures 7(a) and (b)) between the NPs (from 0 to 5 Å). In the range 2–5 Å, SCC-DFTB results are in qualitative agreement with classical electrodynamics. In contrast, below 2 Å, it predicts a strong drop in EET efficiency as demonstrated in figure 7(c), which results from interparticule charge transfer. This is a purely quantum effect that cannot be captured by classical schemes, highlighting the important contribution of SCC-DFTB to probe plasmonic effects at the atomic level.

Figure 7. Schematic representation of two finite chains containing 8 icosahedral Ag55 NPs with two different interparticle (edge-to-edge) distances: (a) 1 Å and (b) 5 Å. (c) Field intensities along silver NP chains with varying interparticle distances. The first NP in each chain is excited at the plasmon resonance energy of the single Ag55 cluster, and the intensity values are computed at the interparticle gaps of the NPs. A drastic drop in the field intensity is seen for NP chains with interparticle spacings less than 2 Å. Adapted from [252] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageVery recently, Alkan and Aikens used the TDDFTB formalism to conduct a study on silver nanorods and nanorod dimers [253]. In this study, the absorption spectra of Agn nanorods with sizes ranging from n = 19 to 67 were first calculated at the TDDFT and TDDFTB levels. TDDFTB produces very similar spectral shapes as compared with TDDFT and predicts the same trends when the nanorod size increases. TDDFTB made possible the calculation of absorption spectra for nanorods up to n = 985. The authors then determined, at the SCC-DFTB level, the evolution of the fractional shifts (defined as the ratio between the change in excitation wavelength and the monomer excitation wavelength) in two different configuration of nanorod dimers, end-to-end or side-to-side, as a function of the separation distance between the nanorods. Three sizes were considered for the monomers: Ag199, Ag391 and Ag799. The results were in good agreement with experimental and classical electrodynamics data previously published in the literature. In addition, the authors unravelled that the change in the excitation wavelength, resulting from the coupling between the two nanorods, depends on the nanorod length.
4.5. Bulk properties
4.5.1. Gold bulk.
The performance of DFTB to address bulk properties has been first evaluated by Fihey et al [246]. They proposed a new parametrization for gold compounds constructed to accurately reproduce the DFT-PBE band structures of three reference gold phases in the energy range around the Fermi level (+/− 4 eV): fcc, body-centered cubic (bcc) and simple cubic. The quality of the high conduction bands and the deep valence bands is poor but only small differences are observed around the Fermi energy (<0.5 eV). Fihey et al have also evaluated the energetic ordering of the three aforementioned phases, complemented by two others, namely A15 and diamond. As expected, the fcc phase has been found as the most stable. The ordering of the other phases is in agreement with the DFT-PBE calculations, the only difference being a reversal of simple cubic and A15 phases. Regarding the structure of the different packings, the bond length deviations are below 2% for all phases. The ability of the Au parametrization proposed by Fihey et al to reproduce relative stabilities and structural properties of gold phases (fcc and hcp closed-packed) has also been evaluated by Oliveira et al [255]. These authors also benchmarked its performance on reproducing elastic properties through the determination of the bulk modulus B0 and the shear elastic moduli  and C44 (and by deduction, two other cubic elastic constants C11 and C12). As mentioned above, another parameter set has been introduced in the latter study, consisting of the Fihey et al parametrization [246] with a shift of the p orbital. Both sets yield the lattice constant and the bulk modulus very close to the experimental ones and slightly underestimate the binding energies when compared to the experimental data. The relative stability of fcc and hcp phases was found similar to that of DFT. However, only poor results were obtained regarding the shear elastic constants and their anisotropy.
and C44 (and by deduction, two other cubic elastic constants C11 and C12). As mentioned above, another parameter set has been introduced in the latter study, consisting of the Fihey et al parametrization [246] with a shift of the p orbital. Both sets yield the lattice constant and the bulk modulus very close to the experimental ones and slightly underestimate the binding energies when compared to the experimental data. The relative stability of fcc and hcp phases was found similar to that of DFT. However, only poor results were obtained regarding the shear elastic constants and their anisotropy.
4.5.2. Silver bulk.
Oliveira et al [255] have conducted a similar evaluation regarding the performance of the parameter set developed for silver by Szűcz et al [248] in reproducing bulk properties. They introduced a slight modification and showed the overall good performance of this parametrization which yields results nearly comparable to those obtained with DFT: the fcc lattice constant differs from the experimental one by only half a percent, the binding energy is slightly overestimated and the phase ordering was correct. Concerning the elastic behavior, despite a quasi-systematic overestimation of the elastic constants, the description of the anisotropy is found to be satisfactory.
5. Functionalized clusters and surfaces
The transferability of the DFTB potential over different chemical systems makes it a valuable tool to study the interaction of molecular species or inorganic materials with metallic surfaces or NPs. The good performance of DFTB for bare gold and silver systems (see section 4) as well as for organic species [268, 269] and inorganic materials [270–272] provide confidence in its ability to model hybrid (Au, Ag)-organic/inorganic systems. As a consequence, over the last 15 years, a number of studies have been devoted to the DFTB study of molecular and inorganic systems in interaction with gold or silver. In the next sections, we give an overview of those studies.
5.1. Gold
The application of DFTB to gold-organic species fall into two main fields: (i) electronic conduction through molecular junctions and simulation of inelastic electron-tunneling spectroscopy (IETS) spectra and (ii) surface chemistry and physics. Both of them take advantage of both DFTB computational efficiency and reasonable accuracy (as compared to DFT) in order to tackle questions hardly tractable at the DFT level.
5.1.1. Electronic conduction through molecular junctions and IETS spectra.
Electronic conduction through molecular junctions has seen considerable progress over the last fifteen years associated to the development of nanoelectronics, optoelectronic and the fabrication of nanodevices [273–278]. Indeed, in this field, where downscaling is of paramount importance, the ability to develop molecular electronics has been a highly appealing research direction motivated by both the small size of molecular components and their tunable electrical characteristics.
Similarly to other atomistic-scale properties, an accurate understanding of molecular conduction has to be developed which necessitates the use of specific theoretical tools. Indeed, modeling electronic transport in molecular junctions is a challenging task which implies a nonequilibrium many-body problem. Although a universal and well established methodology is still missing, various approaches have been proposed to solve this question [275, 277–280]. Among them, the non-equilibrium Green's function (NEGF) technique combined with density functional theory has been widely used as it provides a general framework to compute optical and transport properties in open many-body systems such as metal-molecule-metal junctions. Although it presents some limitations, resulting mainly from the approximate description of exchange-correlation terms [280–282] and the inclusion of vibrational effects [279, 283, 284], the NEGF formalism combined with DFT has allowed to rationalize the conduction properties in terms of electronic structure and to help in interpreting various experimental observations by revealing remarkable trends [275, 277–280].
In this context, Di Carlo and coworkers extended this approach in 2002 to model electron transport in molecular structures at the DFTB level [285]. This was done by coupling the aforementioned non-equilibrium Green's function technique with the DFTB computation of the Hamiltonian leading to the gDFTB approach. gDFTB was first used to compute I–V curves (current–voltage function) in molecular junctions and, in a second step, to model IETS spectra, i.e. simulate  versus
versus  function for a given set of molecules attached on a metallic surface. The theoretical basis of the gDFTB method and its applications have been reviewed in [286–288]. It should be noted that the Green's function formalism was later used by Niehaus and co-workers in order to develop a GW approach based on DFTB energies and orbitals to evaluate reliable optical spectra of solids and molecules [289].
function for a given set of molecules attached on a metallic surface. The theoretical basis of the gDFTB method and its applications have been reviewed in [286–288]. It should be noted that the Green's function formalism was later used by Niehaus and co-workers in order to develop a GW approach based on DFTB energies and orbitals to evaluate reliable optical spectra of solids and molecules [289].
Application of gDFTB in the context of molecular junctions has mostly involved gold contacts on both sides of a given molecule. For instance, its low computational cost enabled to account for thermal vibrations in the conduction properties of di-thio-tolane, dihydroxy–dicyano–diphenylene and alkylthiols between two gold contacts [290–292]. For other alkylthiols, gDFTB provided the basis to rationalize the conduction in terms of contributions of particular orbitals to the transmission channels [293] as well as the role of molecular symmetry in the conduction [294]. In 2008, Pecchia and co-workers also demonstrated how the method can deal with complex boundary conditions, i.e. systems defined by more than two contacts [288]. For instance, this was applied to the case of the tetrathiafulvalene-phenyl-benzoquinone diimine molecule in a three-terminal configuration composed of three gold-contacts. Electron-phonon dephasing effects were further introduced by Penazzi et al in order to account for quantum interference along the junction [295]. The approach was applied in the case of a gold-anthraquinone-gold and gold-anthracene-gold junctions. Finally, transport through oligo(phenylene-ethylene)n molecules coupled to gold was studied as a function of the molecular length and the nature of the anchoring group [296]. By introducing a spectral adjustment in nanoscale transport (SAINT) correction, the authors obtained results in good agreement with experimental data and DFT-based results.
Solomon and co-workers initiated the simulation of IETS spectra with the gDFTB method. They showed that the theoretical IETS spectrum of octanedithiol attached to two gold contacts compares well to the experiments. Such simulation also allowed for an accurate band assignment hardly achievable from experimental data only [297]. This study was extended to other thiols, in particular 1,4-benzenedithiol [298] and cross-conjugated molecules [299]. Finally, one can also note the work by Popov and co-workers which reports the conduction properties of contacts made of molybdenum sulfide nanowires between gold electrodes [300].
5.1.2. Surface chemistry and physics.
In addition to molecular electronics and IETS, DFTB was also used to investigate the structural, energetical and dynamical properties of various surfaces such as molecules attached on gold surface or gold deposited on inorganic surfaces.
Mäkinen et al were the first to develop a DFTB parametrization to describe thiolates attached to gold clusters [244]. Such systems are of primary importance as thiolates are often used to stabilise gold NPs. The DFTB parametrization was validated on three systems: Au25(SMe) , Au102(SMe)44 and Au144(SMe)60 (see optimized DFTB structure in figure 8) for which both experimental and DFT structures were available and on Au102(p-MBA)44 (p-MBA = para-mercaptobenzoic acid). The authors demonstrated that this DFTB parametrization is accurate enough to look for low-energy configurations of other Aun(SMe)m species and to qualitatively describe their electronic structure. Later on, Fihey and co-workers developed another set of DFTB parameters describing the interaction of gold with other atoms: Au-X (X = Au, H, C, S, N, O), in the view to improve the description of the interaction of thiolates and other molecules with gold NPs and surfaces [246]. To test and validate those parameters, the authors considered two model systems: Au3SCH3 and Au25SCH3. For each species, geometrical parameters, density of states, relative energies of four distinct adsorption sites (bridge, hollow hcp, hollow fcc, top) and the potential energy profiles leading from one site to the other, were calculated at both the DFT and DFTB levels of theory. The two sets of data showed good agreement which validates the use of DFTB in the description of gold-thiolate systems.
, Au102(SMe)44 and Au144(SMe)60 (see optimized DFTB structure in figure 8) for which both experimental and DFT structures were available and on Au102(p-MBA)44 (p-MBA = para-mercaptobenzoic acid). The authors demonstrated that this DFTB parametrization is accurate enough to look for low-energy configurations of other Aun(SMe)m species and to qualitatively describe their electronic structure. Later on, Fihey and co-workers developed another set of DFTB parameters describing the interaction of gold with other atoms: Au-X (X = Au, H, C, S, N, O), in the view to improve the description of the interaction of thiolates and other molecules with gold NPs and surfaces [246]. To test and validate those parameters, the authors considered two model systems: Au3SCH3 and Au25SCH3. For each species, geometrical parameters, density of states, relative energies of four distinct adsorption sites (bridge, hollow hcp, hollow fcc, top) and the potential energy profiles leading from one site to the other, were calculated at both the DFT and DFTB levels of theory. The two sets of data showed good agreement which validates the use of DFTB in the description of gold-thiolate systems.

Figure 8. Optimized DFTB structure of Au144(SMe)60. Au atoms in the core (AuC), Au atoms in the thiolate layer (AuL), S, C and H are represented by large yellow, small bright yellow, small orange, small grey and small white spheres, respectively. Reprinted from [244] © EDP Sciences, SIF, Springer-Verlag Berlin Heidelberg 2013. With permission of Springer.
Download figure:
Standard image High-resolution imageIn a similar spirit, Castro and co-workers very recently used the DFTB method to describe the interaction of gold clusters with various amino-acids [247]. They showed that DFTB leads to geometries and adsorption energies in good agreement with DFT results. Furthermore, an analysis of the molecular orbitals suggested an electron-donor character for the proline molecule and an electron-acceptor character for the gold cluster. Analysis for arginine, arginine dipeptide, and TAT peptide were also performed to describe the bonding between those molecules and gold clusters.
Taking advantage of the accuracy and efficiency of the DFTB formalism, Koskinen and Korhonen simulated the dynamical properties of a 2D gold patch suspended in a graphene pore [243]. Aun patches of various sizes were considered: n = 39, 40, 47, 48, 49, 64, 256 and 400. The structure of an Au49 patch is displayed in figure 9(a) as a visual example. Although all patches display a stable solid phase at low temperature, the authors showed that from 49 to ∼256 gold atoms, a stable liquid behaviour of the patch is observed over several hundreds of kelvin (see figure 9(b) a snapshot of the Au49 patch at 900 K). For smaller patches: Au39, Au40, Au47 and Au48, rupture is observed before melting (see a snapshot of Au48 in figure 9(c)), even with fewer atoms in the patch (see the low-temperature structure of Au39 in figure 9(d)). For larger n, large out-of-plane fluctuations lead to the rupture of the gold membrane. This picture was qualitatively confirmed by DFT calculations performed on Au64 only, as extensive molecular dynamics simulation for various patch sizes is computationally too demanding at that level of theory. As already mentioned for small gold clusters (see section 4.1), the two-dimensional character of various gold species results from the impact of relativistic effects on the 6s and 5p orbitals of gold. In the present context, Koskinen and Korhonen showed using both DFT and DFTB simulations that the liquid character of 2D gold patches also partly results from relativistic effects. Consequently, such peculiar behavior is not expected in other metals.

Figure 9. (a) Optimized Au49 patch in a graphene pore of 136 removed C atoms (top and side view). (b) Snapshot of Au49 at 900 K. As a detail, see the Au atoms piled at the upper right hand corner. (c) Snapshot of a ruptured Au48 membrane. (d) Optimized Au39 in a smaller graphene pore of 119 removed C atoms. Adapted from [243] with permission of The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageFinally, one has to mention the coupled experimental/DFTB studies of Nilius and co-workers on the characterization of single layer gold islands (containing up to 200 Au atoms) deposited on a MgO/Ag(001) surface [301, 302]. In those systems, electron transfer occurs between the surface and the gold islands that carry extra electrons which promote the two-dimensional growth of gold. Based on a structural model derived from STM images, DFTB calculations performed on a model Au island demonstrated that the excess charges are localized on the island perimeter in full agreement with the experimental  maps. This peculiar localization explains the formation of elongated belt-like Au structures. From an electronic-structure point of view, DFTB calculations also reproduce the existence and characteristic node structure of quantum-well states in the Au islands [302]. This point was further analysed as a function of the island size and shape which were related to the HOMO-LUMO gap of the gold aggregate and its metallic character [301].
maps. This peculiar localization explains the formation of elongated belt-like Au structures. From an electronic-structure point of view, DFTB calculations also reproduce the existence and characteristic node structure of quantum-well states in the Au islands [302]. This point was further analysed as a function of the island size and shape which were related to the HOMO-LUMO gap of the gold aggregate and its metallic character [301].
5.2. Silver
As stated in section 3.2, a first DFTB potential for silver was developed in 2004 [248]. However, in contrast to gold, fewer studies were further conducted for functionalized silver clusters, NPs or surfaces. Nevertheless, some important DFTB studies have been performed over the last years and are now briefly summarized.
Szűcs and co-workers were the first to use DFTB to study tunneling current between a silver tip and a sulfur-passivated GaAs surface [248]. Two systems were considered: A bare surface and a surface covered by a monolayer of PTCDA (3,4,9,10-perylene tetracarboxylic dianhydride) molecules in order to understand the conduction properties of a representative organic-inorganic semiconductor Schottky-contact. This study was performed using the same gDFTB formalism discussed in the previous section and correctly reproduces the impact of the adsorbate on the tunneling current [248].
Following the methodology applied in section 4.4 for bare silver NPs, Douglas-Gallardo, Berdakin and Sánchez studied the influence of two adsorbates, water and 1,4-benzoquinone, on the SPR band of silver NPs of various sizes [251]. The excitation energy and line width of this SPR band are spectroscopic features of paramount importance in the application of plasmonic NPs as presented in the introduction of the present review. However, those optical properties are strongly sensitive to the size [1, 27], the morphology [1, 27, 28], and the chemical environment [27] of the NP which are properties difficult to probe experimentally. Douglas-Gallardo et al thus intended to rationalise size-effects in five different clusters: Ag55, Ag147, Ag309, Ag561 and Ag923 in three different chemical environments: bare and capped with quinone or water (see for instance the DFTB structure of Ag309 surrounded by 16 1,4-benzoquinones in figure 10(a)). They showed that there exists a linear relationship between the surface plasmon excitation energy and the inverse cube root of the cluster size. Furthermore, the impact of the adsorbate on the spectroscopic features (see figure 10(b) for the case of Ag309 interacting with 1,4-benzoquinones) were analysed in terms of electronic structure changes in the particle resulting from the interaction with the adsorbate. This study thus provides a realistic atomistic scale description of the impact of the chemical environment on the SPR properties of a NP. Of course, real-time excited-state dynamics is rather expensive at the DFT level of theory even for small systems. Consequently, the DFTB approach conducted here provided the correct balance between computational cost and accurate description of the electronic structure to tackle such kind of question that are of high interest in nanoelectronic and optoelectronic.

Figure 10. (a) DFTB optimized structure of 16 1,4-benzoquinones attached on Ag309. (b) Optical absorption spectra of a Ag309 nanocluster with different amounts of adsorbed quinones. Adapted with permission from [251]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution imageAnother appealing study highlighting the contribution of DFTB to better understand surface chemistry of silver is the study of the self-assembly of bisphenol A (BPA) on the Ag(1 1 1) surface by Lloyd and co-workers [303]. In the context of the development of molecular rotors and motors, the authors studied a single-component system built up from BPA molecules displaying a regular two-dimensional network on a silver surface. To provide a complete view of the system, the authors used a synergetic approach coupling STM and NEXAFS experimental measurements with DFTB geometry optimizations of various BPA/Ag(1 1 1) models. The latter were used to rationalize the experimental observations and to provide a description at the atomistic-scale level of the supramolecular arrangement of the BPA molecules. The authors showed that the BPA molecules can arrange differently in dimers and trimers depending on the considered temperature range: from 180 to 250 K and from 250 to 340 K. In the former case, DFTB optimizations confirm that the BPA molecules adopt an L-shape conformation in the dimers with one of the two phenol rings parallel to the metallic surface. In the latter case, the existence of BPA-trimers forming hexagons is also confirmed by the DFTB optimizations. Furthermore, in between 250 and 260 K, both static and rotating trimers are observed by STM (see figure 11(a)) which was interpreted in terms of DFTB optimized structures (see figure 11(b)). The fast development of molecular motors and rotors will make such DFTB calculations more and more attractive and necessary to provide an atomistic-scale understanding of their behavior.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 11. (a) STM image of immobilized caged trimers (colored) coexisting with rotating caged trimers ( nm2,
nm2,  V, I = 0.13 nA, T = 255 K). (b) DFTB optimized trimer structure. The static trimers which are highlighted in green surround the immobilized trimer which is highlighted in orange. The Ag adsorption sites of the trimer motif centers are represented in blue. Adapted with permission from [303]. Copyright (2016) American Chemical Society.
V, I = 0.13 nA, T = 255 K). (b) DFTB optimized trimer structure. The static trimers which are highlighted in green surround the immobilized trimer which is highlighted in orange. The Ag adsorption sites of the trimer motif centers are represented in blue. Adapted with permission from [303]. Copyright (2016) American Chemical Society.
Download figure:
Standard image High-resolution image{kind=link}
6. Outlines and perspectives
In this review, we have attempted to illustrate the applications of DFTB to gold and silver systems, considering bulk, surfaces, clusters and NPs. Although the development of an accurate DFTB parametrization is never an easy task, the joint developments of several groups have provided accurate and transferable DFTB potentials for pure Au and Ag systems, both in the bulk and finite-size domains, as well as for systems interacting with organic molecules and inorganic compounds. This has allowed to perform extensive exploration of the potential energy surfaces of bare gold and silver clusters and NPs with results in agreement with higher-level ab initio simulations. In terms of properties, studies of both electron transport through molecules and plasmonic properties of rather large NPs have been conducted at the DFTB level and have proved to be consistent with DFT calculations or experimental measurements. In near future, one could expect those works to be extended to more complex systems, both in terms of size and chemical complexity. Indeed, the efficiency and accuracy of the DFTB formalism combined to efficient global optimization schemes could allow for the exploration of the potential energy surface of particles containing several hundreds of atoms. Furthermore, in order to keep closer to real compounds, such explorations would benefit from taking surrounding ligands into account as DFTB potentials exist to do so. Finite-temperature effects could also be included by means of molecular dynamics simulations to model defects in the cluster and NP structures, these latter being important for reactivity applications. Those structural studies will also need to be complemented by calculation of quantities experimentally accessible such as absorption and emission spectra through the use of the TDDFTB approach. The recent development of non-adiabatic TDDFTB molecular dynamics will also allow for new applications in the field of photophysics and photochemistry at the surface of gold and silver NPs [232, 304], for instance photoreactivity could be looked at within a dynamical picture. In the plasmonics domain, further efforts would be needed to account for the coupling between nuclear and electronic dynamics to provide new important insights on how a NP structure and chemical environment can affect its absorption and emission properties. Finally, the development of a parameter set to describe mixed [Ag,Au] systems would be of great interest to model AunAg1−n nanoalloys. An increasing interest has been devoted to these particular NPs which composition allows for the fine tuning of their optical properties [305]. As a consequence, their structure has already been subject to various computational studies [306–309] and DFTB has a strong part to play in this domain.
Although the present review focuses on gold and silver only, one has to keep in mind that the DFTB method is general and can be used, a priori, to study any chemical systems. Consequently, a number of DFTB studies were also dedicated to the third element of group 11, i.e. copper. Those studies focused on both the interaction of molecular species such as CO and C60 with a Cu surface [310–312] and the electron transport through Cu nanowires [313]. Application of DFTB to other metals was also conducted but to a lesser extent. For instance, Shi, Koskinen and Ramasubramaniam recently developed a DFTB parametrization for the [Pt,Ru] system in the view to describe PtRu alloys [314]. Using this parametrization to investigate particles made of several tens of atoms, the authors were able to simulate the formation of core-shell-like structures which are observed experimentally. One can also consider to extend such study to other catalytic alloys such as Raney nickel (Ni–Al) for which a proper structural model still does not exist. Such studies will be facilitated by the recent developments of various research groups who attended to provide automatic procedures to generate DFTB potentials [235, 239, 256], at least the electronic part of those potentials [235].
To conclude, the application of DFTB to gold and silver is now at a stage of maturity and is expected to undergo significant expansion over the next years in both the surface and NP fields. Furthermore, a similar development is also expected for other metallic systems, as all the tools are now available for such a rise.
Acknowledgments
The authors would like to thank Cristián G Sánchez, Joachim Reichert, Bryan M Wong and Pekka Koskinen for allowing us to reproduce their pictures. The authors also thank G Seifert and J O Joswig for fruitful discussions. The authors acknowledge the supercomputing facility of CALMIP for generous allocation of computer resources (project P1303, P0059 and P18009). This work was also granted access to the HPC resources of IDRIS under the allocation 2017-i2015087375 attributed by GENCI (Grand Equipement National de Calcul Intensif). It was supported by a CNRS-Inphyniti Grant (ATHENA project), the CNRS-GDR EMIE and the NEXT grants ANR-10-LABX-003 in the framework of the Programme des Investissements d'Avenir (CIM3 and EXTAS projects).