


The morphology of stacked coronene molecules encapsulated in a single-walled carbon nanotube (SWCNT) is investigated using atomistic simulation. First, the minimum energy configuration of coronene molecules in a SWCNT is sought by means of conjugate gradient (CG) minimization. Secondly, encapsulation of coronene molecules into a SWCNT existing in a coronene atmosphere is simulated by means of molecular dynamics (MD). In both of the simulations, the diameter of the SWCNT ranges from 1.35 to 1.69 nm, and the final configurations of coronene molecules within a SWCNT are examined. In a thin SWCNT, coronene molecules tilt against the radial direction of the SWCNT and slide relative to each other, whereas in a thick SWCNT, they do not tilt but rotate relative to each other. In a SWCNT of the intermediate diameter, they tilt, slide, and rotate. For the SWCNT diameter less than or equal to 1.52 nm, the mean tilt angle of the stacked coronene molecules almost linearly decreases with increasing the diameter, whereas for the diameter above 1.52 nm, it is approximately 0∘. To check the validity of the results, the MD simulations are performed changing the density of the coronene atmosphere and the length of the SWCNT; the results prove to be valid. Finally, the effects of temperature on the mean tilt angle and mean intermolecular distance of stacked coronene molecules are examined by a rather simplified simulation, which shows that both of them increase with increasing temperature.
I. INTRODUCTION
It has been almost a quarter of a century since carbon nanotubes (CNTs) were discovered by Iijima.1 Applications of CNTs extend over a wide variety of research fields. The direct applications of CNTs utilize their electronic, thermal and mechanical properties. Researchers have tried to construct devices by using CNTs as building blocks.2–4 A rather indirect application of CNTs is its use as a template or a container to synthesize nanostructures. Synthesis of graphene nanoribbons (GNRs) or columnar stacks of polycyclic aromatic hydrocarbon (PAH) molecules by encapsulation of PAH molecules into CNTs is an example of such usage of CNTs.
Synthesis of GNRs in SWCNTs was succeeded by Talyzin’s group. Talyzin et al.5 synthesized hydrogen-terminated GNRs by polymerization of coronene and perylene molecules encapsulated in SWCNTs; this is a potential method of fabricating GNRs. Their high-resolution transmission electron microscopy (HRTEM) images revealed that the GNRs have helical configurations, which agrees well with the simulation results.6 Furthermore, the electronic structures of the encapsulated GNRs calculated by Mandal et al.7 suggest that these carbon hybrids are applicable to solar cells and hydrogen storage.
On the other hand, synthesis of columnar stacks of PAH molecules was performed first by Okazaki’s group. Okazaki et al.8 obtained one-dimensional structures of coronene molecules encapsulated in SWCNTs through vapor-phase doping. They heated SWCNTs at 500°C to open the ends of the SWCNTs, and confined them into a quartz tube together with coronene molecules. They heated the quartz tube at 450°C. Then, coronene molecules sublimed and entered into the SWCNTs. They washed the SWCNTs encapsulating coronene molecules with toluene, and annealed them at 300°C. From the statistical analysis of the HRTEM images, they found that coronene molecules encapsulated in a SWCNT were stacked in the direction of the SWCNT axis, and that the intermolecular distance and tilt angle of the stacked coronene molecules were 0.35 ± 0.03 nm and about 13∘, respectively. Here the tilt angle is defined as an angle of the normal of the plane of a coronene molecule to the SWCNT axis. Because coronene molecules have a fluorescence property, the columnar stack of coronene molecules in a SWCNT will be applicable as a fluorescence probe which is stable under harsh environments.
Subsequently, Verberck et al.9 performed Monte Carlo (MC) simulations of coronene molecules or dicoronylene molecules in a SWCNT. They assumed coronene and dicoronylene molecules to be rigid. The diameters of their SWCNTs range from 1.4 to 2 nm. They employed a benzene-benzene interaction model10 for intermolecular interactions, whereas they used the smooth-nanotube approximation for molecule-nanotube interactions. According to their simulation results, the morphologies of coronene or dicoronylene molecules are categorized depending on the diameter of the encapsulating SWCNT. When focusing on coronene molecules, ordered one-dimensional stacks of tilted coronene molecules are formed in the SWCNTs of the diameter of from 1.4 to 1.7 nm, which agrees with the experimental observation by Okazaki et al., whereas irregular clusters of coronene molecules are formed in the SWCNTs of the diameter of from 1.8 to 2 nm; the irregular clusters have various orientations. They inferred that formation of such irregular clusters of coronene molecules is a prerequisite for subsequent polymerization of coronene molecules observed by Talyzin et al.5 As for dicoronylene molecules, they also found the two similar regimes depending on the SWCNT diameter. In the diameter range of from 1.5 to 1.7 nm, ordered stacked columns of tilted dicoronylene molecules are observed, whereas in the larger diameter range, disordered configurations are observed, which might form oligomers.
Recently, Kigure et al.11 investigated the energetics and electronic structures of a SWCNT encapsulating PAH molecules, coronene, sumanene, and corannulene. Their first-principles calculation revealed that coronene molecules are likely to be stacked with tilting against the radial direction of the SWCNT, sumanene molecules are likely to be stacked with no tilting, and corannulene are likely to be randomly arranged. They also found that the electronic states of the PAH-SWCNT hybrid depend on the space between the PAH molecules and the SWCNT and on the tilt angle of the PAH molecules, and modulation of the electronic structures of the PAH molecules and the SWCNT is possible by tilting the PAH molecules.
Botka et al.12 tried to realize coronene encapsulation in SWCNTs by vapor phase filling at temperatures 450°C and 385°C and by nanoextraction from supercritical carbon dioxide. In all of their three methods, coronene encapsulation proved to occur. They confirmed the formation of double-walled CNTs by Raman spectroscopy. They recommended the use of the low-temperature method because high temperature vapor filling produces dicoronylene contamination. In the second paper by Botka et al.,13 they used SWCNTs and multi-walled CNTs (MWCNTs) as containers. Encapsulation was realized with the same methods as in their first paper. Characterizing coronene-CNT hybrids by TEM and by Raman and photoluminescence (PL) spectroscopy, they found reaction temperature is the control parameter of the configurations of the final products; at the temperatures 50°C and 385°C, coronene stacks inside SWCNTs were observed, while at 485°C fewer coronene stacks inside SWCNTs were observed. They also concluded that at high temperatures dimerization of coronene molecules and their adhesion to the CNT surfaces occur, and that coronene molecules encapsulated in MWCNTs rarely form a regularly ordered stack and are likely to form dicoronylene molecules. Some authors of the second paper of Botka et al. together with other coauthors wrote a paper14 with a focus on structural characterization of coronene stacks in SWCNTs. They tried to improve imaging of individual atoms by aberration-corrected high-resolution TEM. They showed that the stability of coronene molecules in the electron beam increases by exchanging all the hydrogen atoms of the molecules with deuterium, and for the isotope-substituted coronene molecules, measurements of the inter-molecular spacing and molecular orientation are possible. Their precise analysis yielded the intermolecular distance 0.39 ± 0.03 nm and the tilt angle 10.2 ± 4.6 ∘, which they wrote are consistent with the results of Okazaki et al. However, there are no descriptions about the chiralities and diameters of the SWCNTs in their paper.
Talyzin’s group not only succeeded in creating GNRs inside SWCNTs but also addressed stacking of coronene molecules in SWCNTs. Anoshkin et al.15 reported using TEM that under high-vacuum condition, columnar stacking of coronene molecules and peapod-like moving coronene dimers were observed, whereas under an argon atmosphere, hydrogen-terminated GNRs were identified, and that the columnar stacking agrees with the results of Okazaki et al. They also performed the calculation based on the density functional theory (DFT), and showed that the morphology of encapsulated coronene molecules depends on the diameter of a SWCNT; for the diameter less than 1.2 nm, encapsulation of coronene molecules is not favorable, whereas for the diameters in the range from 1.3 to 1.6 nm, columnar stacking of coronene molecules with tilting is favorable. Chernov et al.16 studied coronene stacking inside SWCNTs using PL spectroscopy, and found that coronene and dicoronylene outside SWCNTs and coronene stacks encapsulated in SWCNTs have their own intrinsic PL spectral features, and these two can be discriminated by the PL spectra. Their molecular mechanics calculation supported their optical observation, and predicted the most favorable configurations of coronene molecules depending on the SWCNT diameter.
Dappe and Martínez17 theoretically investigated coronene encapsulation in a SWCNT by using DFT calculation taking van der Waals interactions into account. They first studied the interaction between two coronene molecules in an open space; these two molecules make a stacked coronene dimer. They found that in the minimum energy structure of the coronene dimer, one coronene rotates with respect to the other by 30∘. Next, they calculated the minimum energy structure of three coronene molecules inside a SWCNT which rotate relative to each other by 30∘. The minimum energy structure was obtained for a tilt angle 35∘ and at an inter-molecular distance 4 Å. Furthermore, they found the calculated many-body excitation spectrum of the coronene molecules inside the SWCNT shows an optical red-shift with respect to that of diluted self-assembled coronene molecules. Dappe18 reviewed theoretical researches of encapsulation of organic molecules in CNTs. After he focused on the theoretical methods to describe van der Waals interactions between organic molecules and CNTs, he presented the structural aspects of fullerene in a SWCNT and in a double-walled CNT (DWCNT), a GNR in a SWCNT, acetylene in a SWCNT, and terthiophene in a SWCNT. He further discussed the magnetic, electrical, and optical properties of encapsulated molecules. He finally presented the confinement of DNA molecules in a SWCNT, and discussed the applications in biomaterials.
Among the above-cited articles regarding encapsulation of PAH molecules into CNTs, the series works of Okazaki et al. are trailblazing in the study of columnar stacking of coronene molecules in CNTs. In this paper, motivated by their series works8,9,11 and Dappe and Martínez’s17 results, morphology of a columnar stack of coronene molecules encapsulated into a SWCNT is studied by means of CG energy minimization and MD simulation. Although Verberck et al. assumed coronene molecules to be rigid in their MC simulation, we will take into account the carbon-carbon and carbon-hydrogen interactions within each of a coronene molecule and a SWCNT and the van der Waals interactions between the atoms of coronene molecules and a SWCNT, and hence our coronene molecules and SWCNTs are deformable. First, we will obtain minimum energy configurations of the nanohybrid consisting of stacked coronene molecules and a SWCNT by means of CG minimization. Next, we will study dynamical encapsulation of coronene molecules into a SWCNT in a coronene atmosphere by MD simulation. We will exclusively focus on how the morphology of the encapsulated coronene molecules depends on the SWCNT diameter.
II. SIMULATION MODEL AND METHOD
The schematic of a coronene molecule is shown in Fig. 1. Coronene is a PAH molecule C24H12 which consists of a central benzene ring and six benzene-type rings around it. Its size is about 1 nm, and its solution emits blue fluorescence. In this paper, the C atoms and their atomic bonds of coronene molecules are colored gray whereas the H atoms are colored blue. If necessary, the C atoms and their atomic bonds of coronene molecules inside SWCNTs are colored red.

Schematic of a coronene molecule. The balls colored with gray are carbon atoms, whereas the balls colored with blue are hydrogen atoms.
Schematic of a coronene molecule. The balls colored with gray are carbon atoms, whereas the balls colored with blue are hydrogen atoms.
Firstly, we seek the minimum-energy structures of columnar stacks of coronene molecules in SWCNTs of different diameters by means of CG minimization. Fourteen SWCNTs are selected so that their diameters may cover the range of from 1.35 to 1.69 nm. We did not care about what chiralities the SWCNTs have, assuming that it is the diameter of the SWCNTs that controls the configurations of the encapsulated coronene molecules. This assumption will prove to be valid from the simulation results in the next section. The chiral indices and diameters of the selected SWCNTs are shown in Tables I, II, and III. The diameters of the SWCNTs are calculated by the equation , where and (n,m) are the chiral indices.19 An example of the initial atomic arrangement of the columnar stacking of coronene molecules in a SWCNT is shown in Fig. 2(a). The length of the SWCNT is 100 Å. Ten coronene molecules are placed at intervals of 3.4 Å in a SWCNT perpendicularly to the SWCNT axis. The length of the SWCNT and the number of the molecules are selected so that the effects of the SWCNT ends may be negligible.
Chiral indices and diameters (nm) of SWNTs of zigzag type.
| chiral indices | (17,0) | (18,0) |
|---|---|---|
| diameter | 1.350 | 1.429 |
| chiral indices | (17,0) | (18,0) |
|---|---|---|
| diameter | 1.350 | 1.429 |
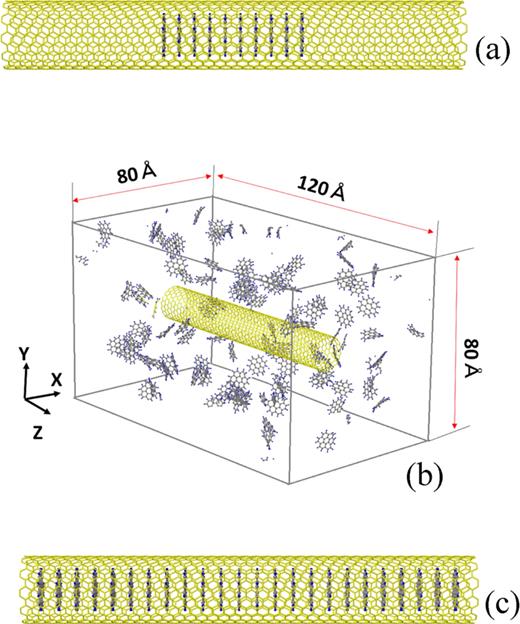
(a)An initial structure of a corenene-SWCNT hybrid for CG energy minimization. (b)Simulation cell for MD simulation of encapsulation of coronene molecules into a SWCNT existing in a coronene atmosphere. The periodic boundary conditions are imposed in all the three directions. (c)An initial structure of a coronene-SWCNT hybrid for the simplified MD simulation to examine the effect of temperature.
(a)An initial structure of a corenene-SWCNT hybrid for CG energy minimization. (b)Simulation cell for MD simulation of encapsulation of coronene molecules into a SWCNT existing in a coronene atmosphere. The periodic boundary conditions are imposed in all the three directions. (c)An initial structure of a coronene-SWCNT hybrid for the simplified MD simulation to examine the effect of temperature.
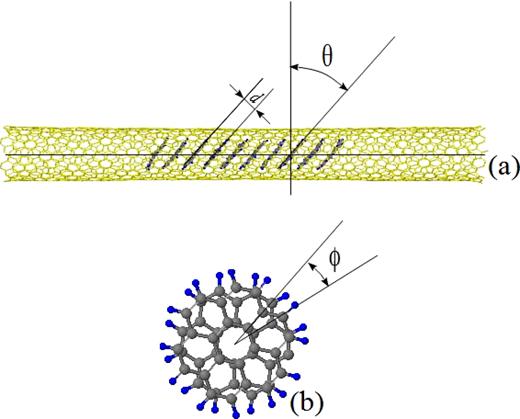
Definitions of the tilt angle θ, distance d, and rotational angle ϕ of the coronene molecules encapsulated in a SWCNT.
Definitions of the tilt angle θ, distance d, and rotational angle ϕ of the coronene molecules encapsulated in a SWCNT.
Secondly, dynamical encapsulation is studied by MD simulation. An initial situation as shown in Fig. 2(b), where a SWCNT is placed in a coronene atmosphere, is created as follows. A fundamental cell is of the sizes 80Å ×80Å ×120Å in the x-, y-, and z-directions, respectively. The periodic boundary conditions are imposed in all the three directions. At first, in this fundamental cell, 104 coronene molecules are orderly placed near the four walls. In addition, four columnar stacks of coronene molecules are placed near the center of the cell so that the projections of the 104 coronene molecules and the four columnar stacks onto an x-y plane may not overlap. Fixing the four columnar stacks, the MD calculation at the constant temperature 700 K is executed during 106 time steps. After that, the four columnar stacks are removed, and a SWCNT of the length 100 Å in Table I, II, and III, which has been equilibrated at 700 K beforehand, is inserted into the space which the removed four columnar stacks have occupied. Then, the MD calculation at 700 K is performed during 3×106 time steps, and dynamical encapsulation of coronene molecules is monitored. A coronene atmosphere created by the above procedure is considerably dense compared with the real atmosphere in experiments. Accordingly, encapsulation of coronene molecules in our simulations must be an accelerated one. However, such acceleration is inevitable because enormous amount of calculation time would be necessary if the density of a real coronene atmosphere was adopted in simulations. To examine the effect of the density, dynamical encapsulation is simulated in a rarefied atmosphere where 52 coronene molecules exist in the fundamental cell shown in Fig. 2(b). In addition, to examine the effect of the CNT length, dynamical encapsulation is simulated for three selected SWCNTS of the length 300 Å.
Chiral indices and diameters (nm) of SWNTs of armchair type.
| chiral indices | (10,10) | (11,11) | (12,12) |
|---|---|---|---|
| diameter | 1.375 | 1.513 | 1.650 |
| chiral indices | (10,10) | (11,11) | (12,12) |
|---|---|---|---|
| diameter | 1.375 | 1.513 | 1.650 |
Chiral indices and diameters (nm) of SWNTs of chiral type.
| chiral indices | (14,6) | (12,9) | (14,7) | (16,5) | (13,9) | (18,3) | (12,11) | (15,8) | (19,4) |
|---|---|---|---|---|---|---|---|---|---|
| diameter | 1.411 | 1.449 | 1.470 | 1.508 | 1.521 | 1.562 | 1.582 | 1.606 | 1.690 |
| chiral indices | (14,6) | (12,9) | (14,7) | (16,5) | (13,9) | (18,3) | (12,11) | (15,8) | (19,4) |
|---|---|---|---|---|---|---|---|---|---|
| diameter | 1.411 | 1.449 | 1.470 | 1.508 | 1.521 | 1.562 | 1.582 | 1.606 | 1.690 |
Finally, the effects of temperature on the tilt angle and intermolecular distances of coronene molecules encapsulated in a SWCNT are examined in a rather simplified simulation because it would take a large amount of computational time to repeat dynamic encapsulation simulations at different temperatures. Temperature of the system consisting of the ‘ready-made’ columnar stack of coronene molecules and the encapsulating SWCNT of the length 100 Å shown in Fig. 2(c) is raised from 1 K to 800K in MD calculation. At first, the raising rate is 99 K per 5×105 time steps, and the second and subsequent raising rates are 100 K per 5×105 time steps. The system is equilibrated at every 100 K during 5×105 time steps. The tilt angle and intermolecular distance at each equilibrated temperature are estimated by the method described in the next after the next paragraph.
Simulations are performed by using Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) code.20 The velocity-Verlet algorithm is adopted for integration of equations. The Nosé-Hoover thermostat21–23 is used for temperature control. The time step is set 1fs. The interactions between C atoms and between C atoms and H atoms are calculated by using the adaptive intermolecular reactive empirical bond order (AIREBO) potential24 which consists of the following three terms. The first term is the Brenner bond order potential for the short-range interactions within hydrocarbons.25 The second term is the Lennard-Jones potential for long-range interactions; this term contains a switching function to make short-range interactions ineffective. The third term is the torsional potential for dihedral-angle interactions in hydrocarbons. Thus the AIREBO potential is valid for representing the intramolecular and intermolecular interactions of coronene molecules and a SWCNT.6
From the minimum-energy structures obtained by the conjugate-gradient minimization and the snap shots at the final time step of the MD simulations, the mean tilt angle, mean intermolecular distances, and mean rotational anlges of stacked coronene molecules encapsulated in a SWCNT are computed. The definitions of the tilt angle θ and the distance d are shown in Fig. 3(a), and the definition of the rotational angle ϕ is shown in Fig. 3(b), where the two coronene molecules whose centers coincide with each other are drawn for example. Using the coordinates of the atoms of each coronene molecule, the equation of the molecular plane is determined by means of the least-squares method. The tilt angle of a coronene molecule is defined as the angle between the normal of the plane of the molecule and the SWCNT axis or in other words, the angle between the plane of the molecule and a plane normal to the SWCNT axis. This definition is the same as of Botka et al. and Kigure et al., while this definition is different from that of Okazaki et al. and Verberck et al. who defined it by 90∘ − θ. For the columnar stack of coronene molecules, the tilt angles of the molecules are averaged. The intermolecular distance between two coronene molecules is obtained by averaging the distances between the points of the 36 atoms consisting the one molecule and the plane of the other molecule. For the columnar stack of coronene molecules, the intermolecular distances between one molecule and its nearest neighbor in the stack are averaged. Furthermore, the individual rotational angle of the neighboring two coronene molecules can be calculated from the angle between the C-C bonds lying on the line passing through each of the centers of the molecules whether or not the centers of the two molecules coincide with each other. The rotational angles are calculated for all pairs of the neighboring molecules constructing the columnar stacks in a SWCNT, and averaging these angles yields the mean rotational angle.
III. RESULTS AND DISCUSSION
A. CG energy minimization
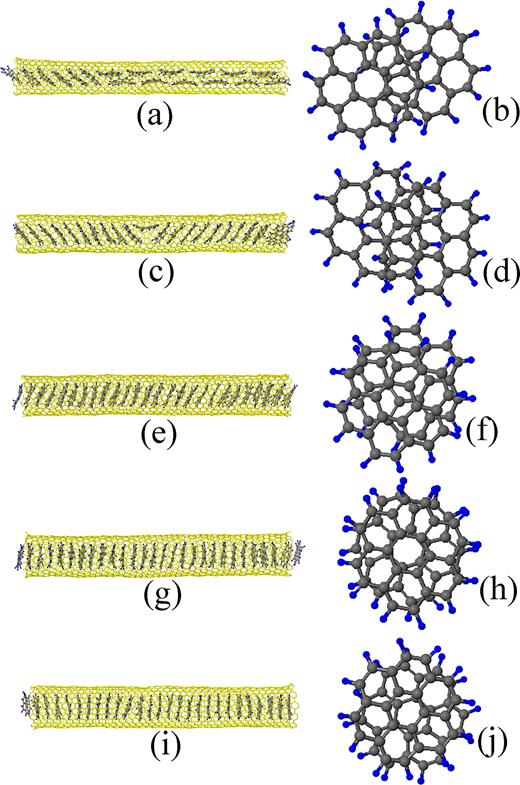
Among the final configurations of the CG energy minimizations for the SWCNTs shown in Tables I, II, and III, the five results for the (10,10), (14,7), (13,9), (18,3), and (12,11) SWCNTs of the diameters 1.375, 1.470, 1.521, 1.562, and 1.582 nm, respectively, are presented in Fig. 4, where (a) and (b) are for the (10,10) SWCNT, (c) and (d) for the (14,7) SWCNT, (e) and (f) for the (13,9) SWCNT, (g) and (h) for the (18,3) SWCNT, and (i) and (j) for the (12,11) SWCNT. Two neighboring coronene molecules near the center of the SWCNT in each of the side views Figs. 4(a), 4(c), 4(e), 4(g), and 4(i) are picked up, and their plan views are shown in the Figs. 4(b), 4(d), 4(f), 4(h), and 4(j), respectively.

Final configurations of the encapsulated coronene molecules in the CG energy minimizations. (a) and (b) are for the (10,10) SWCNT of the diameter 1.375 nm, (c) and (d) for the (14,7) SWCNT of the diameter 1.470 nm, (e) and (f) for the (13,9) SWCNT of the diameter 1.521 nm, (g) and (h) for the (18,3) SWCNT of the diameter 1.562 nm, and (i) and (j) for the (12,11) SWCNT of the diameter 1.582 nm. (a), (c), (e), (g), and (j) are side views. (b), (d), (f), (h), and (j) are plan views of the two neighboring coronene molecules near the centers of each of the coronene stacks.
Final configurations of the encapsulated coronene molecules in the CG energy minimizations. (a) and (b) are for the (10,10) SWCNT of the diameter 1.375 nm, (c) and (d) for the (14,7) SWCNT of the diameter 1.470 nm, (e) and (f) for the (13,9) SWCNT of the diameter 1.521 nm, (g) and (h) for the (18,3) SWCNT of the diameter 1.562 nm, and (i) and (j) for the (12,11) SWCNT of the diameter 1.582 nm. (a), (c), (e), (g), and (j) are side views. (b), (d), (f), (h), and (j) are plan views of the two neighboring coronene molecules near the centers of each of the coronene stacks.
If the side views in these figures are observed, it is found that the tilt angles of the coronene molecules decrease with increasing the diameter of the SWCNT; the mean tilt angles of the coronene molecules shown in Figs. 4(a), 4(c), 4(e), 4(g), and 4(i) are 48.6∘, 32.2∘, 20.4∘, 1.5∘, and 1.3∘, respectively. Although the cross-sectional views of the SWCNTs are not shown, the deformations of the SWCNTs to ellipses are observed at the cross-sections where the coronene molecules exist.
In Fig. 5, the mean tilt angles of stacked coronene molecules for all the SWCNTs shown in Tables I, II, and III except the (17,0) SWCNT are plotted against the diameter. In this figure, the data expressed by the circles are obtained by CG energy minimization, whereas the data expressed by the squares are obtained by dynamic simulation (DS) of encapsulation described in the next subsection III B. The data for the thinnest SWCNT of the chiral indices (17,0) are omitted because the number of the stacked coronene molecules in the dynamic simulation is only one, and its tilt angle cannot be an objective to be compared with. With increasing the diameter of the SWCNT, the mean tilt angle of coronene molecules almost linearly decreases down to about 20.4∘ for the (13,9) SWCNT of the diameter 1.521 nm. If the diameter of the SWCNT is larger than 1.521 nm, the mean tilt angle is almost constant and close to 0∘.

The mean tilt angles of the stacked coronene molecules are plotted against the SWCNT diameter. The data expressed by the circles are calculated from the final configurations by CG energy minimization, whereas the data expressed by the squares are calculated from those by dynamic simulation (DS) of encapsulation.
The mean tilt angles of the stacked coronene molecules are plotted against the SWCNT diameter. The data expressed by the circles are calculated from the final configurations by CG energy minimization, whereas the data expressed by the squares are calculated from those by dynamic simulation (DS) of encapsulation.
Tilting of the coronene molecules in a thin SWCNT is necessary for avoiding steric hindrance and keeping the distances between the atoms consisting of the molecules and the inner wall of the SWCNT constant.11 The mean difference between the radial coordinates of the H atoms constructing coronene molecules and the C atoms constructing the encapsulating thin SWCNT is estimated to be almost a constant 2.7 Å in the SWCNT diameter range of from 1.375 to 1.521 nm. This difference is close to the sum of of the van der Waals radius of a H atom 1.2 Å and that of a C atom 1.7 Å. Accordingly, it is suggested that for the thin SWCNTs, the T-shaped CH/π interactions6,26,27 are prevailing between the CH bonds of the coronene molecules and the six-membered ring of the encapsulating SWCNT.
In Fig. 6, the mean intermolecular distance of stacked coronene molecules is plotted against the diameter of the SWCNT. The data are obtained only from the final configurations by CG energy minimization because the data calculated from those by dynamic simulation of encapsulation vary widely. From Fig. 6, it can be said the intermolecular distance increases very slightly or is considered to be almost a constant of 3.4 Å except the one for the thinnest (17,0) SWCNT.
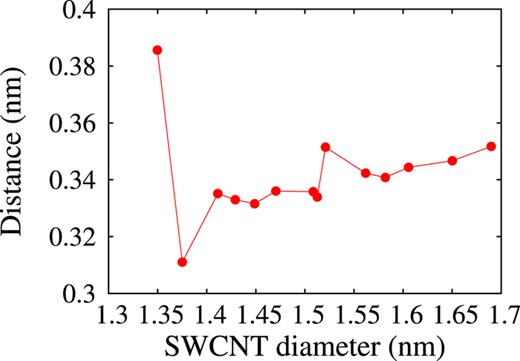
The mean distances between the stacked coronene molecules in the final configurations obtained by CG energy minimization are plotted against the SWCNT diameter.
The mean distances between the stacked coronene molecules in the final configurations obtained by CG energy minimization are plotted against the SWCNT diameter.
Returning now to Fig. 4 and referring to Figs. 5 and 6, let us discuss the morphology of the coronene molecules. Observation of the plan views shown in Fig. 4 reveals that if the diameter of the SWCNT is small (see Figs. 4(b) and 4(d)), the coronene molecules accommodate themselves to the inner space of the SWCNT by sliding relative to each other and making a stacking similar to ‘AB stacking’ under tilting. Here, ‘AB stacking’ is such a way of stacking of two graphene sheets that the carbon atoms of one graphene sheet are positioned at the centers of the six-membered rings of the other graphene sheet. Podeszwa28 named this stacking ‘graphite’ structure for the coronene dimer. He suggested that the ‘graphite’ structure cannot be the global minimum for the coronene dimer owing to the edge effects, and the ‘shifted-graphite’ structure where one coronene molecule in the ‘graphite’ structure is a little further shifted relative to the other molecule is likely to be the structure of the global minimum energy for the coronene dimer. Although in our simulation results, the detailed stacking features of the two neighboring coronene molecules encapsulated into SWCNTs depend on their positions in the column and slightly fluctuate, it is probable that in thin SWCNTs, the tilted coronene molecules are likely to be stacked taking the graphite or shifted-graphite structure. If the diameter of the SWCNT is large (see Figs. 4(h) and 4(j)), the coronene molecules accommodate themselves to the inner space of the SWCNT by rotating relative to each other under no tilting. In Fig. 4(j), one coronene molecule rotates relative to the other by about 20∘; Podeszwa and Szalewicz29 named this stacking ‘crossed’ for the coronene dimer, while Zhao and Truhlar30 named it ‘twisted sandwich.’ In the intermediate case of the (13,9) SWCNT in Fig. 4(f), coronene molecules both slide and rotate relative to each other under tilting.
In Fig. 7, the mean rotational angles of coronene molecules relative to each other in the final configurations obtained by CG energy minimization are plotted against the diameter of the SWCNT. It can be seen the rotational angle tends to increase on the whole and gets closer to 30∘ with increasing the diameter of the SWCNT, although some data are out of this tendency.

The mean rotational angles between the neighboring coronene molecules are plotted against the SWCNT diameter.
The mean rotational angles between the neighboring coronene molecules are plotted against the SWCNT diameter.
Dappe and Martínez17 sought the stable structure of three coronene molecules inside a (19,0) SWCNT by using a calculation scheme based on the density functional theory (DFT) incorporating van der Waals interaction. They concluded that a rotation of every coronene molecule relative to the neighboring molecules is essential to the stable structures of coronene molecules, and the most stable structure is obtained for a tilt angle of 35∘ at an inter-coronene distance of 4 Å under a rotation of 30∘. The diameter of the (19,0) SWCNT is 1.508 nm which is the same as the diameter of our (16,5) SWCNT. However, the final configuration of our CG energy minimization for the (16,5) SWCNT is almost equal to that shown in Figs. 4(c) and 4(d) where no relative rotation of coronene molecules is necessary to formation of its configuration and only relative sliding suffices for it. As shown in Fig. 5 and Fig. 6, the tilt angle and intermolecular distance for the (16,5) SWCNT are 29.8∘ and 3.36 Å, respectively. The reason why the configuration by our CG minimization for the (16,5) SWCNT is different from the most stable one Dappe and Martínez obtained is that they calculated the cohesive energy of the structures on the condition that one coronene molecule rotates relative to each other by 30∘, and they did not take its sliding relative to each other into account. It would be confirmed by first-principles calculation that the stacking structures such as shown in Figs. 4(b) and 4(d) are energetically favorable for the thin SWCNTs. However, it is beyond the scope of this paper.
On the other hand, the configuration shown in Fig. 4(j) is close to the most stable one Dappe and Martínez obtained for an isolated coronene dimer in a free space. For the (12,11) SWCNT of the relatively large diameter 1.582 nm, the interaction between the coronene molecules and SWCNT is weak, and it is as if the coronene molecules were in an free space.
The statistical analysis of the HRTEM images by Okazaki et al. yielded the tilt angle 13∘ and intermolecular distance 0.35 ± 0.03 nm for their encapsulated coronene molecules. They made no mention of the detailed relative positioning of the neighboring coronene molecules in their experiments and DFT calculation. They compared the tilt angle 13∘ with the tilt angle 25∘ of the optimum structure of coronene molecules in the (19,0) SWCNT obtained by DFT calculation, in which they had to use the fixed intermolecular distance 0.42 nm. In the fourteen SWCNTs selected in our simulation, the diameter 1.508 nm of the (16,5) SWCNT is the same as of the (19,0) SWCNT, and the diameter 1.513 nm of the (11,11) SWCNT are close to that of the (19,0) SWCNT. According to Fig. 5, the tilt angles for the (16,5) and (11,11) SWCNTs are 29.8∘ and 24.6∘, respectively, and according to Fig. 6, the intermolecular distances are 0.336 nm and 0.334 nm, respectively. Th tilt angle 24.6∘ is almost the same as the result of Okazaki et al.’s DFT calculation. Although the tilt angles 29.8∘ and 24.6∘ are larger than their experimental value 13∘, the intermolecular distance 0.336 nm and 0.334 nm are close to the experimental value 0.35 ± 0.03 nm.
Verberck et al.9 also made no mention of relative positioning of the neighboring coronene molecules in their columnar stacks obtained by their MC simulation. They obtained the average tilt angle 11.1∘ for the SWCNT of the diameter 1.5 nm at 300 K. In addition, the average intermolecular distance which can be read from their figure is 0.365 nm at 300K. Their tilt angle and intermolecular distance are very close to the experimental values of Okazaki et al. Such agreements are remarkable if it is considered that Verberck et al. assumed coronene molecules to be rigid, and used a model of benzene-benzene interactions10 for intermolecular interactions. From the side views shown in Fig. 4, our coronene molecules are warped even though our results obtained by CG energy minimization correspond with the results of MD simulation at 0 K. Accordingly, it is conjectured that the deformations of coronene molecules could not be neglected.
Finally in this subsection, the following should be noticed. The fact that the mean tilt angle and mean rotational angle of the coronene molecules encapsulated in a SWCNT almost linearly dependent on the SWCNT diameter in the smaller half of the diameter range of our SWCNTs and the mean intermolecular distance is almost constant implies that such geometrical factors determining the morphology of the stacked coronene molecules do not depend on the chirality of the encapsulating SWCNT. Thus, our assumption on selecting the SWCNTs prove to be valid.
B. Dynamic simulation of encapsulation
Dynamic simulations of encapsulation of coronene molecules from a coronene atmosphere at 700 K were performed for the SWCNTs shown in Tables I, II, and III. In all the simulations, coronene molecules were spontaneously encapsulated into SWCNTs. According to the DFT calculations by Kigure et al.,11 encapsulation of coronene molecules into the (18,0), (19,0), and (20,0) SWCNTs is exothermic, and encapsulation of coronene molecules into the (16,0) and (17,0) SWCNTs is endothermic. They estimated the threshold diameter for encapsulation to be 1.36 nm, which is slightly larger than the diameter 1.35 of the (17,0) SWCNT. However, this estimation was done on the condition that coronene molecules are stacked with no tilting. As they pointed out, for thin SWCNTs, tilted coronene molecules yield a decrease of the total energy of the hybrid. Hence, spontaneous encapsulation of coronene molecules into the (17,0) SWCNT in our dynamic simulation does not contradict their result. The final snapshot of coronene molecules encapsulated into the (17,0) SWCNT will be shown in the next subsection III C.
As typical examples of the encapsulation, two series of snapshots for the (10,10) and (11,11) SWCNTs are shown in Figs. 8 and 9, respectively. Furthermore, video movies of these two simulations are provided; each of the video movies is divided into ten segments, from which the five segments are selected. In the snapshots shown in Figs. 8 and 9, the C atoms and their atomic bonds of the coronene molecules existing inside a SWCNT are colored red in order to discriminate them from the coronene molecules existing outside the SWCNT. As shown in Figs. 8 and 9, coronene molecules swarm around and move along the surfaces of SWCNTs.
![FIG. 8. The snapshots in the dynamic simulation of encapsulation of coronene molecules into the (10,10) SWCNT of the diameter 1.375 nm; (a) is at 0th time step, (b) at 38000th time step, (c) at 225000th time step, (d) at 278000th time step, (e) at 408000th time step, (f) at 814000th time step, (g) at 996000th time step, and (h) at 1039000th time step. (Multimedia view) [URL: http://dx.doi.org/10.1063/1.4935482.1] [URL: http://dx.doi.org/10.1063/1.4935482.2] [URL: http://dx.doi.org/10.1063/1.4935482.3] [URL: http://dx.doi.org/10.1063/1.4935482.4] [URL: http://dx.doi.org/10.1063/1.4935482.5]](https://aipp.silverchair-cdn.com/aipp/content_public/journal/adv/5/11/10.1063_1.4935482/3/m_117113_1_f8.jpeg?Expires=1716405946&Signature=UgB6VkCgimOgYvSkmpCH6yGW-JqnSdr4osnfhHyUZes0dxTdXjBzy38HUW~u7~4nXi7qcCf8-7d9nWNVSXn~gquLXvk89fF3AG6yfaU2oxByZMNAizCUVmvdHskPpn~WSb9AJNiGExLk-YB~Oz9L~Bz3gUkFggqiuIPFD6mguok2Q6sEw2cNvcih01JM9Crg2Ra64X4xYALICHfusogwW7AZuYdFiKsB6s2Bwme~4ysU77BpvUmDN8Cbq6yCGP7iVTNlakKiO6IE4pUaZinXdvgOeTQeCGqURV2WMAVnv9OqWvcWTN2AgQN8792XVL056zoqLg7C7MVHE84DY3HLZQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The snapshots in the dynamic simulation of encapsulation of coronene molecules into the (10,10) SWCNT of the diameter 1.375 nm; (a) is at 0th time step, (b) at 38000th time step, (c) at 225000th time step, (d) at 278000th time step, (e) at 408000th time step, (f) at 814000th time step, (g) at 996000th time step, and (h) at 1039000th time step. (Multimedia view) [URL: http://dx.doi.org/10.1063/1.4935482.1] [URL: http://dx.doi.org/10.1063/1.4935482.2] [URL: http://dx.doi.org/10.1063/1.4935482.3] [URL: http://dx.doi.org/10.1063/1.4935482.4] [URL: http://dx.doi.org/10.1063/1.4935482.5]
The snapshots in the dynamic simulation of encapsulation of coronene molecules into the (10,10) SWCNT of the diameter 1.375 nm; (a) is at 0th time step, (b) at 38000th time step, (c) at 225000th time step, (d) at 278000th time step, (e) at 408000th time step, (f) at 814000th time step, (g) at 996000th time step, and (h) at 1039000th time step. (Multimedia view) [URL: http://dx.doi.org/10.1063/1.4935482.1] [URL: http://dx.doi.org/10.1063/1.4935482.2] [URL: http://dx.doi.org/10.1063/1.4935482.3] [URL: http://dx.doi.org/10.1063/1.4935482.4] [URL: http://dx.doi.org/10.1063/1.4935482.5]
![FIG. 9. The snapshots in the dynamic simulation of encapsulation of coronene molecules into the (11,11) SWCNT of the diameter 1.513 nm; (a) is at 0th time step, (b) at 231000th time step, (c) at 290000th time step, (d) at 732000th time step, (e) at 1100000th time step, (f) at 1108000th time step, (g) at 1492000th time step, and (h) at 1502000th time step. (Multimedia view) [URL: http://dx.doi.org/10.1063/1.4935482.6] [URL: http://dx.doi.org/10.1063/1.4935482.7] [URL: http://dx.doi.org/10.1063/1.4935482.8] [URL: http://dx.doi.org/10.1063/1.4935482.9] [URL: http://dx.doi.org/10.1063/1.4935482.10]](https://aipp.silverchair-cdn.com/aipp/content_public/journal/adv/5/11/10.1063_1.4935482/3/m_117113_1_f9.jpeg?Expires=1716405946&Signature=u4nN0LL4YYL-R9b3t3FzZg9PQscy5FulfcVUCgu5ZvfUCNBhIg7yzfCRcvTB~xJX0uzQmT2swYgg-SyzF9WP0P2-gG8Lfs1gfOw7-dZvmLb3M2RpWkxka~3Vngo9pw6AZ4bPcMMfZNgqp41XCUnKL8hwOLz2h9JPIXtSR0yv7fEmhyWRKsPu0gv4VSn9xrjkxx5NxFbYY8~xTs0TFNFIJhPTbJTzllzneALwMuCAHYiPx~28tiQpM9PdAZKVyHJtUdXOA-eKz-Uosbmyx5-891rFCgFNf-TqrEtVpTVZwGWYEFVjLij53Waa2YS~AlXDAJf30yw908VprzUS9SyOZQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The snapshots in the dynamic simulation of encapsulation of coronene molecules into the (11,11) SWCNT of the diameter 1.513 nm; (a) is at 0th time step, (b) at 231000th time step, (c) at 290000th time step, (d) at 732000th time step, (e) at 1100000th time step, (f) at 1108000th time step, (g) at 1492000th time step, and (h) at 1502000th time step. (Multimedia view) [URL: http://dx.doi.org/10.1063/1.4935482.6] [URL: http://dx.doi.org/10.1063/1.4935482.7] [URL: http://dx.doi.org/10.1063/1.4935482.8] [URL: http://dx.doi.org/10.1063/1.4935482.9] [URL: http://dx.doi.org/10.1063/1.4935482.10]
The snapshots in the dynamic simulation of encapsulation of coronene molecules into the (11,11) SWCNT of the diameter 1.513 nm; (a) is at 0th time step, (b) at 231000th time step, (c) at 290000th time step, (d) at 732000th time step, (e) at 1100000th time step, (f) at 1108000th time step, (g) at 1492000th time step, and (h) at 1502000th time step. (Multimedia view) [URL: http://dx.doi.org/10.1063/1.4935482.6] [URL: http://dx.doi.org/10.1063/1.4935482.7] [URL: http://dx.doi.org/10.1063/1.4935482.8] [URL: http://dx.doi.org/10.1063/1.4935482.9] [URL: http://dx.doi.org/10.1063/1.4935482.10]
In Fig. 8(b) for the (10,10) SWCNT of the diameter 1.375 nm, a coronene molecule keeping its plane parallel to the SWCNT axis enters into the SWCNT from the left end. This molecule moves to and fro within the SWCNT. In Fig. 8(c), one more molecule has entered the SWCNT. In Fig. 8(d), this molecule and the previously-encapsulated one make a pair of coronene molecules, which also moves to and fro within the SWCNT. After that, one more molecule enters the SWCNT, and the three molecules move together to and fro; no snapshots representing such a situation are selected in Fig. 8. The number of coronene molecules forming such a group gradually increases as the calculation proceeds (see Fig. 8(e)). After half of the SWCNT is filled with coronene molecules, there appear stacked coronene molecules which make an angle with the SWCNT axis (see Fig. 8(f)). When the SWCNT is completely filled with coronene molecules, the tilted stack of coronene molecules and the bilayer of the molecules parallel to the SWCNT axis coexist (see Fig. 8(g)).
Also for the (11,11) SWCNT of the diameter of 1.513 nm (see Fig. 9), coronene molecules enter into the SWCNT keeping their planes parallel to the SWCNT axis, however, tilted coronene stacks are likely to be created because single coronene molecule or a pair of coronene molecules can easily rotate around the axis perpendicular to the SWCNT axis. As shown in Fig. 9(d), three coronene molecules stand up and make an angle with the SWCNT axis after five coronene molecules have entered the SWCNT. Encapsulated coronene molecules successively stand up (see Fig. 9(e) and 9(f)). When the SWCNT is completely filled with coronene molecules, all the molecules stand up and make an angle with the SWCNT axis (see Fig. 9(g) and 9(h)). Consequently, the number of coronene molecules encapsulated in the (11,11) SWCNT becomes larger than the number of coronene molecules encapsulated in the (10,10) SWCNT.
Representative examples of the final snapshots of the dynamic encapsulation simulations are shown in Fig. 10, where only the five results for the (10,10), (14,7), (13,9), (18,3), and (12,11) SWCNTs in Tables I, II, and III are also presented as in Fig. 4. In Figs. 10(a), 10(c), 10(e), 10(g), and 10(i), where the side views are shown, the coronene molecules existing outside the SWCNT are not displayed. Two neighboring coronene molecules which stand up and make an angle with the SWCNT axis are picked up from the coronene molecules shown in each of the side views Figs. 10(a), 10(c), 10(e), 10(g), and 10(i), and their plan views are shown in the Figs. 10(b), 10(d), 10(f), 10(h), and 10(j), respectively.
Final snapshots of the encapsulated coronene molecules at 3000000th time step in the dynamic simulations. (a) and (b) are for the (10,10) SWCNT of the diameter 1.375 nm, (c) and (d) for the (14,7) SWCNT of the diameter 1.470 nm, (e) and (f) for the (13,9) SWCNT of the diameter 1.521 nm, (g) and (h) for the (18,3) SWCNT of the diameter 1.562 nm, and (i) and (j) for the (12,11) SWCNT of the diameter 1.582 nm. (a), (c), (e), (g), and (j) are side views. (b) is a plan view of the two neighboring coronene molecules at the right end of the six stacked coronene molecules in (a), (d) is a plan view of those near the center of the right-side coronene stack in (c), and (f), (h), and (j) are plan views of those near the centers of the coronene stacks in (e), (g), and (i), respectively.
Final snapshots of the encapsulated coronene molecules at 3000000th time step in the dynamic simulations. (a) and (b) are for the (10,10) SWCNT of the diameter 1.375 nm, (c) and (d) for the (14,7) SWCNT of the diameter 1.470 nm, (e) and (f) for the (13,9) SWCNT of the diameter 1.521 nm, (g) and (h) for the (18,3) SWCNT of the diameter 1.562 nm, and (i) and (j) for the (12,11) SWCNT of the diameter 1.582 nm. (a), (c), (e), (g), and (j) are side views. (b) is a plan view of the two neighboring coronene molecules at the right end of the six stacked coronene molecules in (a), (d) is a plan view of those near the center of the right-side coronene stack in (c), and (f), (h), and (j) are plan views of those near the centers of the coronene stacks in (e), (g), and (i), respectively.
If the morphologies at the final snapshots of all the dynamic simulations for the SWCNTs shown in Tables I, II, and III are examined, it is found both of the coronene molecules whose planes are parallel to the SWCNT axis and the ones making angles with the axis coexist for the SWCNT diameter in the rage of from 1.350 to 1.470 nm. For the diameter less than 1.411 nm, the number of the coronene molecules whose planes are parallel to the axis is larger than the number of the coronene molecules making angles with the axis (see Fig. 10(a), whereas, for the diameter larger than 1.411 nm, the situation is reversed (see Fig. 10(c)). For the SWCNT diameter in the range of from 1.508 nm to 1.606 nm, all coronene molecules stand up and stack in columnar shapes (see Figs. 10(e), 10(g), and 10(i)). For the SWCNT diameter equal to or larger than 1.650 nm, stacks of three coronene molecules whose planes are parallel to the SWCNT axis are observed in places, although the corresponding figure is not displayed.
Now let us see the plan views of the two neighboring coronene molecules. Figs. 10(b) and 10(d) reveal that the two coronene molecules accommodate themselves to the inner spaces of the thin SWCNTs by relative slidings under tilting. The sliding lengths are larger than those in Figs. 4(b) and 4(d) by about half of the size of the six-membered ring, and the stackings are not so similar to AB stack as the stacks shown in Figs. 4(b)) and 4(d). On the other hand, in Figs. 10(h) and 10(j), the two coronene molecules accommodate themselves to the inner spaces of the thick SWCNTs by relative rotations under no tilting. In Fig. 10(f), the two coronene molecules accommodate themselves by both relative sliding and rotation under tilting. Thus, it is confirmed that the SWCNT diameter-dependent stacking manner of the coronene molecules resulting from dynamics simulation is consistent with that resulting from CG energy minimization.
The tilt angles of the columnar stacks of coronene molecules are also calculated from the final snapshots of the dynamic simulations; the tilt angles are calculated only for the coronene molecules which stand up and make angles with the SWCNT axis. The result is shown with the squares in Fig. 5. The tendency that the tilt angle almost linearly decreases in the range of the SWCNT diameter down to 1.521 nm and is approximately constant above the SWCNT diameter 1.521 nm is the same as of the result by CG energy minimization. However, the tilt angles obtained by the dynamic simulation are larger than those obtained by CG energy minimization. It must be the effect of temperature. The dynamic simulations are executed under 700 K, whereas the results of conjugate-gradient minimization correspond with the results under 0 K. The temperature effect will be confirmed again by a rather simplified simulation in the next subsection III C.
Although the intermolecular distances and rotational angles relative to the neighboring molecules can be calculated also from the final configurations obtained by dynamic simulation, the obtained data vary too widely. Accordingly, averaging such data is meaningless.
C. Effects of coronene density, CNT length, and temperature
Firstly, to examine the effect of the density of coronene molecules, a coronene atmosphere of half the density of the atmosphere adopted in the dynamic simulations described in the previous subsection III B is prepared; the rarefied atmosphere consists of 52 coronene molecules in the cell.
The snapshots for the (17,0) SWCNTs of the diameter of 1.350 nm in the rarefied and dense atmospheres are shown in Figs. 11(a) and 11(b), respectively. In the dense atmosphere, the SWCNT is filled with coronene molecules already at 1915000th time step (see Fig. 11(b)), whereas in the rarefied atmosphere, the SWCNT is filled with coronene molecules just at 4811000th time step (see Fig. 11(a)). In both of Figs. 11(a) and 11(b), the encapsulated coronene molecules take the analogous morphologies where almost all the coronene molecules are aligned parallel to the SWCNT axis in bilayers, and a few coronene molecules stand up and make angles with the SWCNT axis.
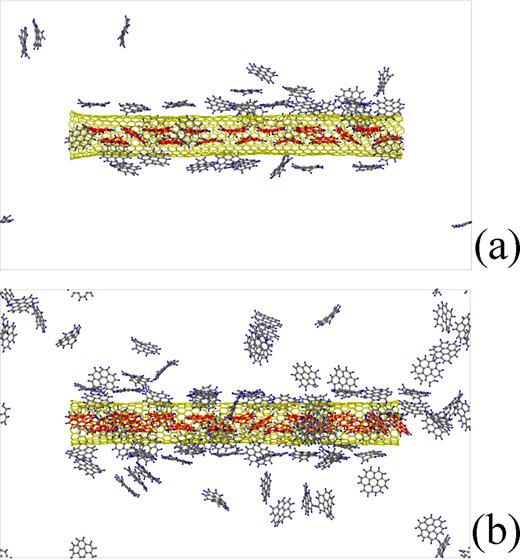
(a) is a snapshot at 4811000th time step of the dynamic simulation for the (17,0) SWCNT of the diameter 1.350 nm in the rarefied atmosphere, whereas (b) is a snapshot at 1915000th time step of the dynamic simulaton for the same SWCNT in the dense atmosphere. In both of the snapshots, the SWCNTs have just been filled with coronene molecules.
(a) is a snapshot at 4811000th time step of the dynamic simulation for the (17,0) SWCNT of the diameter 1.350 nm in the rarefied atmosphere, whereas (b) is a snapshot at 1915000th time step of the dynamic simulaton for the same SWCNT in the dense atmosphere. In both of the snapshots, the SWCNTs have just been filled with coronene molecules.
The snapshots for the (11,11) SWCNTs of the diameter of 1.513 nm in the rarefied and dense atmosphere are also shown in Figs. 12(a) and 12(b), respectively. As shown in Fig. 12(b), the SWCNT in the dense atmosphere has been filled with coronene molecules already at 1502000th time step. However, as shown in Fig. 12(a), the SWCNT in the rarefied atmosphere has not been filled with coronene molecules at 107th time steps yet. From Fig. 12(a), it is found that almost all the coronene molecules in the cell exist in the interior of or in the outer neighborhood of the SWCNT, and the number of the coronene molecules is not enough to fill the SWCNT. With the increase of the diameter of the SWCNT, the surface area of its side wall increases, and more coronene molecules swarm around and move along the surface of the SWCNT. Accordingly, more coronene molecules are necessary to fill the SWCNT. The encapsulated coronene molecules in Fig. 12(a) are on the way of making a columnar stack, and it is guessed the final configuration would be the same as shown in Fig. 12(b).

(a) is the final snapshot at 10000000th time step of the dynamic simulation for the (11,11) SWCNT of the diameter 1.513 nm in the rarefied atmosphere, whereas (b) is a snapshot at 1502000th time step of the dynamic simulation for the same SWCNT in the dense atmosphere. In (b), the SWCNT has just been filled with coronene molecules.
(a) is the final snapshot at 10000000th time step of the dynamic simulation for the (11,11) SWCNT of the diameter 1.513 nm in the rarefied atmosphere, whereas (b) is a snapshot at 1502000th time step of the dynamic simulation for the same SWCNT in the dense atmosphere. In (b), the SWCNT has just been filled with coronene molecules.
Secondly, in order to examine the effect of the SWCNT length, dynamic encapsulation simulations are performed for the (17,0), (10,10), and (11,11) SWCNTs of the length 300 Å. The snapshots at the final time step for these three SWCNTs are shown in Fig. 13, where the coronene molecules existing outside the SWCNTs are not displayed. The corresponding snapshot at the final time step for the (10,10) SWCNT of the length 100 Å is already shown in Fig. 10(a). In addition, the corresponding snapshots at the final time step for the (17,0) and (11,11) SWCNTs of the length of 100 Å are shown in Fig. 14(a) and 14(b), respectively. Comparison of these figures reveals that there is no difference between the fundamental features of the morphologies of coronene molecules encapsulated in the SWCNTs of the lengths 300 Å and 100 Å. In Figs. 13(a) and 14(a), almost all the coronene molecules are aligned parallel to the SWCNT axis, and one coronene molecule per 100 Å stands up and makes an angle with the SWCNT axis. In Figs. 13(b) and 10(a), the number of coronene molecules which stand up and make angles with the SWCNT axis increases, however is less than the number of coronene molecules whose planes are parallel to the SWCNT axis. In Figs. 13(c) and 14(b), all the coronene molecules stand up and make angles to the SWCNT axis. Consequently, it can be said the results obtained by dynamic simulation for the SWCNTs of the length 100 Å are reliable on investigating the morphology of the encapsulated coronene molecules.

The final snapshots of the dynamic simulation of encapsulation of coronene molecules into the SWCNTs of the length 300 Å. (a) is for the (17,0) SWCNT of the diameter 1.350 nm, (b) for the (10,10) SWCNT of the diameter 1.375 nm, and (c) for the (11,11) SWCNT of the diameter 1.513 nm.
The final snapshots of the dynamic simulation of encapsulation of coronene molecules into the SWCNTs of the length 300 Å. (a) is for the (17,0) SWCNT of the diameter 1.350 nm, (b) for the (10,10) SWCNT of the diameter 1.375 nm, and (c) for the (11,11) SWCNT of the diameter 1.513 nm.
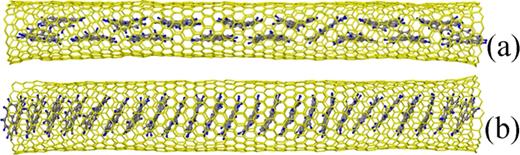
The final snapshots of the dynamic simulation of encapsulation of coronene molecules into the SWCNTs of the length 100 Å. (a) is for the (17,0) SWCNT of the diameter 1.350 nm, whereas (b) for the (11,11) SWCNT of the diameter 1.513 nm.
The final snapshots of the dynamic simulation of encapsulation of coronene molecules into the SWCNTs of the length 100 Å. (a) is for the (17,0) SWCNT of the diameter 1.350 nm, whereas (b) for the (11,11) SWCNT of the diameter 1.513 nm.
Finally, the effects of temperature are studied in the simplified MD simulation for ‘ready-made’ columnar stacks of coronene molecules in SWCNTs of the length 100 Å shown in Fig. 2(c). Raising the temperature from 1 K to 800 K, the system is equilibrated at every 100 K. The mean tilt angle and mean intermolecular distance are calculated from the last snapshot at each equilibrated temperature. The calculations are performed for (18,0), (14,7), (11,11), and (12,11) SWCNTs, the diameters of which are 1.429, 1.470, 1.513, and 1.582 nm, respectively. In Fig. 15, the mean tilt angle of the coronene molecules are plotted against temperature. With increasing temperature, the tilt angle gradually increases little by little. Such a tendency is consistent with the result already shown in Fig. 5 where the mean tilt angles obtained by dynamic simulation at 700 K are larger than those obtained by CG energy minimization. In addition, the values of the tilt angles for the four SWCNTs at 700 K in Fig. 15 almost agree with the corresponding values obtained by dynamic simulation in Fig. 5. Hence, it is expected that such rough estimations of the tilt angles at the other temperatures are also not so bad. In Fig. 16, the mean distance between coronene molecules is plotted against temperature. With increasing temperature, the distance almost linearly increases. The rough estimations of the mean intermolecular distances at different temperatures will supplement the incompleteness of the data shown in the subsection III B that the data of the intermolecular distances obtained by dynamic simulation vary too widely, and averaging them is meaningless. It should be mentioned that these tendencies of the temperature effects show qualitative agreement with the results obtained by Verberck et al.9
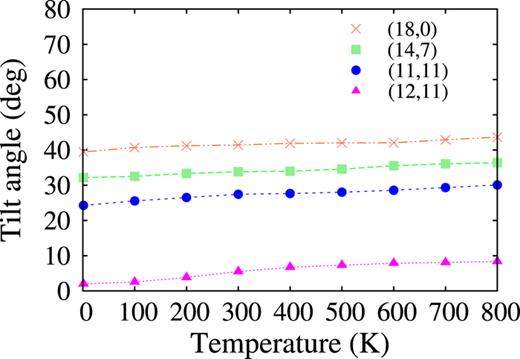
The mean tilt angles of the stacked coronene molecules in the final configurations obtained by the simplified MD simulation are plotted against temperature. The data represented by the crosses, squares, circles, and triangles are for the (18,0) SWCNT of the diameter 1.429 nm, (14,7) SWCNT of the diameter 1.470 nm, (11,11) SWCNT of the diameter 1.513 nm, and (12,11) of the diameter 1.582 nm, respectively.
The mean tilt angles of the stacked coronene molecules in the final configurations obtained by the simplified MD simulation are plotted against temperature. The data represented by the crosses, squares, circles, and triangles are for the (18,0) SWCNT of the diameter 1.429 nm, (14,7) SWCNT of the diameter 1.470 nm, (11,11) SWCNT of the diameter 1.513 nm, and (12,11) of the diameter 1.582 nm, respectively.

The mean intermolecular distances of the stacked coronene molecules in the final configurations obtained by the simplified MD simulation are plotted against temperature. The data represented by the crosses, squares, circles, and triangles are for the (18,0) SWCNT of the diameter 1.429 nm, (14,7) SWCNT of the diameter 1.470 nm, (11,11) SWCNT of the diameter 1.513 nm, and (12,11) of the diameter 1.582 nm, respectively.
The mean intermolecular distances of the stacked coronene molecules in the final configurations obtained by the simplified MD simulation are plotted against temperature. The data represented by the crosses, squares, circles, and triangles are for the (18,0) SWCNT of the diameter 1.429 nm, (14,7) SWCNT of the diameter 1.470 nm, (11,11) SWCNT of the diameter 1.513 nm, and (12,11) of the diameter 1.582 nm, respectively.
IV. CONCLUDING REMARKS
The morphology of stacked coronene molecules encapsulated in a SWCNT of the diameter range of from 1.35 to 1.69 nm was addressed by means of CG energy minimization and MD simulation. Both of the simulations yielded the same features of their morphology. Coronene molecules accommodate themselves to the inner space of a thin SWCNT by tilting against the radial direction of the SWCNT and by sliding relative to the neighboring molecules; they are likely to form a stack similar to AB stack. On the other hand, they accommodate themselves to the inner space of a thick SWCNT by rotating relative to the neighboring molecules under no tilting; they are likely to form a crossed stack. For a SWCNT of the intermediate diameters, they accommodate themselves by tilting, sliding, and rotating. For the SWCNT diameter less than or equal to 1.52 nm, the mean tilt angle of the stacked coronene molecules almost linearly decreases with increasing the diameter, whereas for the SWCNT diameter above 1.52nm, it is close to 0∘. Because the electronic structures of coronene-SWCNT hybrids can be modulated by changing the tilt angles,11 it is expected their electronic structures will be intendedly modulated by selecting the diameters of the encapsulating SWCNTs.
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI Grant Number 15K05674.

